
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhen the inhibitor tells more than the substrate: the cyanide-bound state of a carbon monoxide dehydrogenase†
Alexandre
Ciaccafava‡
*a,
Daria
Tombolelli‡
a,
Lilith
Domnik
b,
Jochen
Fesseler
b,
Jae-Hun
Jeoung
b,
Holger
Dobbek
b,
Maria Andrea
Mroginski
*a,
Ingo
Zebger
*a and
Peter
Hildebrandt
a
aTechnische Universität Berlin, Institut für Chemie, Sekretariat PC 14, D–10623 Berlin, Germany. E-mail: alex.ciacca@gmail.com; andrea.mroginski@tu-berlin.de; ingo.zebger@tu-berlin.de
bHumboldt-Universität zu Berlin, Institut für Biologie, Unter den Linden 6, D–10099 Berlin, Germany
First published on 27th January 2016
Abstract
Carbon monoxide dehydrogenase (CODH) is a key enzyme for reversible CO interconversion. To elucidate structural and mechanistic details of CO binding at the CODH active site (C-cluster), cyanide is frequently used as an iso-electronic substitute and inhibitor. However, previous studies revealed conflicting results on the structure of the cyanide-bound complex and the mechanism of cyanide-inhibition. To address this issue in this work, we have employed IR spectroscopy, crystallography, site directed mutagenesis, and theoretical methods to analyse the cyanide complex of the CODH from Carboxydothermus hydrogenoformans (CODHIICh). IR spectroscopy demonstrates that a single cyanide binds to the Ni ion. Whereas the inhibitor could be partially removed at elevated temperature, irreversible degradation of the C-cluster occurred in the presence of an excess of cyanide on the long-minute time scale, eventually leading to the formation of [Fe(CN)6]4− and [Ni(CN)4]2− complexes. Theoretical calculations based on a new high-resolution structure of the cyanide-bound CODHIICh indicated that cyanide binding to the Ni ion occurs upon dissociation of the hydroxyl ligand from the Fe1 subsite of the C-cluster. The hydroxyl group is presumably protonated by Lys563 which, unlike to His93, does not form a hydrogen bond with the cyanide ligand. A stable deprotonated ε-amino group of Lys563 in the cyanide complex is consistent with the nearly unchanged C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching in the Lys563Ala variant of CODHIICh. These findings support the view that the proton channel connecting the solution phase with the active site displays a strict directionality, controlled by the oxidation state of the C-cluster.
N stretching in the Lys563Ala variant of CODHIICh. These findings support the view that the proton channel connecting the solution phase with the active site displays a strict directionality, controlled by the oxidation state of the C-cluster.
Introduction
The O2-sensitive [Ni–Fe] carbon monoxide dehydrogenase (CODH) is a homodimeric enzyme responsible for the biological interconversion between CO and CO2 according to the reaction: CO + H2O ↔ CO2 + 2H+ + 2e−. CODHs play crucial roles in various metabolic pathways, including the Wood-Ljungdahl pathway that allows anaerobic microorganisms, such as acetogenic firmicutes Moorella thermoacetica (Mt) and hydrogenogenic firmicutes Carboxydothermus hydrogenoformans (Ch) to grow on H2 and CO2 as electron and carbon source, or to oxidise CO using it as a source of electrons.1The CODH hosts a unique [NiFe4S4OHx] cluster, called C-cluster, which efficiently catalyses the reversible CO oxidation reaction.2 Briefly, the C-cluster resembles a classical cubane shape [4Fe–4S] from which one Fe is pulled out and left to dangle. The dangling Fe, so called Fe1, presumably binds a hydroxyl ligand while the vacant Fe position has been filled by a Ni atom. The C-cluster adopts at least two different oxidation states during catalysis. The Cred1 redox state is able to bind CO while Cred2, obtained by a further, two-electron reduction, specifically targets CO2. The midpoint potential of the Cred1/Cred2 couple (−530 mV vs. SHE) almost matches the midpoint potential of the substrates (E0 CO/CO2 = −558 mV, pH 7).3 Moreover, turnover frequencies up to 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 and Michaelis constants of ∼20 μM have been reported for CO oxidation by two CODHs from thermophilic bacteria Ch, whereas the corresponding values for CO2 reduction by the CODH of Rhodospirillum rubrum were found to be ca. 45 s−1 and 200 μM, respectively.4,5
000 s−1 and Michaelis constants of ∼20 μM have been reported for CO oxidation by two CODHs from thermophilic bacteria Ch, whereas the corresponding values for CO2 reduction by the CODH of Rhodospirillum rubrum were found to be ca. 45 s−1 and 200 μM, respectively.4,5
Important information about the catalytic cycle was obtained by X-ray crystallography.2,6–8 Particularly, the recent atomic-resolution structures of CODHIICh (dmin ≤ 1.1 Å) provided insight into the binding and activation of CO2 (and its inhibitory counterpart NCO).9 Notably, both CO2 and NCO had undergone a two-electrons reduction step and the respective products were stabilised via π-backbonding. Such a detailed picture is yet not available for CO binding to the C-cluster, although structural analysis pointed to a bent geometry of CO (or formyl) bound to the terminal Ni in the CODH–CO complex of Methanosarcina barkeri.10 This finding was consistent with the view that a bent Ni–CO geometry would lower the energetic activation barrier for CO oxidation. In a complementary crystallographic study, cyanide (CN−), a structural analogue of CO and inhibitor of CODH, was found to possess a similar bent conformation.7 Recently, however, an independent re-evaluation of the electron density maps of these two structures (resolution ≥ 2 Å) did not provide clear evidence for the orientation of the CO or the CN− ligand.11 In addition, another crystal structure presenting a different binding mode of CN− has been reported for CODHIICh8 (Fig. 1).
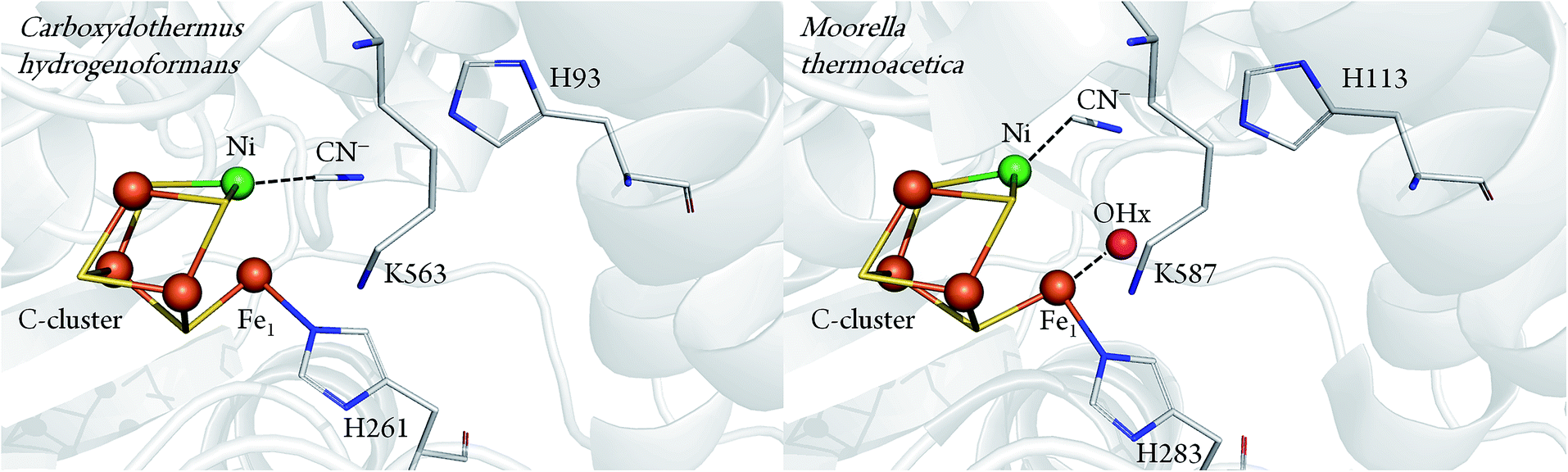 | ||
| Fig. 1 Comparison between CN− bound C-cluster of CODHIICh (pdb 3I39) and CODHMt (pdb 3I04), according to ref. 7 and 8. | ||
In principle, spectroscopic techniques such as IR spectroscopy may contribute to rationalise these partially conflicting results. In particular, IR spectroscopy is an ideal tool since it probes the CN stretching mode in a spectral region free of any interfering bands of the protein. However, the results reported in previous IR spectroscopic studies on Clostridium thermoaceticum CODH (CODHCt) revealed multiple signals in the CN stretching region12,13 that cannot readily be reconciled with the current structural data. In this work, we have therefore set up an integral approach including IR spectroscopic, crystallographic, site directed mutagenesis and theoretical analysis. Spectroscopic measurements were carried out with the wild-type and engineered CODHIICh variant to assess the parameters that control the CN stretching frequency. This analysis served as a reference for the quantum-mechanical/molecular mechanics (QM/MM) calculations which were performed on the basis of new high-resolution crystal structures to elucidate details of the active site structure of the CODHIICh–CN complex.
Materials and methods
Sample preparation
Buffers and reagents were anoxically prepared in a glovebox. Inhibition procedures described below were performed under the same anoxic conditions. In addition to the inherent safety risks while handling cyanide, it is worth noticing that controlling precisely cyanide concentration is not straightforward, especially when using buffer in the physiological relevant range, i.e. pH 7–8, as the pKa value for HCN is 9.21. Therefore, the band position of the CN stretching mode ranges from 2080 cm−1 (aqueous CN−) to 2093 cm−1 (dissolved HCN), when the pH is decreased below the pKa. This is the pH range, where the enzyme exhibits its highest activity. Thus, the absolute concentration effect of CN− has to be estimated with caution. Protein expression and purification were performed as described earlier.2 Protocol A: 6 μL of CODHIICh 400 μM were diluted in 500 μL of buffer A (Tris–HCl 50 mM pH 8, NaCl 100 mM) containing either dithionite (DT) 4 mM or titanium citrate (Ti(III) citrate) 5 mM and incubated for 45 min. Cyanide (1 mM) was added to the sample and allowed to react with CODHIICh for 30 min under gentle stirring. The CODHIICh sample was subsequently washed several times using Microcon® Centrifugal Filters (cut-off 100 kDa) with buffer A and reductant in order to remove excess cyanide and finally concentrated to 10 μL prior to injection into the transmission cell. Protocol B: 6 μL of CODHIICh 400 μM were added to 6 μL of an inhibition mixture containing cyanide (ranging from 2 to 20 mM) and DT (4 mM) in buffer A. Finally, 10–12 μL of the inhibited CODHIICh sample, which was prepared according to procedures described above, was injected into a gas-tight transmission cell. Protocol C: crystals of CODHIICh were incubated in buffer A′ (25% PEG3350, 50 mM Tris–HCl pH 8.0 and 5 mM Ti(III) citrate or DT 4 mM). Crystals were then transferred into a buffer B solution (25% PEG3350, 50 mM Tris–HCl pH 8.0, 7 mM DTT), and 0.5 mM of oxidised methyl viologen (MVox) as electron acceptor for reoxidation. After no further reduction of MVox to the blue colored reduced methyl viologen (MVred) was detectable, the crystals were incubated in buffer B solution containing 7 mM DTT for 2 h. Finally, crystals were transferred into a buffer B solution containing 5 mM KCN and loaded into the transmission cell.
Enzymatic assays
Inhibited aliquots of CODHIICh were reactivated under CO atmosphere at 70 °C for 30 min. Then the CO oxidation activity was assayed as described before.4Fourier transform infrared spectroscopy
FTIR spectra were recorded on a Bruker Tensor 27 spectrometer equipped with a liquid nitrogen-cooled MCT detector applying a spectral resolution of 2 cm−1. The sample compartment was purged with dried air, and the sample was held in a temperature-controlled (10 °C) gas-tight IR-cell for liquid samples (volume ∼ 10 μL, path length = 50 μm) with CaF2 windows. Spectra were base-line corrected by using the OPUS software from Bruker.Crystallization and manipulation of crystals
CODHIICh crystals and the −320 mV state were produced as described earlier.2,8 Crystals in the −320 mV state were incubated with 70 mM KCN for 30 min. CN−-soaked crystals were shock-frozen in liquid nitrogen, generating the −320 mV CODHIICh–CN state, with 15% (v/v) 2R,3R-butanediol (Sigma) serving as cryoprotectant.Data collection, structure solution and refinement
Diffraction data were collected at 100 K at a wavelength of 0.9184 Å at BESSY II (Berlin, Germany).14 The synchrotron data set was integrated and scaled using XDSAPP.15 Data statistics are reported in Table S1 (see ESI†). Initial rigid body refinement was used to solve phasing, followed by a simulated annealing refinement to minimise model bias using PHENIX.16 Models were built and refined with COOT.17 Positional and temperature factor refinements were carried out using Refmac5.18,19 New parameters for the C-cluster were generated by iterative cycles of refinement using Refmac5. Individual occupancies of atoms from the C-cluster were adjusted until comparable B factors of atoms were obtained. In the final refinement cycles the B factors were refined anisotropically and alternative conformations of several side chains and Fe1 were included. The final refinement statistics are shown in Table S1 of ESI.† The atomic coordinates have been deposited in the Protein Data Bank, http://www.pdb.org (PDB ID 5FLE).Theoretical methods
The eight structural models listed in Table 2, were built starting from two crystal structures of the CODHIICh in the −320 mV state. The first one, with pdb 5FLE, represents the CN− bound state refined in this work, while the second structure is inhibitor free (pdb 3B53). In the latter, a starting geometry for the CN− bound state was generated by attaching a CN− ligand to the Ni atom. The protein was solvated in an explicit TIP3P water box of dimension 130 × 130 × 130 Å3. Hydrogens were inserted and protonable side chains were set to their standard states at pH 7. Protonation states of some specific residues, e.g. those in the vicinity of the C-cluster or involved in suggested proton transfer pathways, required special attention based on chemical sense and visual inspection.20 Although the His93 was doubly protonated on Nδ and on Nε, further investigation was required to test different protonation states of this residue together with Lys563, since they are interacting via hydrogen bonds with the non-proteic ligands at the active site. First, 20000 steps of energy minimization were performed using NAMD21 with the CHARMM22 force field.22 Hydrogen positions and orientation of water molecules were optimised. Heavy atoms were kept fixed to their initial positions, in order to obtain a good starting point for the subsequent geometry optimization and, at the same time, to remain as close as possible to the crystal structure conformation. Due to the lack of specific force field parameters, the five metal clusters (three standard [4Fe–4S] and two [NiFe3S4–Fe]) were initially kept fixed during the energy minimization steps. Charges for the metal clusters were computed with DFT methods while van der Waals parameters were taken from values reported for similar systems in the literature.23 The structure resulting from the last minimization step was then refined through hybrid QM/MM calculations, performed within the ChemShell environment.24 The program interfaces to a variety of QM and MM codes. In our work, Turbomole was chosen as the quantum chemical program while the MM uses DL_POLY with CHARMM22 force field. The system was divided in three layers, each of which was treated at a different level of theory and accuracy. The QM part consists of (1) the [NiFe3S4] C-cluster and the iron Fe1; (2) Cys295, Cys333, Cys446, Cys476 and Cys526, which coordinate the Fe atoms of the cluster; (3) His261, which is interacting with Fe1; (4) all the other non-proteic ligands that includes cyanide on Ni in different positions and hydroxyl on Fe1 in some of the models; (5) His93, Lys563 interacting directly with the non-proteic ligands, Gln332 interacting via hydrogen bonds with Lys563 and Ile567 whose role is important regarding the steric hindrance exerted on the cyanide ligand. DFT was employed in the optimization of the QM part, applying the pure density functional BP86 combined with the resolution-of-the-identity (‘RI’) technique.25 The def2-TZVP basis set was used for metals (Ni and Fe), while the 6-31G* basis set was employed for all other atoms of the QM part. This combination of a high quality basis set and BP86 functional is able to give both accurate molecular structures and harmonic vibrational frequencies, due to an error compensation that allows direct comparison of calculated harmonic frequencies with experimental ones.26 The second layer was made up of protein residues and water molecules included in a sphere of 15 Å around the Ni atom of the C-cluster in monomer A. All the atoms in this section were treated in an explicit manner at the MM level and allowed to move during the optimization cycles. For this purpose the HDLC optimiser was employed.27 The third layer includes the remaining atoms not included in the previous two. This layer was held fixed during the QM/MM geometry optimization. Covalent bonds at the QM/MM border were cut and saturated by hydrogen link atoms. The coupling between QM and MM was computed using electrostatic embedding with a charge-shift scheme.24 Once optimised, the structures were analysed and compared with the crystal structure. The latter was based on some fundamental geometrical parameters such as bond lengths and bond angles. In addition, a tilt angle was defined as the angle between a unit vector with origin in the center of the plane formed by Ni–S–Fe4–S and a unit vector directed along the Ni–CN bond axis.
N stretching frequencies observed experimentally for CODHIICh–CN adducts according to protocols A and B. Control experiments and published data are also reporteda
| Wavenumber/cm−1 | ||||
|---|---|---|---|---|
| Sample | ν(CN) | ν(13CN) | Δν (CN–13CN) | |
| a Data taken from literature are written in italic. | ||||
| Controls | CN− (aq.) | 2080 | 2036 | 44 |
| HCN (gas.) | 2093 | 2061 | 32 | |
| [Fe(CN)6]4− | 2037 | 1995 | 42 | |
| [Ni(CN)4]2− | 2124 | 2078 | 46 | |
| Protocol A | CODHIICh | 2110 | 2065 | 45 |
| Protocol B | CODHIICh | 2037 | 1995 | 42 |
| CODHIICh | 2093 | 2061 | 32 | |
| CODHIICh | 2124 | 2078 | 46 | |
| Published data | CODHCt | 2079 | 2037 | 42 |
| CODHCt | 2037 | 1995 | 42 | |
| Structures | Distances/Å | Angles/° | Presence of | ν(CN)/cm−1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | Specificity | Origin | Ni–Fe1 | Ni–CCN | NCN–NK563 | NCN–NεH93 | CN–tilt | Ni–C–N | OH− | Calc. | Δ (exp. − calc.) |
| a Abbreviations used: calc.: calculated, exp.: experimental, dep.: deprotonated. The corresponding structure names, in the text, are defined by assembling the column name and specificity. | |||||||||||
| CODHIICh–CN pdb(5FLE) | — | Exp. | 2.67 | 1.92 | 3.00 | 2.66 | 5.98 | 168 | No | — | — |
| — | Calc. | 2.65 | 1.81 | 2.98 | 2.72 | 6.40 | 169 | No | 2074 | −36 | |
| /tilt | Calc. | 2.66 | 1.82 | 3.79 | 2.69 | 18.42 | 171 | No | 2107 | −3 | |
| /tilt/OH− | Calc. | 2.94 | 1.85 | 4.59 | 2.82 | 29.07 | 165 | Yes | 2054 | −56 | |
| /K563dep. | Calc. | 2.73 | 1.83 | 3.97 | 2.72 | 24.44 | 173 | No | 2109 | −1 | |
| /K563A | Calc. | 2.71 | 1.83 | — | 2.70 | 23.52 | 173 | No | 2112 | +10 | |
| CODHIICh–Cred1–CN*pdb(3B53) *: CN− was added in silico | — | Calc. | 2.63 | 1.80 | 2.88 | 2.74 | 3.03 | 175 | No | 2058 | −52 |
| /tilt | Calc. | 2.68 | 1.81 | 3.94 | 2.66 | 17.71 | 175 | No | 2129 | +19 | |
| /tilt/OH− | Calc. | 2.86 | 1.83 | 4.65 | 2.76 | 37.35 | 165 | Yes | 2054 | −56 | |
Finally the vibrational frequencies of the quantum mechanical part in the optimised conformation were computed in the presence of the point charges of the surrounding atoms of the second and third layers. For this purpose the Gaussian09 program was used with the same DFT functional and basis set employed during the optimization.28
Results
IR spectroscopic identification of the CN− inhibited state
After exposure of CODHIICh to CN− according to protocol A (see Materials and methods), a sharp band immediately appears in the CN stretching region at 2110 cm−1 (Fig. 2A). The frequency upshift with respect to the free CN− anion (2080 cm−1) is expected since Ni is a weak π-donor metal.29 The band position and intensity are stable for hours. The frequency at 2110 cm−1 is significantly higher than for cyanide-bound Fe2+ heme proteins such as horseradish peroxidase, microperoxidase and myoglogin (between 2029 and 2057 cm−1), illustrating the rule of electronegativity (Ni > Fe) discussed previously.30,31 The origin of the band was further proven with labelled 13CN− (2065 cm−1), revealing the expected isotopic shift of 45 cm−1 (Fig. 2C).
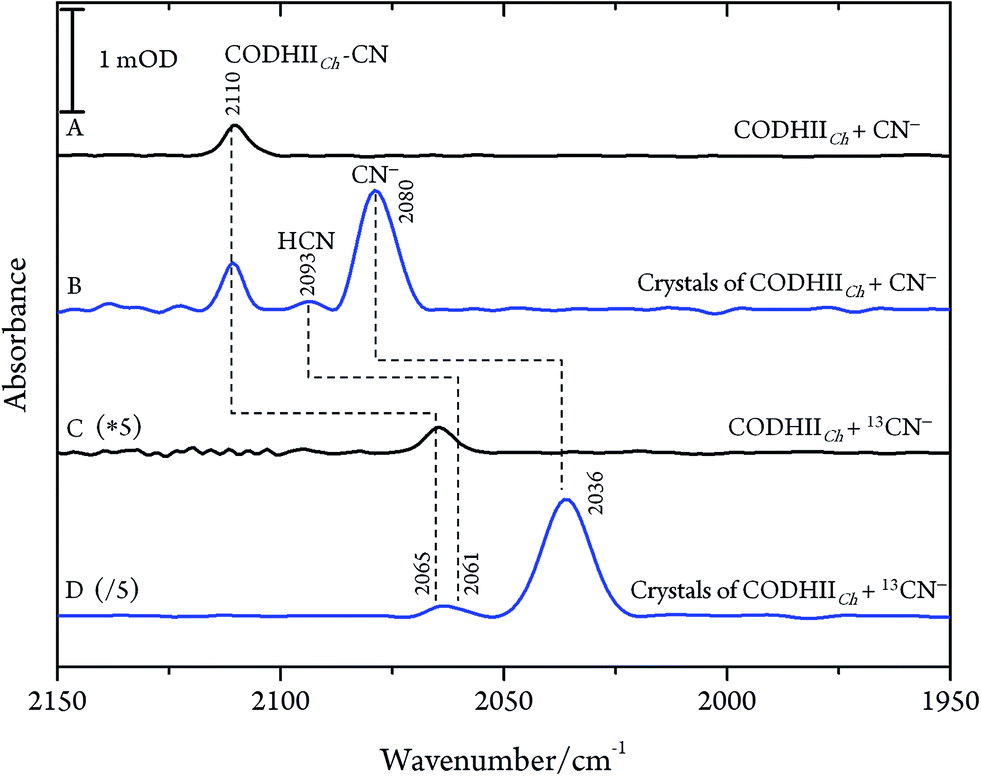 | ||
| Fig. 2 IR spectra of the CODHIICh–CN complex at pH 8, 10 °C. (A) CODHIICh in solution after 30 min of exposure to CN−; (B) crystalline state of CODHIICh after treatment with CN−; (C) CODHIICh in solution after 30 min of exposure to 13CN−; (D) crystalline state of CODHIICh after treatment with 13CN−. | ||
Next, we studied the CODHIICh–CN complex in the crystalline state which was prepared following the same protocol as used for crystallographic analysis. Several of these crystals were loaded into an IR transmission cell (Fig. 2B and D). The IR spectra obtained in this way revealed a band at 2110 cm−1 that shifts down by 45 cm−1 to 2065 cm−1 when crystals were incubated with 13CN−. Thus, the frequency (and isotopic shift) of the Ni-bound cyanide of the crystalline complex is the same as for the sample in solution.
Reversibility of CN− binding
In order to prove the reversibility of the inhibition process,32 we prepared a batch of inhibited CODHIICh that we used in parallel for enzymatic activity assay and IR spectroscopy. A classical enzymatic assay for CO oxidation by CODHIICh based on reduction of methyl viologen was performed to evaluate the initial enzymatic activity (see Materials and methods). Subsequently, the enzyme was inhibited following protocol A, and afterwards a new enzymatic assay for CO oxidation was performed. The inhibited protein exhibited not more than 5% of the initial CO oxidation activity (Fig. 3). In parallel, the corresponding enzyme was also loaded into a transmission cell for IR measurement and revealed the characteristic CN stretching band at 2110 cm−1 (Fig. 3 inset). Finally, the enzyme was reactivated at 70 °C under a CO atmosphere for 30 min and tested for CO oxidation afterwards. More than 60% of CO oxidation was recovered thereby proving a high reversibility of the CN− binding (Fig. 3).
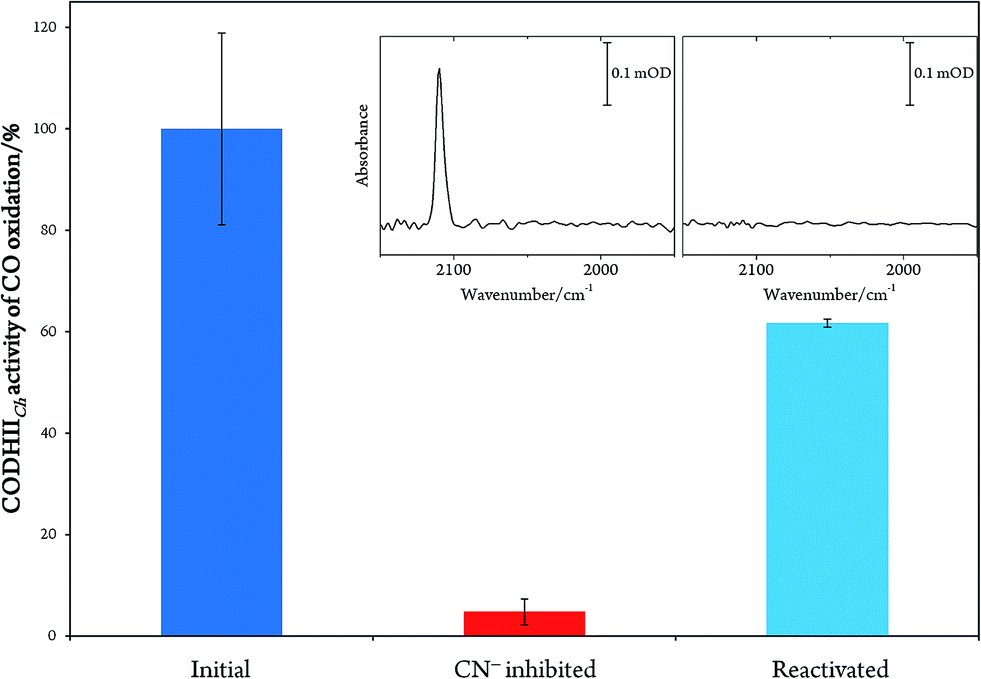 | ||
| Fig. 3 Plot representing the percentage of CO oxidation activity by CODHIICh before inhibition (blue column), after inhibition (red column) and after reactivation (light blue column). Inset: IR spectra of the inhibited CODHIICh before and after reactivation. | ||
These results are, however, not in agreement with previous IR data published for CN− inhibition of CODHCt.12,13 These authors observed a band at 2037 cm−1 (1995 cm−1 with 13CN−) and a second absorption appearing only at higher CN− concentration at 2079 cm−1 (2037 cm−1 with 13CN−). However, the preparation protocol employed in that work did not include the removal of excess cyanide. In fact, when we repeated the experiments upon omitting the final buffer exchange step (protocol B, see Materials and methods) we observed several bands at 2124, 2093 and 2037 cm−1 shifting to 2078, 2061 and 1995 cm−1 upon exposure to 13CN−, respectively (Fig. 4A and B).
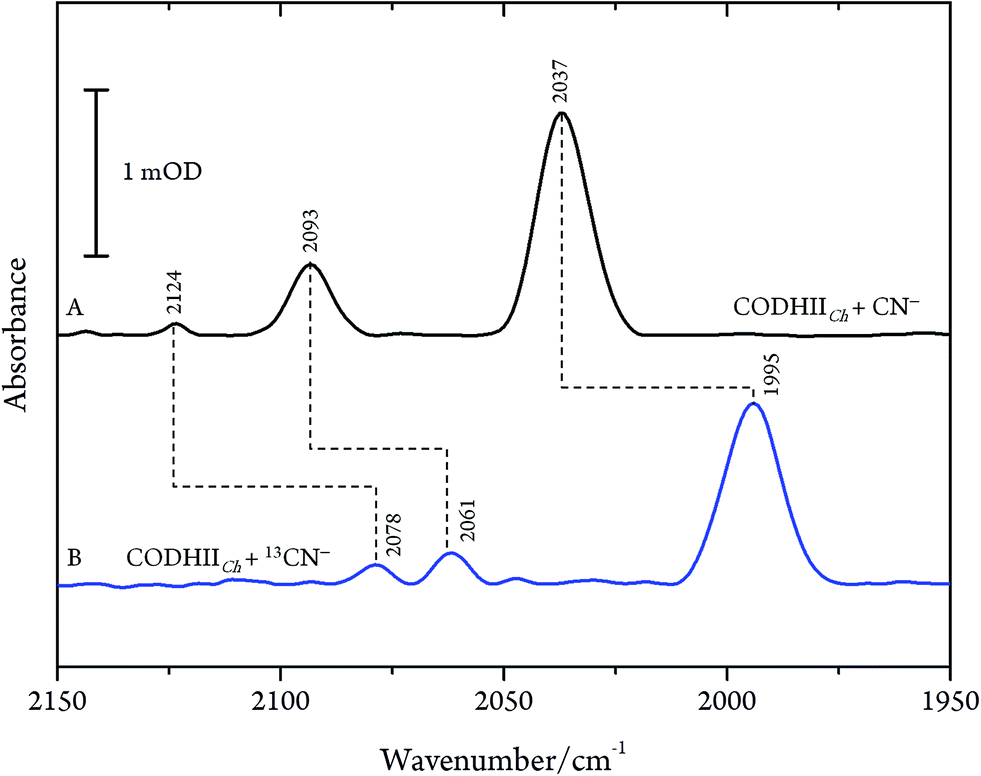 | ||
| Fig. 4 Infrared spectra of CODHIICh at pH 8, 10 °C left with (A) CN− and (B) 13CN− with a CN−/CODHIICh ratio of 100/1 measured at 10 °C. | ||
While the assignment of the band at 2093 cm−1 and its isotopically 32 cm−1 shifted counterpart at 2061 cm−1 to HCN and H13CN is straightforward,33 the origin of the two other bands is less obvious. Since only one binding site for CN− is available in the CODHIICh structure,8 the presence of two extra bands is surprising. Moreover, these two features appear with a very slow rising time (≥5 h) (Fig. 5). In contrast, formation kinetics of the cofactor-cyanide adduct according to protocol A (removal of excess cyanide) and reflected by the 2110 cm−1 band is not resolved on the time scale of these experiments and, hence, must occur on the minute time scale or faster (<30 min), (Fig. 5).
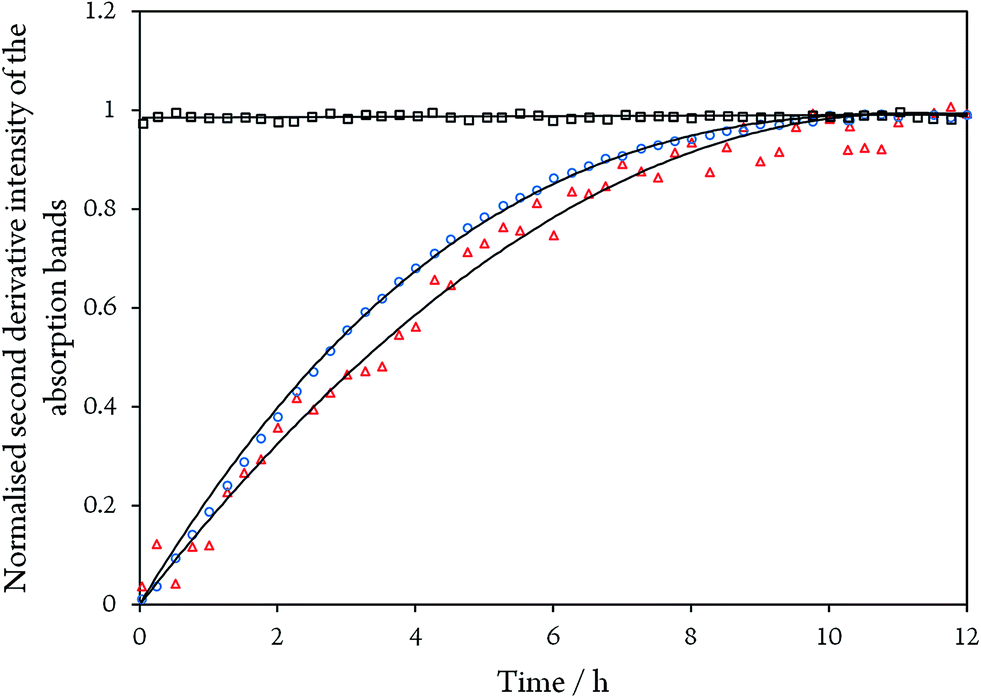 | ||
| Fig. 5 Time evolution of the normalised second derivative intensity of the absorption bands at 2110 cm−1 for CODHIICh inhibited with CN− according to protocol A (black squares), and of the bands at 1995 cm−1 (blue circles) and 2078 cm−1 (red triangles) for CODHIICh inhibited with 13CN− according to protocol B, respectively. Note that bands at 1995 and 2078 cm−1 did not start at zero intensity and were normalised to unity for better comparison. | ||
CN−-induced degradation of the C-cluster
Even though CN− has been described as a slow reversible inhibitor, CODHIICh is fully inhibited in less than one minute at 70 °C with 1 mM KCN under turnover conditions and in less than 80 min at 23 °C with 0.1 mM KCN under non-turnover conditions, as described in a previous study.32 Furthermore, CN− is known to have a high affinity towards metal ions including Fe and Ni which are the main components of the C-cluster, therefore, we suspected that prolonged exposure of CODHIICh to CN− might lead to the formation of various cyano–metallic complexes. Therefore, we performed control experiments in order to verify the possible CN− induced degradation of the metal clusters due to prolonged exposure of CODHIICh to CN−.The corresponding IR spectra were measured under the same experimental conditions as used for CODHIICh inhibition studies (Fig. 6). Thus, nickel chloride (NiCl2) has been incubated with CN−, resulting in the appearance of an absorption band at 2124 cm−1 (2078 cm−1 with 13CN−) (Fig. 6B, red line), which can be assigned to [Ni(CN)4]2− complex.29 In addition, the corresponding cyano complex of iron, [Fe(CN)6]4−, exhibits an absorption band at 2037 cm−1 (1995 cm−1 for 13CN−) (Fig. 6B, blue line).30,34,35
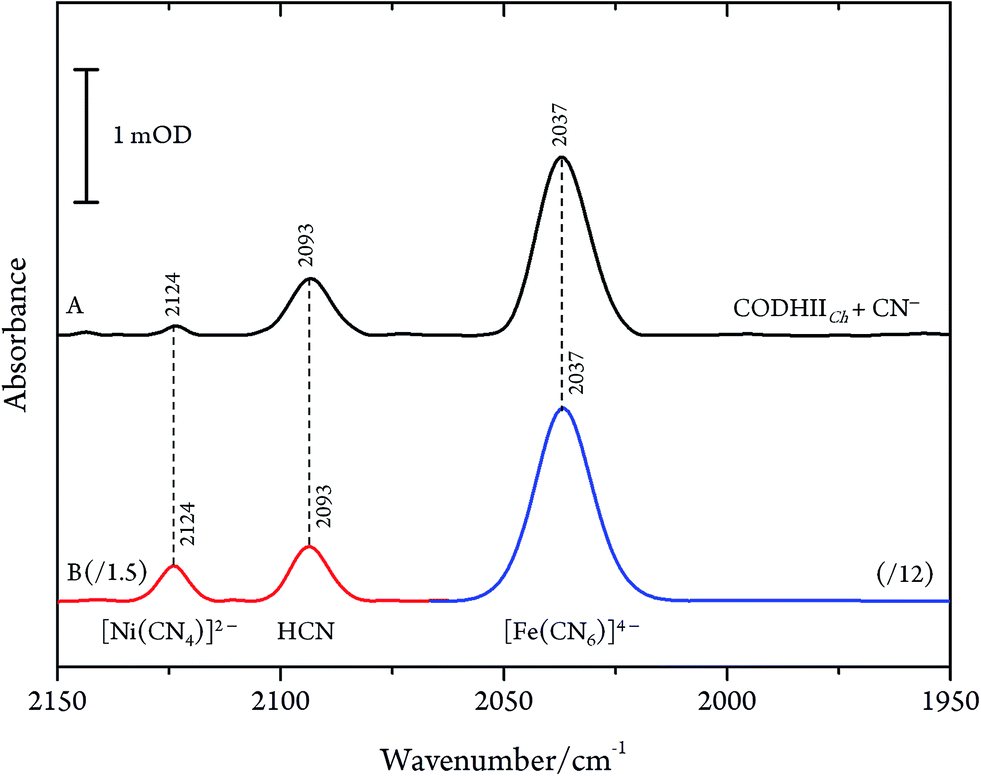 | ||
| Fig. 6 (A) Infrared spectra of CODHIICh left with CN− with a CN−/CODHIICh ratio of 100/1 and (B) NiCl2 0.2 mM incubated with CN− 1 mM (red line) and [Fe(CN)6]4− 1 mM (blue line) measured at 10 °C, pH 8. Spectra of cyano–metallic complexes are scaled down for better comparison. | ||
Accordingly, the control experiments allowed an assignment of the three bands observed for CODHIICh inhibition according to the alternative protocol B. Two of these bands, i.e. at 2080 cm−1 (2036 cm−1 for 13CN−) and 2037 cm−1 (1995 cm−1 for 13CN−) at pH 8, are readily attributed to aqueous CN− and [Fe(CN)6]4−, respectively. The frequencies agree very well with those reported by Qiu et al. for CODHCt although the assignment suggested by these authors can be ruled out.13 The third band at 2124 cm−1 (2078 cm−1 for 13CN−) was not reported by these authors although a weak band at ca. 2078 cm−1 can be identified in the published spectrum of the CODHCt13CN− adduct in the corresponding figure. This band at 2124 cm−1 band is now assigned to the CN stretching of [Ni(13CN)4]2− (Table 1).
Parameters controlling the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) N stretching mode
N stretching mode
It was suggested previously that, depending on the concentration, cyanide may bind in different modes to the C-cluster.36 IR experiments were, therefore, carried out using different CN−/CODHIICh molar ratios from large excess (1000/1) to substoichiometric (1/2) amounts. These experiments did not reveal any additional band nor any frequency shifts of the only observed band at 2110 cm−1 (data not shown). Furthermore, another, alternative binding mode was proposed to involve a hydroxyl ligand on the dangling Fe1,1,36,37 which should be sensitive to the local proton concentration. Thus, cyanide inhibition was studied as a function of the pH.
Whereas the CN frequency at 2110 cm−1 remained unaffected in the pH range from 6 to 10, the related band intensities displayed a bell-like shape with a maximum at pH 8 (Fig. 7). Again, these findings argue against any pH-dependent changes of ligand binding at the C-cluster. Instead, the intensity changes most likely reflect pH-dependent alterations of the active site accessibility for CN−.
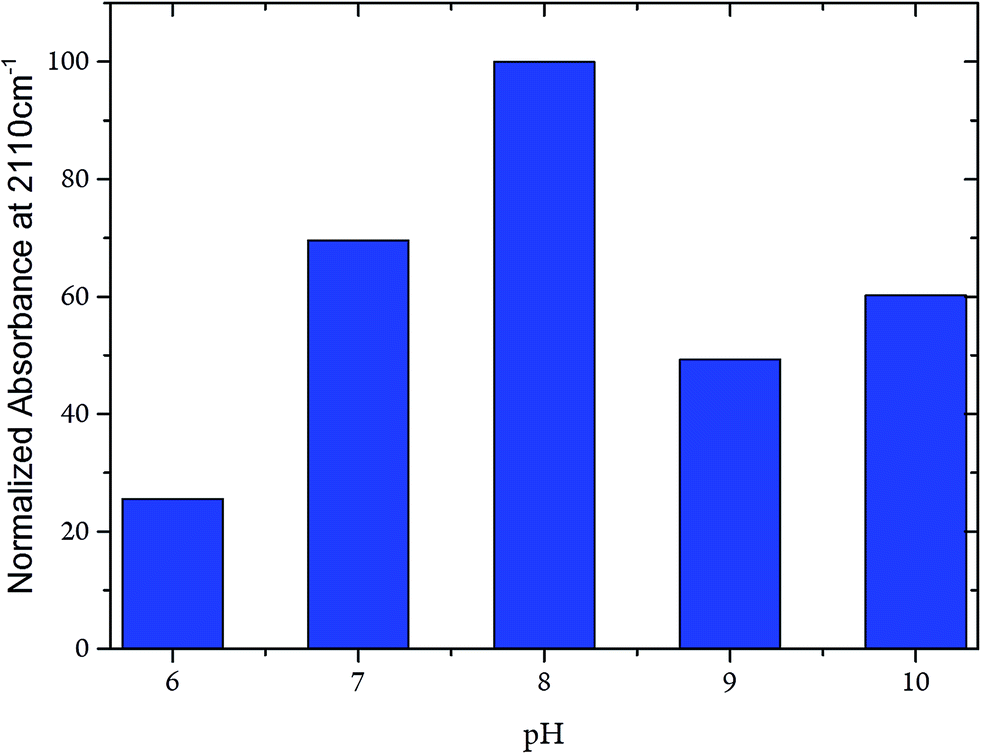 | ||
| Fig. 7 Intensity of the CN stretching mode at 2110 cm−1 of the CODHIICh–CN adduct plotted as a function of the pH. Intensities were normalised with respect to the intensity of the corresponding amide II band. | ||
There are also no qualitative changes of cyanide binding depending on the temperature.
The CN stretching frequency and the band intensity decrease only slightly (by 2.0 cm−1 and ∼30%, respectively) upon raising the temperature from 10 to 60 °C (Fig. 8). Conversely, in the frozen state (100 K) the frequency is increased by only 5 cm−1. These spectral variations can be rationalised in terms of subtle conformational changes of the protein in the vicinity of the C-cluster. Consistent with this explanation is the observation that the temperature-dependent changes are fully reversible, underpinning the remarkable stability of the CODHIICh–CN adduct.
 | ||
| Fig. 8 (A) Frequency and (B) intensity changes of the CN mode of the CODHIICh–CN adduct as a function of the temperature. The arrow tip points to the direction of the temperature steps. | ||
Environmental effects on the CN stretching mode
According to X-ray data, Ni-bound cyanide is in close vicinity to His93 and Lys563 which may interact with the cyanide-bound metal center. We therefore extended the studies to the CODHIICh variant with alanine substitution at the corresponding position. Addition of cyanide to the Lys563A variant displayed a wild-type like IR spectrum since the ν(CN) undergoes a downshift from 2110 to 2102 cm−1.
Crystal structures of CN−-inhibited CODHIICh
We have determined a new crystal structure of CN− bound CODHIICh (pdb 5FLE) at a resolution of 1.23 Å. As expected, the 2009 structure (pdb 3I39, dmin = 1.35 Å) and the new CODHIICh–CN structure are nearly identical with a rmsd for core Cα atoms of 0.14 Å. The main difference between the two structures is the degree of multiple conformations. While the 2009 structure revealed dual conformations for residues starting from the Fe1-coordinating His261 down to residue 266 as well as residues in their vicinity, the electron density of these parts agree now with only a single conformation, indicating a more homogenously CN− bound state.8QM/MM calculations
Two different experimentally determined structures served as starting points for the QM/MM calculations. First, we used the crystal structure of the CODHIICh–CN inhibited state determined in this work (pdb 5FLE). Geometry optimization did not affect the main structural parameters of the C-cluster and residues surrounding the CN− ligand except for Ni–C(CN) bond length, which was predicted to be shorter by 0.11 Å compared to the experimental value (Table 2). A comparably short Ni–C bond length was also reported in a previous QM/MM study by Amara et al.11 and recently observed in CO2 and NCO bound structures.9 Also in the previous CODHIICh–CN structure, solved at lower resolution (1.35 Å), a shorter bond length was determined,8 in line with the results of an earlier EXAFS study.32 In agreement with the experimental structure, the calculations predict the Ni atom in a square-planar geometry with a tilt angle of 6.40° of the CN− ligand (see Materials and methods, section QM/MM for tilt angle definition). In this configuration, the CN− ligand is in hydrogen bonding distance to the side chains of His93 (2.72 Å) and Lys563 (2.98 Å).Second, calculations were carried out starting with the CODHIICh structure of the −320 mV reduced state (pdb 3B53) to which we added a CN− ligand in silico (CODHIICh–Cred1–CN). Prior to optimization, we have removed the water molecule in the vicinity of the CN− binding site to keep in line with the experimental structure of CODHIICh–CN. Between both optimised structures a very good agreement was obtained with respect to the geometries of the C-cluster and CN− environment (Table 2). Also for the optimised CODHIICh–Cred1–CN structure a small tilt angle (3.03 Å) for the CN− ligand and similar distances to His93 (2.74 Å) and Lys563 (2.88 Å) were obtained.
The CN stretching frequencies were calculated to be 2074 and 2058 cm−1 for CODHIICh–CN and CODHIICh–Cred1–CN, respectively. They are both significantly lower than the experimental value of 2110 cm−1.
As postulated previously,7,36 CN− may initially bind to complete a distorted tetrahedral coordination geometry at the Ni. Thus, an alternative geometry optimization was started by setting the initial tilt angle to 60.71°. In fact, the subsequent energy minimization afforded different minima for both CODHIICh–CN (CODHIICh–CN/tilt) and CODHIICh–Cred1–CN (CODHIICh–Cred1–CN/tilt) with somewhat higher tilt angles of ∼18°. In these configurations, the nitrogen-to-nitrogen distance of the CN− ligand to Lys563 is significantly increased to 3.8–3.9 Å, whereas that to His93 is nearly unchanged. The corresponding calculated stretching frequencies are now much closer to the experimental value (Table 2).
The two optimised active site structures with the more tilted CN− geometries exhibited an open space, which allowed binding of an OH− to Fe1. To assess the influence of such a ligand on the CN stretching frequency and to get an equivalent structure of the CN− inhibited state observed in CODHMt (pdb 3I04),7 an OH− ligand was introduced. In these structures (CODHIICh–CN/tilt/OH−; CODHIICh–Cred1–CN/tilt/OH−), geometry optimizations caused the tilt angle to increase from ∼18 to ∼35° because of increased steric interactions, consistent with the experimentally determined tilt angle in CODHMt (∼30°). In addition to the hydrogen bond formed with the Nε of His93, the hydroxyl ligand also provides an extra hydrogen bond to the CN− molecule thereby contributing to a significant decrease of the stretching frequency down to 2054 cm−1 (Table 2).
Whereas in all geometries, His93 remains in H-bond distance, the tilt angle geometry seems to depend on the interactions of the CN− ligand with Lys593. To assess the role of Lys593, we have generated the CODHIICh–CN/K563A structure in silico, starting from that of the wild-type protein complex (pdb 5FLE). Optimization leads to a tilt angle of ca. 24° and the resultant calculated CN stretching frequency (2112 cm−1) agrees well with the experimental frequency of 2102 cm−1 (Table 2).
Additional information about the potential interactions of Lys593 with the cyanide ligand was provided by the optimised CODHIICh–CN (wild-type) structure including a deprotonated Lys593 side chain. Also in this case, calculated (2109 cm−1) and experimental (2110 cm−1) CN stretching frequencies nearly coincide (Table 2).
Discussion
Cyanide-inhibition of Ni-dependent CODHs has been investigated for many years using a variety of experimental approaches. The analysis of the CODH–CN complex is thought to contribute to a better understanding of the catalytic mechanism but the previous studies did not yet provide a consistent view about the structure of the cyanide-bound active site and its degradation pathway.The cyanide binding site
Early IR spectroscopic studies revealed two different CN stretching modes that, due to their dependence on the cyanide concentration, were interpreted in terms of different modes of cyanide binding. Although the previously proposed bridging position between Fe1 and Ni has been meanwhile discarded, the view of two different binding geometries was re-animated on the background of slightly different active geometries derived from X-ray crystallography.7,8 In an attempt to reconcile crystallographic7,8 and spectroscopic data (ENDOR, Mössbauer),38,39 a two-step inhibition mechanism was proposed.36 In the first step, CN− was suggested to bind reversibly to Ni in a distorted tetrahedral geometry of the coordinated Ni ion as suggested by the crystal structure of CODHMt. This step may be similar to CO binding whereas in the second step, the release of an OH− ligand of Fe1, previously suggested to bind to Cred1,38 is followed by a slow reorganization of the cofactor that allows CN− to bind more tightly to the C-cluster. This is thought to be reflected by the square-planar coordination geometry of Ni observed in CODHIICh. The different CN− concentrations used for inhibition of both the CODHIICh and CODHMt crystals in soaking solutions were suggested to be the origin of these two binding modes.
In the present work, we have now shown that the previously reported frequencies of 2037 and 2079 cm−1 do not refer to the cyanide bound to the cofactor but originate from free CN− (2080 cm−1) and the degradation product [Fe(CN)6]4− (2037 cm−1). Instead, the intact cofactor-CN adduct gives rise to a band at 2110 cm−1. This band is observed at the same position for the enzyme in solution and in the crystalline state. Accordingly, the IR spectroscopic data should originate from the same state to which the crystallographic model refers to and which served as the starting point for the theoretical approaches. Specifically, QM/MM calculations indicate that the cofactor-CN adduct is characterised by the lack of an OH− ligand at the Fe1 as the hydrogen bonding interactions would cause a substantial downshift of this mode by ca. 50 cm−1 compared to the experimental value (2110 cm−1). A band at such a low frequency that could be ascribed to a Fe1(OH−)Ni(CN−) adduct is not observed at any experimental condition employed in this work. This negative result does not per se rule out the existence of such an intermediate state. However, if it exists, it is not accumulated even at sub-stoichiometric CODHIICh/CN− ratios and must, instead, decay quite rapidly on the sub-minute time scale to the stable Fe1Ni(CN) adduct.
Mechanism of the cyanide inhibition and degradation of the C-cluster
Once the CODHIICh–CN complex was formed according to protocol A, i.e. after removal of excess cyanide, it remained stable for hours at 10 °C but the cyanide ligand can be removed at elevated temperature in the presence of CO, accompanied with a recovery of the enzymatic activity. Conversely, a large excess of cyanide accelerates the degradation of the cofactor that is observed on the time scale of hours and eventually leads to the formation of the stable [Fe(CN)6]4− and [Ni(CN)4]2− complexes. These findings point to a two-step inhibition mechanism. First, cyanide rapidly binds to the Ni ion and induces the release of the OH− ligand from Fe1. This process can be reversed at high temperature in the presence of CO. In the second step, an additional cyanide binds to Fe1, albeit with a much slower rate, such that this step and thus the irreversible degradation can be efficiently blocked upon removal of excess cyanide after CN− binding to Ni (protocol A). Since the IR spectra provide no indication for a C-cluster coordinated by two cyanides, CN− binding to Fe1 appears to be followed by the rapid decomposition of the cofactor into the [Fe(CN)6]4− and [Ni(CN)4]2− complexes.Structure of the CN−-cofactor adduct
As already shown in previous studies on hydrogenases, the stretching frequency of the metal-bound cyanide ligand depends sensitively on the geometry of the cofactor, its electronic configuration, as well as on the electrostatics in the binding pocket, including hydrogen bonding interactions.40,41 To assess the importance of these parameters and to relate the observed frequency with the crystallographic data, we have employed QM/MM calculations. Starting with the coordinates of the high-resolution structural model determined in this work, the QM/MM-optimised cofactor structure displays a very good agreement, but the corresponding CN stretching frequency is distinctly underestimated by 36 cm−1. This deviation is much larger than the inherent inaccuracy of the theoretical approach (Table S2 of ESI†). In contrast, a good frequency match was obtained for the optimised structure of the Lys563A variant in which the positively charged side chain in the vicinity of the cyanide ligand is removed. Furthermore, the optimised geometry of the C-cluster displays a large tilt angle (ca. 24°) which is also obtained upon optimizing the high-resolution wild-type structure but using a deprotonated Lys563. Here the increase of the tilt angle is accompanied by an increased nitrogen–nitrogen distance of the cyanide ligand to the Lys563 from 3 to 4 Å, suggesting a substantial weakening of the (electrostatic) interactions with the cyanide ligand. Also in this case, the calculated frequency agrees very well with the experimental value. The same large nitrogen–nitrogen distance but a somewhat smaller tilt angle is obtained for the wild-type protein upon raising the initial value for the tilt angle prior to optimization. Again, an excellent agreement with the experimental CN stretching frequency was obtained. In all optimised structures, the Nε atom of His93 remains at a distance of 2.7 Å which, together with the nearly linear (Ni)CN–H–Nε (His93) geometry, points to strong hydrogen bonding interactions. Conversely, the comparison of calculated and experimental CN stretching frequencies indicates that the interactions between the cyanide ligand and Lys563 must be weak, corresponding to a relatively large nitrogen-to-nitrogen distance (4 Å). Such weak interactions point to a deprotonated side chain of Lys563. The potential proton acceptor is readily identified as the hydroxyl ligand of Fe1 which is released upon cyanide binding to the Ni. Furthermore, the time-independent frequency and intensity of the CN stretching indicates that Lys563 remains deprotonated over the time scale of hours, implying that the active site of the cyanide-bound Cred1 state is not accessible for protons from the solution side via the proton channel identified in CODHMt.20
It might be that structural changes along the proton channel in the cyanide-bound complex may impair reprotonation of Lys563. However, the present structure of the cyanide-complex does not permit an unambiguous conclusion (vide infra). As an alternative interpretation, one may invoke a redox-state dependent proton transfer. This view is consistent with the high turnover number for CO oxidation at the Cred1 state which requires a rapid and unidirectional proton translocation from the C-cluster to the solution. The reverse proton translocation occurs during enzymatic CO2 reduction which, in turn, is based on the Cred2 state. Accordingly, the oxidation state of the C-cluster controls the directionality of the proton transfer such that reprotonation of Lys563 is inhibited in the cyanide complex of Cred1.
A deprotonated Lys563, however, is not consistent with the present high-resolution structure of the CODHIICh–CN complex which displays a distance of only 3 Å, concomitant to a smaller tilt angle, and thus points to appreciable Lys563–cyanide interactions. Whereas X-ray crystallography reflects the protein including a protonated side chain, the IR spectra that are identical in the crystalline state at low temperature and in solution at ambient temperature indicate a deprotonated Lys563 residue. To reconcile these findings, we suggest that, during accumulation of the diffraction data, radiation damage at the C-cluster may cause generation of a hydroxyl radical from a water molecule in the vicinity of Lys563, which is associated with a distinctly lower pKa of ca. 11.42 As a consequence, protonation of the Lys side chain could occur.
Conclusions
In the present study we have unambiguously identified the CN stretching mode of the cyanide bound complex of CODHIICh, confirming that a single cyanide is bound to the Ni ion. Additional IR bands observed in previous investigations could be attributed to free cyanide or to degradation productions of the C-cluster. This complete cyanide-induced degradation of the Fe–Ni center only occurs in the presence of an excess of cyanide. In the absence of excess cyanide, the CODHIICh–CN complex is stable for hours at ambient temperature and enzymatic activity towards CO oxidation can even be recovered at elevated temperature. On the basis of QM/MM calculations the CODHIICh–CN complex, cyanide binding to the Ni occurs upon release of the hydroxyl ligand from the Fe1. The hydroxyl ligand captures a proton from the side chain of Lys563. On the other hand, the present crystallographic data point to a protonated Lys563, which is not compatible with the IR spectroscopic data obtained from CODHIICh–CN crystals. This discrepancy is suggested to be due to reprotonation of Lys563 via a water radical, generated in situ upon X-ray irradiation. The IR spectroscopic data, however, indicate that in the cyanide complex Lys563 remains deprotonated for hours. This finding prompted us to suggest a redox-state dependent directionality of proton translocation through the proton channel connecting the solution phase with the active site. This interpretation is consistent with the enzymatic processes that require proton release from and proton uptake by the active site in the Cred1 state (CO oxidation) and Cred2 state (CO2 reduction), respectively.
The present results and the proposed interpretations demand for further experimental and theoretical investigations of the proton translocation mechanisms and the protonation pattern of the different active site states during the enzymatic reaction sequences.
Acknowledgements
This research has been financed by the Deutsche Forschungs-gemeinschaft, Cluster of Excellence “Unicat”. The authors thank the Alexander-von-Humboldt Foundation (to Alexandre Ciaccafava) and the Federation of European Biochemical Societies (to Alexandre Ciaccafava) for financial support.References
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- J. H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS PubMed.
- J. Heo, C. R. Staples, C. M. Halbleib and P. W. Ludden, Biochemistry, 2000, 39, 7956–7963 CrossRef CAS PubMed.
- V. Svetlitchnyi, C. Peschel, G. Acker and O. Meyer, J. Bacteriol., 2001, 183, 5134–5144 CrossRef CAS PubMed.
- S. A. Ensign, Biochemistry, 1995, 34, 5372–5381 CrossRef CAS PubMed.
- J. H. Jeoung and H. Dobbek, J. Biol. Inorg. Chem., 2012, 17, 167–173 CrossRef CAS PubMed.
- Y. Kung, T. I. Doukov, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Biochemistry, 2009, 48, 7432–7440 CrossRef CAS PubMed.
- J. H. Jeoung and H. Dobbek, J. Am. Chem. Soc., 2009, 131, 9922–9923 CrossRef CAS PubMed.
- J. Fesseler, J.-H. Jeoung and H. Dobbek, Angew. Chem., Int. Ed., 2015, 54, 8560–8564 CrossRef CAS PubMed.
- W. Gong, B. Hao, Z. Wei, D. J. Ferguson, T. Tallant, J. A. Krzycki and M. K. Chan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9558–9563 CrossRef CAS PubMed.
- P. Amara, J. M. Mouesca, A. Volbeda and J. C. Fontecilla-Camps, Inorg. Chem., 2011, 50, 1868–1878 CrossRef CAS PubMed.
- D. Qiu, M. Kumar, S. W. Ragsdale and T. G. Spiro, J. Am. Chem. Soc., 1997, 119, 11134 Search PubMed.
- D. Qiu, M. Kumar, S. W. Ragsdale and T. G. Spiro, J. Am. Chem. Soc., 1996, 118, 10429–10435 CrossRef CAS.
- U. Mueller, N. Darowski, M. R. Fuchs, R. Forster, M. Hellmig, K. S. Paithankar, S. Puhringer, M. Steffien, G. Zocher and M. S. Weiss, J. Synchrotron Radiat., 2012, 19, 442–449 CrossRef CAS PubMed.
- M. Krug, M. S. Weiss, U. Heinemann and U. Mueller, J. Appl. Crystallogr., 2012, 45, 568–572 CrossRef CAS.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubak, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- E. J. Kim, J. Feng, M. R. Bramlett and P. A. Lindahl, Biochemistry, 2004, 43, 5728–5734 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- L. Yu, C. Greco, M. Bruschi, U. Ryde, L. De Gioia and M. Reiher, Inorg. Chem., 2011, 50, 3888–3900 CrossRef CAS PubMed.
- S. R. Billeter, A. J. Turner and W. Thiel, Phys. Chem. Chem. Phys., 2000, 2, 2177–2186 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.1), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, Hoboken, N. J., 2009 Search PubMed.
- S. Yoshikawa, D. H. O'Keeffe and W. S. Caughey, J. Biol. Chem., 1985, 260, 3518–3528 CAS.
- K. S. Reddy, T. Yonetani, A. Tsuneshige, B. Chance, B. Kushkuley, S. S. Stavrov and J. M. Vanderkooi, Biochemistry, 1996, 35, 5562–5570 CrossRef CAS PubMed.
- S. W. Ha, M. Korbas, M. Klepsch, W. Meyer-Klaucke, O. Meyer and V. Svetlitchnyi, J. Biol. Chem., 2007, 282, 10639–10646 CrossRef CAS PubMed.
- S. Kim, D. C. Sorescu and J. T. Yates, J. Phys. Chem. C, 2007, 111, 5416–5425 CAS.
- P. Yu, F. Yang, J. Zhao and J. Wang, J. Phys. Chem. B, 2014, 118, 3104–3114 CrossRef CAS PubMed.
- G. M. Sando, Q. Zhong and J. C. Owrutsky, J. Chem. Phys., 2004, 121, 2158–2168 CrossRef CAS PubMed.
- Y. Kung and C. L. Drennan, Curr. Opin. Chem. Biol., 2011, 15, 276–283 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
- V. J. DeRose, J. Telser, M. E. Anderson, P. A. Lindahl and B. M. Hoffman, J. Am. Chem. Soc., 1998, 120, 8767–8776 CrossRef CAS.
- Z. G. Hu, N. J. Spangler, M. E. Anderson, J. Q. Xia, P. W. Ludden, P. A. Lindahl and E. Munch, J. Am. Chem. Soc., 1996, 118, 830–845 CrossRef CAS.
- M. Horch, Y. Rippers, M. A. Mroginski, P. Hildebrandt and I. Zebger, ChemPhysChem, 2013, 14, 185–191 CrossRef CAS PubMed.
- Y. Rippers, M. Horch, P. Hildebrandt, I. Zebger and M. A. Mroginski, ChemPhysChem, 2012, 13, 3852–3856 CrossRef CAS PubMed.
- G. A. Poskrebyshev, P. Neta and R. E. Huie, J. Phys. Chem. A, 2002, 106, 11488–11491 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1. Statistics on data collection and structure refinement. See DOI: 10.1039/c5sc04554a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |