
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular trap for a transient corannulene trianion†
Alexander V.
Zabula
ab,
Sarah N.
Spisak
a,
Alexander S.
Filatov
a,
Andrey Yu.
Rogachev
*c,
Rodolphe
Clérac
de and
Marina A.
Petrukhina
*a
aDepartment of Chemistry, University at Albany, State University of New York, Albany, NY 12222, USA. E-mail: mpetrukhina@albany.edu
bDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA 19104, USA
cDepartment of Chemistry, Illinois Institute of Technology, Chicago, IL 60616, USA. E-mail: arogache@iit.edu
dCNRS, Centre de Recherche Paul Pascal (CRPP), Pessac, F-33600, France
eUniversité de Bordeaux, Pessac, F-33600, France
First published on 17th December 2015
Abstract
The first structural characterization of the transient triply-reduced state of corannulene (C20H10) is accomplished. The X-ray crystallographic study reveals that the C20H10˙3− trianions, generated by corannulene reduction with metallic cesium, form a novel type of supramolecular sandwich-type assembly, [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+]. In the product, two triply-charged corannulene decks encapsulate a rectangle of four cesium ions with the external concave bowl cavities being filled by one cesium ion each. The structural investigation is augmented by in-depth theoretical calculations to provide insights into the geometrical features and electronic structure of this unique organometallic self-assembly.
Introduction
Multi-electron reduction of planar and non-planar polyaromatic hydrocarbons (PAHs), structures of the resulting carbanions as well as their supramolecular aggregation with alkali metal ions have been the focus of great attention in the last two decades.1–3 The discoveries of curved and bent π-conjugated molecules, such as fragments of fullerenes and nanotubes,4 further reinvigorated this field with the focus on aromaticity, reactivity, magnetism and electronic properties of the resulting charged carbon-rich species.5–7 Special interest in non-planar radicals with extended π-surfaces8 has arisen from their charge transport abilities,9 magnetic properties,10 interesting coupling pathways11 as well as prospective applications in organic microelectronics and energy storage.12 Considering a broad family of polycyclic hydrocarbons, monoanion-radicals are well-documented and have been characterized both in solution and solid state for a great number of polyaromatic scaffolds.13,14 In contrast, the number of isolated and structurally characterized trianion-radicals is limited. Only for C60-fullerene, the trianions are well-known,15 stemming from the triply degenerate nature of its low-lying unoccupied molecular orbital (LUMO) that can accept up to six electrons.16 For the extended family of PAHs, the trianions have been isolated and crystallographically characterized for large bisanthryl17 and decacyclene18 only. Overall, the controlled preparation of trianion-radicals of PAHs, which are often transient species on the way to more highly reduced carbanions, is challenging, while their electronic structures, properties, stability, and reactivity are especially intriguing.Bowl-shaped polyaromatic hydrocarbons, representing curved fragments of fullerenes, are known to readily uptake multiple electrons. For example, corannulene (C20H10, Scheme 1), which maps a 1/3 of the C60-fullerene surface, can acquire up to four electrons, owing to the doubly degenerate nature of its LUMO.19 For the family of corannulene anions, products of mono-,20 di-,20b and highly reduced tetraanions21 (C20H10n−, n = 1, 2, 4) have been recently isolated with different alkali metal counterions and characterized by X-ray crystallography. This allowed us to follow structural perturbations of the corannulene core upon addition of one, two, and four electrons. These studies also revealed a tendency of very electron-rich tetrareduced corannulene to form remarkable supramolecular products with the high number of encapsulated alkali metal ions, including unprecedented heterobimetallic combinations.22 This behavior clearly differentiates the corannulene tetraanion, C20H104−, bearing the record charge per carbon atom (one electron per five C-atoms vs. one electron per ten C-atoms in the C60-fullerene hexaanion, for comparison) from mono- and doubly-charged corannulene, which can be isolated in their “naked” forms.23 Notably, until now the triply-reduced corannulene remained missing in the above series of structurally characterized corannulene anions.
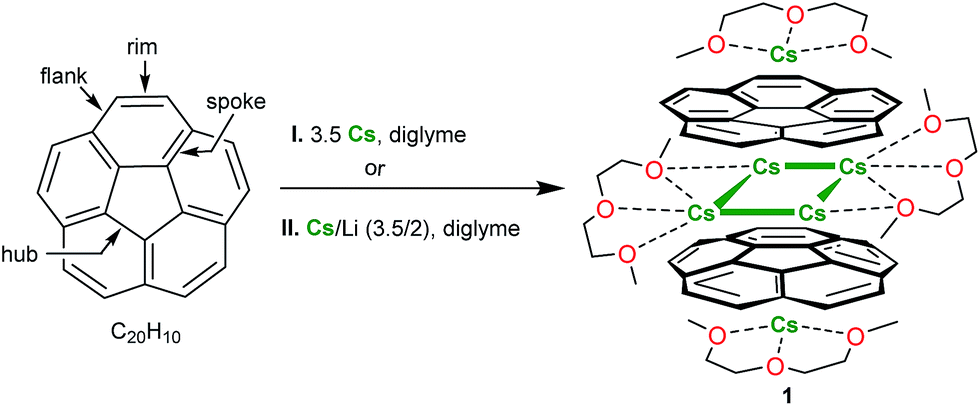 | ||
| Scheme 1 Preparation of 1. | ||
Previously, the paramagnetic C20H10˙3− radical was detected by ESR spectroscopy using the in situ reduction reaction of corannulene with lithium metal in THF.24 The ESR data provided the only evidence of existence of this transient species in solution, as the initially reported UV-vis spectroscopic data for the C20H10˙3− anion24a have been later re-assigned to the tetrareduced state of corannulene.25 The question whether the corannulene trianion engages in self-assembly with alkali metal ions or can exist in discrete form has remained open. Herein, we prove that the transient C20H10˙3− anion can be isolated in the solid state in the form of its cesium salt. We also reveal a remarkable supramolecular structure formed by C20H10˙3− anions with multiple cesium counterions based on single crystal X-ray diffraction study and provide full characterization of this novel organometallic product. Moreover, comprehensive theoretical evaluation of unique geometric and electronic features of the new sandwich-type assembly has been accomplished.
Results and discussion
Corannulene reacts with cesium metal (3.5 equiv.) in diglyme with the initial formation of an intense green solution characteristic of the monoanion, C20H10˙−.20 The subsequent fast reduction of the monoanion leads to the appearance of the bright purple dianion in the reaction mixture.26 The prolonged reaction time (up to 60 h) at ambient conditions resulted in the formation of the red-purple solution attributed to the C20H10˙3− trianion. The UV-vis spectrum of this reaction mixture exhibits a very broad absorbance band around 500 nm and a very intense band, λmax = 388 nm, characteristic of the C20H10˙3− anion (ESI, Fig. S2 and S3†). The latter absorbance maximum is hypsochromically shifted compared to the most intense band of C20H104− (λmax = 460 nm in diglyme21b and 429 nm in THF21a). Notably, no further reduction of C20H10˙3− to the C20H104− state was observed in this work even when an excess of metallic cesium was used over a very extended reaction time period (more than 2 months).We found that the target carbanion was very difficult to isolate in the crystalline form. Multiple initial attempts to crystallize corannulene trianion from the above systems led to the precipitation of oily or amorphous powders. Ultimately, we observed that the addition of dicyclohexano-18-crown-6 to the reaction mixture followed by its layering with hexanes resulted in the formation of several dark-red crystals of [Cs+3(diglyme)2(C20H103−)] (1) (Scheme 1). Surprisingly, crown ether is not incorporated into the final product although its presence in the reaction solution somehow facilitated crystallization. We later found that the same crystalline product 1 can also be obtained when a mixture of cesium and lithium metals (in a 3.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio with respect to corannulene) is used for the reduction reactions. Notably, the resulting crystals are very difficult to handle due to their extreme air- and moisture-sensitivity. Crystals of 1 are soluble in diglyme only and have very limited solubility in THF.
:2 ratio with respect to corannulene) is used for the reduction reactions. Notably, the resulting crystals are very difficult to handle due to their extreme air- and moisture-sensitivity. Crystals of 1 are soluble in diglyme only and have very limited solubility in THF.
The X-ray crystallographic study revealed that 1 crystallizes in the C2/c space group with the asymmetric unit consisting of one corannulene trianion, three cesium ions, and two diglyme molecules (Fig. 1A, see ESI† for more details). In the molecular structure residing on the inversion center, four cesium ions are sandwiched between two triply-reduced corannulene bowls to yield a novel type of supramolecular assembly, [(C20H103−)/4Cs+/(C20H103−)]2− (Fig. 1B). A similar arrangement of four cesium ions between two tetrareduced corannulene moieties has been suggested by theoretical calculations.27
 | ||
| Fig. 1 Asymmetric unit (A), sandwich view (B) and depiction of the solvated Cs+ ions within the sandwich, superimposed with the space filling model (C) for 1. | ||
It has been previously proven that the highly reduced corannulene tetraanions form the triple-decker supramolecular aggregates with lithium counterions, [(C20H104−)/5Li+/(C20H104−)]3−.21a,b In the latter, five small lithium ions are encapsulated between the convex faces of two corannulene anions (Fig. 2A). Based on earlier NMR investigations,28 it was speculated that such aggregation is not favored for C20H104− anions in the presence of the heavier congeners of lithium.
 | ||
| Fig. 2 Side and top views of [(C20H104−)/5Li+/(C20H104−)]3− (A) and [(C20H103−)/4Cs+/(C20H103−)]2− (B) based on their X-ray diffraction studies along with the optimized geometry of the Cs4-sandwich based on DFT calculations (C, exterior Cs ions are not shown). | ||
In contrast to tetrareduced corannulene, NMR spectroscopy could not be used for predicting the self-aggregation pattern of the trianion-radical, leaving single crystal X-ray diffraction as the only source of structural information in this case.
In 1, a convex-to-convex arrangement of two bowls is found with their shortest separation of 4.952(5) Å. However, in contrast to an almost ideally eclipsed bowl overlap in [(C20H104−)/5Li+/(C20H104−)]3−, the [(C20H103−)/4Cs+/(C20H103−)]2− aggregate exhibits a staggered conformation of two C20H10˙3− anions, which are slipped in respect to each other by 1.843(5) Å (Fig. 2B). The sandwiched Cs2, Cs2′, Cs3, and Cs3′ ions form a rectangle with the Cs⋯Cs separations of 4.181(4) and 4.995(4) Å. The Cs2 ions are bound to both anionic bowls with the Cs2⋯C interatomic distances of 3.275(5)–3.740(5) Å (Cs2⋯C6(centroid) 3.229(5) and 3.265(5) Å). The Cs3 ions exhibit noticeably shorter Cs3⋯C distances of 3.231(5)–3.775(5) Å (Cs3⋯C6(centroid) is 3.188(5) Å). The binding of the sandwiched alkali metal ions and corannulene trianions can be best described as the electrostatic gluing of the Cs44+-unit between two highly-charged polyaromatic surfaces, as shown below by theoretical calculations. The coordination spheres of the encapsulated Cs2 and Cs3 ions are completed by the chelating diglyme molecules (Fig. 1C). The corresponding Cs⋯O bond lengths (3.069(4)–3.466(4) Å) are close to those previously reported for the salts of aromatic ligands with cesium ions solvated by O-donors.20a,29
The extraneous Cs1 cation occupies the concave cavity of the anionic corannulene dish and is located above its five-membered ring with the Cs⋯Chub distances measured at 3.116(5)–3.328(5) Å (Fig. 1B). The concave placement of metals into π-bowls is rare.30 For corannulene, the first endo-bound complex was reported in 2011 for the cesium salt of mono-anion, [Cs+(18-crown-6)(C20H10−)].20a
The preferential endo-binding of cesium ions to the corannulene bowl has been later observed in several other products.26,31 In 1, the endo Cs⋯Chub contacts with the triply-charged bowl are much shorter than those with C20H10˙− in the above complex (3.424(3)–3.573(3) Å).20a In contrast to a very symmetrical cesium coordination in [Cs+(18-crown-6)(C20H10−)], the Cs1 ion in 1 is side-shifted toward the benzene rings of the corannulene trianion. The resulting intramolecular contacts between Cs1 and peripheral C-atoms of the bowl range from 3.275(5) to 3.662(5) Å. In addition, this Cs1 ion also shows binding (Cs⋯C 3.542(5)–3.638(5) Å) to the exterior of C20H10˙3− from the neighboring unit and shares a diglyme molecule with the sandwiched Cs2 cation. The resulting Cs1⋯C and Cs1(2)⋯O interactions lead to the formation of a hybrid 2D polymeric network in the solid state of 1 (Fig. 3).
 | ||
| Fig. 3 Fragment of a 2D polymeric network in 1. Corannulene trianions and cesium cations are depicted using the space-filling model. | ||
Interestingly, the acquisition of three electrons by C20H10 and aggregation with multiple cesium ions does not induce any significant flattening of the resulting trianion. The bowl depth of C20H10˙3− in 1 (0.850(7) Å) remains almost unchanged compared to that in C20H100 (0.875(2) Å).32 Although the addition of the first and second electrons to C20H10 is known to cause only minor core flattening (to 0.850(3) and 0.811(3) Å, respectively),20,23,26 a significant bowl depth reduction is observed upon four-electron acquisition (0.283(5)/0.329(5) Å,21Table 1). Thus, the carbon framework of C20H10˙3−, being more curved than those of the dianion and tetraanion of corannulene, stands out from the previously expected trend of a gradual flattening of the bowl upon consecutive electron addition.33 It is worth mentioning here that the theoretical value of the C20H10˙3− bowl depth (0.41 Å) calculated earlier34 is twice smaller than the experimentally determined value found in 1.
| n = | 0a | 1b | 2c | 3 (in 1) | 4d |
|---|---|---|---|---|---|
| C20H100 | C20H10˙− | C20H102− | C20H10˙3− | C20H104− | |
| a According to literature.32 b For [Li+(DME)3][C20H10−].23 c For [Li+(diglyme)2]2[C20H102−].23 d For the naked [Li+5(C20H104−)2]3− sandwich.21a | |||||
| Hub | 1.411(2)–1.417(2) | 1.389(5)–1.425(5) | 1.390(3)–1.427(3) | 1.401(7)–1.429(7) | 1.391(5)–1.403(5) |
| Spoke | 1.376(2)–1.381(2) | 1.392(5)–1.417(5) | 1.400(3)–1.418(3) | 1.418(8)–1.443(7) | 1.424(5)–1.432(5) |
| Flank | 1.441(2)–1.450(2) | 1.388(5)–1.462(5) | 1.404(3)–1.475(3) | 1.421(7)–1.439(7) | 1.429(5)–1.443(5) |
| Rim | 1.377(2)–1.387(2) | 1.396(5)–1.453(6) | 1.375(3)–1.435(3) | 1.416(7)–1.438(8) | 1.453(5)–1.462(5) |
| Bowl depth | 0.875(2) | 0.841(5) | 0.811(3) | 0.850(7) | 0.283(5)/0.329(5) |
The structural characterization of the previously missing corannulene trianion allowed us to compare for the first time the induced geometrical changes of C20H10˙3− with those of C20H102− and C20H104− (Table 1). The central hub C–C bond lengths in trianion (1.401(7)–1.429(7) Å) are comparable to those measured in C20H102− (1.390(3)–1.427(3) Å)20,26 and slightly elongated than in C20H104− (1.391(5)–1.403(5) Å).21 The spoke C–C bond lengths in C20H10n− (n = 2–4) species are almost equidistant. The rim C–C bonds of C20H10˙3− (1.416(7)–1.438(8) Å) are notably shorter than in C20H104− (1.453(5)–1.462(5) Å). In general, all C–C bonds of C20H10˙3− demonstrate some equalization of the bond lengths compared to corannulene, its mono- and di-anions.
Computational studies
Due to extreme sensitivity of crystalline product 1 toward traces of water and/or oxygen, many important questions regarding its electronic structure and even its ground state remained unanswered. In order to provide insights into the geometry and electronic structure of the title product, computational modeling was performed at the PBE0/def2-TZVP+ECP(Cs)//cc-pVDZ(C,H,O) level of theory (see the ESI† for details).Geometry
The discrete trianionic species, C20H10˙3−, is expected to have a doublet ground state with one unpaired electron. In the sandwich-type aggregate (Fig. 2C), two such bowl-shaped radicals can exhibit ferromagnetic or antiferromagnetic coupling (so-called open-shell singlet or OS-singlet state), or alternatively show no coupling. First, the model system [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+] was optimized in its triplet state. The direct comparison of this discrete calculated structure, [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+], with the extended solid state structure of 1 immediately revealed a significantly smaller shift along the sandwich axis in the former (0.453 vs. 1.843(5) Å or, in terms of the distance between the bowl centroids: 4.89 Å vs. 5.28 Å, respectively). As a result, the encapsulated Cs4-unit exhibits an essentially square geometry in the calculated structure with the Cs⋯Cs contacts of 4.668 and 4.805 Å (Δ = 0.137 Å). For comparison, the corresponding Cs⋯Cs distances in the X-ray crystal structure differ by ca. 0.814 Å. The observed differences between the geometries of calculated and experimental sandwich structures illustrate the importance of additional intermolecular interactions existing in the 2D network in the solid state of 1. This is the first time when the role of intermolecular interactions at the sandwich exterior is clearly observed, as in previous cases such effects were found negligible.21 The significant deviation of the theoretical model from the experimental X-ray crystal structure prompted us to consider four different models, namely (i) the simplest fully-optimized [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+] model (1-small), (ii) the same model, but with the core structure taken from the X-ray experiment and kept unchanged, while positions of hydrogen atoms were optimized (1H-small), (iii) the fully-optimized model, in which all solvent molecules were considered explicitly (1-full), and (iv) the same system as in 1-full, but with only hydrogen atom positions been optimized (1H-full), while the rest was taken from the crystal structure and kept frozen. Selecting the above four models (further details and all model structures are provided in the ESI†), we planned to evaluate the influence of coordinated solvent molecules and crystal packing on the ground state and electronic structure of the supramolecular sandwich-type aggregates formed by C20H10˙3− with cesium ions.Indeed, the fully relaxed geometry of a system with all coordinated solvent molecules (1-full) shows much closer resemblance with that of the X-ray crystal structure of 1. For instance, the distance between the bowl centroids in 1-full is equal to 5.38 Å and that is close to the experimental value of 5.28 Å. The difference in Cs⋯Cs contacts was found to be ca. 0.8 Å, which is essentially the same as in the experimental X-ray sandwich structure.
Notably, the calculated bowl depth values of corannulene trianion in the discrete model sandwich complexes with cesium ions are only slightly smaller than the experimental value (0.832 Å in 1-small and 0.845 Å in 1-full vs. 0.850(7) Å, respectively). This illustrates good approximation provided by the selected level of DFT calculations but also raises questions about the reasons for such unexpectedly high curvature of C20H10˙3− in the [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+] type of sandwiches. We have found that replacement of the concave bound Cs ions by much smaller Li ions to form the [Li+//(C20H103−)/4Cs+/(C20H103−)//Li+] sandwich results only in minor flattening of the bowl depth (0.775 Å), thus indicating that the effect of the outside cations is rather weak and the size of the sandwiched belt comprised of four large cesium ions may be the main reason for the observed curvature of corannulene trianion.
Ground state
For the next step, we looked into the ground state of this unique sandwich-type assembly. Both abovementioned possible states, triplet and open-shell singlet, were examined. Broken-symmetry DFT (BS-PBE0 in our case) solution reveals small preference of the triplet state for the 1H-small (J = +3.84 cm−1), 1-full (+2.75 cm−1), and 1H-full (+0.54 cm−1) models, whereas for 1-small the OS-singlet state was found to be slightly lower in energy (−2.39 cm−1). However, the accuracy of these calculations does not allow an unambiguous conclusion about the actual ground state to be made. Therefore, we turned to the highly accurate multireference Møller–Plesset perturbation theory of the second order (MRMP2). The active space for the reference CASSCF wavefunction included six electrons over four orbitals (CASSCF(6,4) approach, see the ESI† for details). Calculations performed at the MRMP2 level revealed that the energy gap between the OS-singlet and triplet states is only −0.61 cm−1 for 1-small and −0.02 cm−1 for 1H-small. Subsequent extension of the active space to 14 electrons and 8 orbitals, CASSCF(14,8), resulted in a very similar outcome (+0.002 cm−1 and −0.01 cm−1, respectively). These findings revealed the absence of any significant magnetic coupling between the two C20H10˙3− bowls within the triple-decker sandwich. It now could explain why the shift of two triply-reduced corannulene decks in respect to each other does not lead to changes in the nature of the ground state of the sandwich.Notably, for this large and complex system the BS-DFT approach gives reasonably good results, correlating well with the more accurate MRPT2 method, and thus it can be considered as computationally “cheap” alternative to usually more accurate but significantly more demanding multireference approaches. Although the use of the multireference technique for full models (1-full and 1H-full) is not feasible, we believe that the trend (no magnetic coupling between bowls as defined by BS-DFT method) is still valid and the main conclusion remains unchanged.
Electronic structure
Based on our previous findings, only triplet electronic configuration is considered hereafter for the sandwich aggregates. To begin with, the direct comparison of the electronic structure of neutral corannulene (singlet ground state) with the “naked” triply-reduced corannulene species was performed. In the discrete C20H10˙3− anion, one unpaired electron is delocalized over the bowl core in accordance with topology of LUMO+1 of the neutral C20H10 bowl, thus making the curved polyaromatic surface to be uniformly more negatively charged (Fig. 4). | ||
| Fig. 4 Molecular electrostatic potentials (MEPs) of the neutral C20H10 molecule (left) and discrete C20H10˙3− anion (right). | ||
Comparison of charge distribution in the “naked” C20H10˙3− bowl with that in the supramolecular sandwich revealed a notable redistribution of electron density or polarization of the corannulene trianions in the latter. Specifically, the interior part of the bowl (hub C-atoms) becomes more negatively charged, whereas the exterior part (rim C-atoms) appears to show negative charge depletion. For instance, the calculated atomic charges of the hub C-atoms range from −0.134 to −0.176 and from −0.144 to −0.171 for models 1-small and 1H-small, respectively, vs. −0.10 charges in the discrete C20H10˙3− species. This effect can naturally be assigned to the presence of the positively charged belt of cesium cations jammed between two triply-reduced corannulene bowls. Accounting for coordinated solvent molecules resulted in a slightly less pronounced redistribution of the negative charge (−0.126 to −0.153 and −0.126 to −0.151 in 1-full and 1H-full models). These observations indicate that coordination of solvent molecules by metal cations, albeit influencing the geometry, does not significantly disturb the electronic structure of the supramolecular aggregate.
Despite the redistribution of atomic charges when going from the “naked” C20H10˙3− species to [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+] aggregate, the spin density in the latter remains essentially the same (Fig. 5), showing the presence of two uncoupled corannulene trianion-radicals in the system. Moreover, the topology of spin density is not notably influenced by the presence of solvent molecules and/or by crystal packing effects (as shown by comparison of models 1H-small, 1-full and 1H-full, Fig. 5). This finding is in complete agreement with the previous conclusion about ground state of such supramolecular aggregates.
 | ||
| Fig. 5 Spin density distribution (0.002 a.u. isosurface) for the discrete C20H10˙3− anion (top left), 1H-small model (top right), and 1-full and 1H-full models (bottom). | ||
Supramolecular assemblies formed by corannulene trianion vs. tetraanion
This work revealed for the first time an ability of C20H10˙3− to form sandwich-type supramolecular assembly with alkali metals, allowing us to provide the direct comparison of electronic structures and bonding in the remarkable organometallic aggregates formed by trianion vs. tetraanion of corannulene. The highly reduced C20H104− anion was previously shown to afford triple-decker supramolecular assemblies with encapsulated belts of alkali metal cations, several of which were crystallographically characterized and their core electronic structures were probed by theoretical methods.21a,b Notably, the C20H104− anions in such sandwich products were found to be strongly coupled with a singlet ground state, showing significant delocalization of corresponding molecular orbitals (MOs) between the two polyaromatic bowls. Moreover, it was found that such coupling is responsible for the record 7Li-NMR negative chemical shift observed for the internally encapsulated lithium cation in the heterobimetallic alkali metal sandwiches.22 In contrast, the aggregates formed by C20H10˙3− show no coupling between the corannulene moieties, as confirmed by the high-level theoretical modeling in this study. In this regard, a direct comparison of these two types of supramolecular sandwich-type assemblies should be interesting and informative.Careful checking of MOs in [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+] (all models) indeed revealed the absence of any orbital responsible for the coupling between the two triply-reduced corannulene bowls, in contrast to the coupling previously found in the triple-decker sandwiches formed by the tetraanions.
Subsequent analysis of charge distribution in 1H-small and 1-small models and their comparison with a sandwich formed by C20H104− anions, [Cs+//(C20H104−)/5Cs+/(C20H104−)//Cs+]− (14−-small), revealed that the interior part of the latter aggregate is significantly more negatively charged with a very narrow range of atomic charges ranging from −0.179 to −0.180 for hub C-atoms. Similar equalization of atomic charges was also observed for rim C-atoms (from −0.387 to −0.388) as well as for spoke ones (from −0.182 to −0.183), which can be attributed to the electron density sharing due to coupling between two tetrareduced corannulene bowls. In the [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+] systems, such equalization was not found and the range of atomic charges for carbon atoms of the same type is notably wider. This observation is illustrated graphically in Fig. 6, which unambiguously shows that non-compensated negative charge is still localized on the bowl-shaped fragments in [Cs+//(C20H103−)/4Cs+/(C20H103−)//Cs+] aggregates. Interestingly, both models, 1-small and 1H-small, show the same features in MEP maps, again confirming the absence of notable coupling between the C20H10˙3− bowls.
 | ||
| Fig. 6 Molecular electrostatic potentials (MEPs) for the 1H-small (left), 1-small (center), and 14−-small (right) models. | ||
At the same time, the NBO charge distribution analysis revealed the positive charge of cesium centers to be very close to 1 (average value for four sandwiched cations is +0.92 and for two exterior ions is +0.96) in all considered Cs4-sandwich models. Interestingly, the charges of interior cesium ions in the Cs5-sandwich (14−-small model, calculated at the same level of theory) were also found to be +0.93. The total charge of the computed Cs5-sandwich of −3 also shows essentially no charge transfer or charge delocalization to/from the encapsulated cesium ions. Thus, the positively charged belt of alkali metal cations plays the role of “electrostatic glue” in both types of sandwich aggregates. The main difference between the trianion- and tetraanion-based supramolecular products is the coupling of two negatively charged corannulene decks, which is not observed in the former and found to be significant in the latter.
In order to evaluate the coupling and thus to provide further insights into the electronic structure and bonding in these unusual supramolecular systems, the Energy Decomposition Analysis (EDA) has been performed. The results are summarized in Table 2.
| Parameters | 1-small | 1-full | 1 4−-small |
|---|---|---|---|
| DEint | −942.57 | −786.63 | −1536.03 |
| DEelstat | −853.45 | −760.52 | −1452.97 |
| DEorb | −241.89 | −200.84 | −341.41 |
| DEPauli | +152.77 | +174.73 | +258.35 |
In the EDA analysis we used the following fragmentation scheme. For 1-small, three interacting fragments were considered, namely two [(Cs)(C20H10)]2− and one [Cs4]4+ species. For 1-full model, all metal cations (including the external Cs+ ions as well as ions jammed between two bowls) were coordinated by solvent molecules, similar to the X-ray crystal structure. In the case of 14−-small, these three fragments included two [(Cs)(C20H10)]3− and one [Cs5]5+ species. Such fragmentation allows one to tentatively evaluate interactions in the supramolecular aggregates as well as to get estimates of coupling between the negatively charged bowls. As shown in Table 2, the electrostatic contribution is dramatically greater in 14−-small system in comparison with that in 1-small and/or 1-full ones. This result should be expected due to the difference in charges of bowl-shaped fragments (4- vs. 3-) and the number of Cs+ ions sandwiched in-between. At the same time, the orbital term, which is usually interpreted as a covalent contribution, is also greater by ca. 100 kcal mol−1 in 14−-small system. This large difference can be naturally attributed to the presence of a notable coupling between the two polyaromatic bowls in the latter. Altogether, these contributions (DEelstat and DEorb) make supramolecular systems based on tetrareduced corannulene significantly more stable than those based on C20H10˙3− anions. Note, this general trend is not altered even with some increase in Pauli repulsion in the same direction (DEPauli term in Table 2). This conclusion is in perfect agreement with experimental findings of this work as well as with previous studies.
Conclusions
The triply-reduced corannulene, that was previously missing in the family of consecutively generated negatively charged C20H10n− anions (n = 1–4), has now been isolated as the crystalline cesium salt, [Cs+3(diglyme)2(C20H103−)]. The X-ray crystallographic characterization of the product demonstrated that corannulene trianions form a novel supramolecular aggregate with four large cesium ions sandwiched between two bowls in [(C20H103−)/4Cs+/(C20H103−)]2−. The double negative charge of the resulting triple-decker sandwich is compensated by two extraneous Cs+ cations that fill the concave cavities of both corannulene bowls. This supramolecular trapping of triply-reduced corannulene in the “cesium-sealed” self-assembly seems required in order to catch this transient species, as multiple previous attempts to isolate C20H10˙3− with small lithium ions have been unsuccessful so far.This first crystallographic characterization of the C20H10˙3− anion allowed us to follow the effect of adding three electrons to a π-bowl and to provide structural comparison for the whole series of successively generated C20H10n− anions.
The isolation of the crystalline product 1 also provided the first reliable UV-vis spectroscopic characterization of C20H10˙3− based on comparison of the spectra for dissolved crystals with the in situ generated trianion. These data can now be used for the detection of the triply-reduced state of corannulene in solutions. Finally, these new results expand the unique organometallic and supramolecular chemistry of the highly charged bowl-shaped polyaromatic carbanions.21,22,35 For the first time, we show that the triply-reduced corannulene also has an ability to stabilize a high-nuclearity alkali metal layer, as previously observed for the very electron-rich corannulene tetraanions.
Acknowledgements
Financial support of this work from the National Science Foundation (CHE-1212441 and MRI-1337594) and ACS PRF is gratefully acknowledged by M. A. P., while A. Yu. R. greatly acknowledges support from the Illinois Institute of Technology (IIT) through startup funding. We thank the University at Albany for supporting the X-ray Center, Dr A. A. Granovsky and Firefly team for providing the developing version of the Firefly program package, Dr V. M. Grigoryants for ESR measurements, and the International Centre for Diffraction Data (ICDD) for the Ludo Frevel Crystallography Scholarship to S. N. S.References
- (a) M. Rabinovitz, I. Willner and A. Minsky, Acc. Chem. Res., 1983, 16, 298–304 CrossRef CAS; (b) M. Rabinovitz, Top. Curr. Chem., 1988, 146, 99–169 CrossRef CAS; (c) R. Benshafrut, E. Shabtai, M. Rabinovitz and L. T. Scott, Eur. J. Org. Chem., 2000, 1091–1106 CrossRef CAS.
- (a) J. D. Smith, Adv. Organomet. Chem., 1999, 43, 267–348 CrossRef; (b) A. V. Zabula and M. A. Petrukhina, Adv. Organomet. Chem., 2013, 61, 375–462 CrossRef CAS.
- D. Eisenberg and R. Shenhar, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 527–545 CrossRef.
- Fragments of Fullerenes and Carbon Nanotubes: Designed Synthesis, Unusual Reactions, and Coordination Chemistry, ed. M. A. Petrukhina and L. T. Scott, John Wiley & Sons, New Jersey, 2012, p. 413 Search PubMed.
- I. Hirosawa, K. Prassides, J. Mizuki, K. Tanigaki, M. Gevaert, A. Lappas and J. K. Cockcroft, Science, 1994, 264, 1294–1297 CAS.
- S. Aoyagi, Y. Sado, E. Nishibori, H. Sawa, H. Okada, H. Tobita, Y. Kasama, R. Kitaura and H. Shinohara, Angew. Chem., Int. Ed., 2012, 51, 3377–3381 CrossRef CAS PubMed.
- K. Y. Amsharov, Y. Krämer and M. Jansen, Angew. Chem., Int. Ed., 2011, 50, 11640–11643 CrossRef CAS PubMed.
- (a) Y. Morita, A. Ueda, S. Nishida, K. Fukui, T. Ise, D. Shiomi, K. Sato, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2008, 47, 2035–2038 CrossRef CAS PubMed; (b) S. Nishida, Y. Morita, A. Ueda, T. Kobayashi, K. Fukui, K. Ogasawara, K. Sato, T. Takui and L. Nakasuji, J. Am. Chem. Soc., 2008, 130, 14954–14955 CrossRef CAS PubMed; (c) A. Ueda, S. Nishida, K. Fukui, T. Ise, D. Shiomi, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Angew. Chem., Int. Ed., 2010, 49, 1678–1682 CrossRef CAS PubMed; (d) A. Ueda, K. Ogasawara, S. Nishida, T. Ise, T. Yoshino, S. Nakazawa, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Angew. Chem., Int. Ed., 2010, 49, 6333–6337 CrossRef CAS PubMed.
- (a) A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193–1202 CrossRef CAS PubMed; (b) K. Shi, T. Lei, X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. Sci., 2014, 5, 1041–1045 RSC; (c) S. Sanyal, A. K. Manna and S. K. Pati, ChemPhysChem, 2014, 15, 885–893 CrossRef CAS PubMed; (d) B.-T. Wang, M. A. Petrukhina and E. R. Margine, Carbon, 2015, 94, 174–180 CrossRef CAS.
- (a) Y. Takabayashi, A. Y. Ganin, M. J. Rosseinsky and K. Prassides, Chem. Commun., 2007, 870–872 RSC; (b) D. Arcon, A. Y. Ganin, Y. Takabayashi, M. J. Rosseinsky and K. Prassides, Chem. Mater., 2008, 20, 4391–4397 CrossRef CAS; (c) A. Y. Ganin, Y. Takabayashi, P. Jeglic, D. Arcon, A. Potocnik, P. J. Baker, Y. Ohishi, M. T. McDonald, M. D. Tzirakis, A. McLennan, G. R. Darling, M. Takata, M. J. Rosseinsky and K. Prassides, Nature, 2010, 466, 221–225 CrossRef CAS PubMed; (d) N. V. Kozhemyakina, K. Y. Amsharov, J. Nuss and M. Jansen, Chem.–Eur. J., 2011, 17, 1798–1805 CrossRef CAS PubMed; (e) D. V. Konarev, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, New J. Chem., 2011, 35, 1829–1835 RSC.
- (a) T. Kato and T. Yamabe, J. Chem. Phys., 2002, 117, 2324–2331 CrossRef CAS; (b) I. Aprahamian, R. E. Hoffman, T. Sheradsky, D. V. Preda, M. Bancu, L. T. Scott and M. Rabinovitz, Angew. Chem., Int. Ed., 2002, 41, 1712–1715 CrossRef CAS; (c) N. V. Kozhemyakina, K. Yu. Amsharov, J. Nuss and M. Jansen, Chem.–Eur. J., 2011, 17, 1798–1805 CrossRef CAS PubMed; (d) H. Phan, K. Lekin, S. M. Winter, R. T. Oakley and M. Shatruk, J. Am. Chem. Soc., 2013, 135, 15674–15677 CrossRef CAS PubMed; (e) Z. Mou, K. Uchida, T. Kubo and M. Kertesz, J. Am. Chem. Soc., 2014, 136, 18009–18022 CrossRef CAS PubMed; (f) M. Yamada, H. Kurihara, M. Suzuki, M. Saito, Z. Slanina, F. Uhlik, T. Aizawa, T. Kato, M. M. Olmstead, A. L. Balch, Y. Maeda, S. Nagase, X. Lu and T. Akasaka, J. Am. Chem. Soc., 2015, 137, 232–238 CrossRef CAS PubMed.
- (a) J. P. Lemmon, Nature, 2015, 525, 447–449 CrossRef CAS PubMed; (b) K. E. Moore, D. D. Tune and B. S. Flavel, Adv. Mater., 2015, 27, 3105–3137 CrossRef CAS PubMed; (c) F. Meng, W. Lu, Q. Li, J.-H. Byun, Y. Oh and T.-W. Chou, Adv. Mater., 2015, 27, 5113–5131 CrossRef CAS PubMed.
- (a) H. Bock, C. Arad, C. Näther and Z. Havlas, J. Chem. Soc., Chem. Commun., 1995, 2393–2394 RSC; (b) C. Näther, H. Bock and R. F. C. Claridge, Helv. Chim. Acta, 1996, 79, 84–91 CrossRef; (c) H. Bock, C. Arad and C. Näther, J. Organomet. Chem., 1996, 520, 1–13 CrossRef CAS; (d) H. Bock, T. Hauck and C. Näther, Organometallics, 1996, 15, 1527–1529 CrossRef CAS; (e) C. Näther, H. Bock, Z. Havlas and T. Hauck, Organometallics, 1998, 17, 4707–4715 CrossRef; (f) H. Bock, Z. Havlas, K. Gharagozloo-Hubmann and M. Sievert, Angew. Chem., Int. Ed., 1999, 38, 2240–2243 CrossRef CAS; (g) H. Bock, K. Gharagozloo-Hubmann, M. Sievert, T. Prisner and Z. Havlas, Nature, 2000, 404, 267–269 CrossRef CAS PubMed; (h) H. Bock, Z. Halvas, K. Gharagozloo-Hubmann, S. Holl and M. Sievert, Angew. Chem., Int. Ed., 2003, 42, 4385–4389 CrossRef CAS PubMed.
- (a) S. Neander, U. Behrens and F. Olbrich, J. Organomet. Chem., 2000, 604, 59–67 CrossRef CAS; (b) S. V. Rosokha and J. K. Kochi, J. Org. Chem., 2006, 71, 9357–9365 CrossRef CAS PubMed; (c) A. Jaeanschke, F. Olbrich and U. Behrens, Z. Anorg. Allg. Chem., 2009, 635, 2550–2557 Search PubMed.
- (a) C. Janiak, S. Mühle, H. Hemling and K. Köhler, Polyhedron, 1996, 15, 1559–1563 CrossRef CAS; (b) T. F. Fässler, R. Hoffmann, S. Hoffmann and M. Wörle, Angew. Chem., Int. Ed., 2000, 39, 2091–2094 CrossRef; (c) M. B. Boeddinghaus, M. Salzinger and T. F. Fässler, Chem.–Eur. J., 2009, 15, 3261–3267 CrossRef CAS PubMed.
- Q. Xie, E. Pérez-Cordero and L. Echegoyen, J. Am. Chem. Soc., 1992, 114, 3978–3980 CrossRef CAS.
- H. Bock, Z. Havlas, D. Hess and C. Näther, Angew. Chem., Int. Ed., 1998, 37, 502–504 CrossRef CAS.
- H. Bock, K. Gharagozloo-Hubmann, S. Holl and M. Sievert, Z. Naturforsch., B: J. Chem. Sci., 2000, 55, 1163–1178 CAS.
- A. Ayalon, M. Rabinovitz, P.-C. Cheng and L. T. Scott, Angew. Chem., Int. Ed. Engl., 1992, 31, 1636–1637 CrossRef.
- (a) S. N. Spisak, A. V. Zabula, A. S. Filatov, A. Yu. Rogachev and M. A. Petrukhina, Angew. Chem., Int. Ed., 2011, 50, 8090–8094 CrossRef CAS PubMed; (b) A. V. Zabula, S. N. Spisak, A. S. Filatov, V. M. Grigoryants and M. A. Petrukhina, Chem.–Eur. J., 2012, 18, 6476–6484 CrossRef CAS PubMed.
- (a) A. V. Zabula, A. S. Filatov, S. N. Spisak, A. Yu. Rogachev and M. A. Petrukhina, Science, 2011, 333, 1008–1011 CrossRef CAS PubMed; (b) A. V. Zabula, S. N. Spisak, A. S. Filatov and M. A. Petrukhina, Organometallics, 2012, 31, 5541–5545 CrossRef CAS; (c) A. V. Zabula, S. N. Spisak, A. S. Filatov and M. A. Petrukhina, Angew. Chem., Int. Ed., 2012, 51, 12194–12198 CrossRef CAS PubMed.
- (a) A. S. Filatov, A. V. Zabula, S. N. Spisak, A. Yu. Rogachev and M. A. Petrukhina, Angew. Chem., Int. Ed., 2014, 53, 140–145 CrossRef CAS PubMed; (b) A. S. Filatov, S. N. Spisak, A. V. Zabula, J. McNeely, A. Yu. Rogachev and M. A. Petrukhina, Chem. Sci., 2015, 6, 1959–1966 RSC.
- S. N. Spisak, A. V. Zabula, M. V. Ferguson, A. S. Filatov and M. A. Petrukhina, Organometallics, 2013, 32, 538–543 CrossRef CAS.
- (a) M. Baumgarten, J. L. Gherghel, M. Wagner, A. Weitz, M. Rabinovitz, P.-C. Cheng and L. T. Scott, J. Am. Chem. Soc., 1995, 117, 6254–6257 CrossRef CAS; (b) G. Zilber, V. Rozenshtein, P.-C. Cheng, L. T. Scott, M. Rabinovitz and H. Levanon, J. Am. Chem. Soc., 1995, 117, 10720–10725 CrossRef CAS.
- R. Shenhar, I. Willner, D. V. Preda, L. T. Scott and M. Rabinovitz, J. Phys. Chem. A, 2000, 104, 10631–10636 CrossRef CAS.
- S. N. Spisak, A. V. Zabula, A. S. Filatov and M. A. Petrukhina, J. Organomet. Chem., 2015, 784, 69–74 CrossRef CAS.
- T. E. Bauert, L. Zoppi, G. Koller, J. S. Siegel, K. K. Baldridge and K.-H. Ernst, J. Am. Chem. Soc., 2013, 135, 12857–12860 CrossRef CAS PubMed.
- (a) A. Ayalon, A. Sygula, P.-C. Cheng, M. Rabinovitz, P. W. Rabideau and L. T. Scott, Science, 1994, 265, 1065–1067 CAS; (b) E. Shabtai, R. E. Hoffman, P.-C. Cheng, E. Bayrd, D. V. Preda, L. T. Scott and M. Rabinovitz, J. Chem. Soc., Perkin Trans. 2, 2000, 129–133 RSC.
- S. Neander, J. Körnich and F. Olbrich, J. Organomet. Chem., 2002, 656, 89–96 CrossRef CAS.
- (a) T. Amaya, H. Sakane and T. Hirao, Angew. Chem., Int. Ed., 2007, 46, 8376–8399 CrossRef CAS PubMed; (b) M. A. Petrukhina, Angew. Chem., Int. Ed., 2008, 47, 1550–1552 CrossRef CAS PubMed; (c) T. Amaya and T. Hirao, Chem. Rec., 2015, 15, 310–321 CrossRef CAS PubMed.
- N. J. Sumner, S. N. Spisak, A. S. Filatov, A. Yu. Rogachev, A. V. Zabula and M. A. Petrukhina, Organometallics, 2014, 33, 2874–2878 CrossRef CAS.
- M. A. Petrukhina, K. W. Andreini, J. Mack and L. T. Scott, J. Org. Chem., 2005, 70, 5713–5716 CrossRef CAS PubMed.
- C. Bruno, R. Benassi, A. Passalacqua, F. Paolucci, C. Fontanesi, M. Marcaccio, E. A. Jackson and L. T. Scott, J. Phys. Chem. B, 2009, 113, 1954–1962 CrossRef CAS PubMed.
- D. Eisenberg, J. M. Quimby, L. T. Scott and R. Shenhar, J. Phys. Org. Chem., 2013, 26, 124–130 CrossRef CAS.
- S. N. Spisak, Z. Wei, N. J. O'Neil, A. Yu. Rogachev, T. Amaya, T. Hirao and M. A. Petrukhina, J. Am. Chem. Soc., 2015, 137, 9768–9771 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of preparation, characterization, X-ray diffraction study, and theoretical calculations. CCDC 948063. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04385a |
| This journal is © The Royal Society of Chemistry 2016 |