
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular mechanism of cardiolipin-mediated assembly of respiratory chain supercomplexes†
C.
Arnarez‡
,
S. J.
Marrink
and
X.
Periole
*
Groningen Biomolecular Sciences and Biotechnology Institute, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands. E-mail: x.periole@rug.nl; Fax: +31-503634398; Tel: +31-503632462
First published on 15th March 2016
Abstract
Mitochondria produce most of the ATP consumed by cells through the respiratory chain in their inner membrane. This process involves protein complexes assembled into larger structures, the respiratory supercomplexes (SCs). Cardiolipin (CL), the mitochondrial signature phospholipid, is crucial for the structural and functional integrity of these SCs, but it is as yet unclear by what mechanism it operates. Our data disclose the mechanism for bulk CL in gluing SCs, steering their formation, and suggest how it may stabilize specific interfaces. We describe self-assembly molecular dynamics simulations of 9 cytochrome bc1 (CIII) dimers and 27 cytochrome c oxidase (CIV) monomers from bovine heart mitochondria embedded in a CL-containing model lipid bilayer, aimed at mimicking the crowdedness and complexity of mitochondrial membranes. The simulations reveal a large diversity of interfaces, including those of existing experimental CIII/CIV SC models and an alternative interface with CIV rotated by 180°. SC interfaces enclose 4 to 12 CLs, a ∼10 fold enrichment from the bulk. Half of these CLs glue complexes together using CL binding sites at the surface of both complexes. Free energy calculations demonstrate a larger CL binding strength, compared to other mitochondrial lipids, that is exclusive to these binding sites and results from non-additive electrostatic and van der Waals forces. This study provides a key example of the ability of lipids to selectively mediate protein–protein interactions by altering all ranges of forces, lubricate protein interfaces and act as traffic control agents steering proteins together.
Introduction
In the inner membrane of mitochondria, the respiratory chain synthesizes most of the ATP used by our cells. During this process the energy associated with the exchange of electrons is used to build a proton gradient across the membrane, triggering the conversion of ADP in ATP by ATP synthase. Two main ligands (ubiquinone and cytochrome c) and three protein complexes (NADH dehydrogenase, complex I, CI; cytochrome bc1, complex III, CIII; and cytochrome c oxidase, complex IV, CIV) are involved in the formation of a transmembrane potential.Blue-native gel electrophoresis (BN-PAGE) and electron microscopy have been particularly helpful in showing higher order structural organization of complexes into structures named supercomplexes together defining the “respirasome”.1 Supercomplexes (SCs) involving CI, CIII and CIV have been extracted from bovine heart2–4 and potato,5,6 and from yeast with different stoichiometry for CIII and CIV7,8 and other organisms.9 A series of models have been proposed that take into account the existence of these SCs.10–13 They range from stochastic and dynamic contacts between free complexes in the membrane10 to more static arrangement into SCs. More recently a combination of both resulted in a plasticity model.12–14 The balance between static and dynamic (re)arrangements of SCs could be used as a switching mechanism as observed for the photosynthetic complexes in the thylakoid membrane.15 But the fact is that strong evidence supporting the existence and functionality of supercomplexes in vivo is still missing due to the experimental challenges entailed. It has, however, been shown for isolated SCs.12,16 Alternatively, SCs might have evolved to offer a set of weak and reversible interactions to preclude irreversible strong interactions in this high protein concentration medium.17
As for today, it remains unclear which factors determine the structure and stoichiometry of the supercomplexes, but the composition of the mitochondrial membrane plays an active role.18 In particular cardiolipin (CL),19 a phospholipid present in a large concentration in the inner membrane of mitochondria, has been shown to play an essential role in the stability of the supercomplexes20–22 and it has been suggested that it acts as a glue holding the supercomplexes together.20 In recent simulation studies we described a set of CL binding sites on the membrane-exposed surface of two complexes, CIII and CIV,23,24 complementing the other sites found buried in the protein.25,26 Other modelling experiments have revealed a similar picture.27 The fast exchange (∼μs) of bound CL with the bulk membrane precludes their detection in crystal structures28,29 but other approaches have suggested their presence.22 The existence of CL binding sites led us to two hypotheses on the role of CL; either the complexes interact through the CLs located in these binding sites (“bridging” hypothesis) or the complexes use these CLs on the protein surface to define unfavorable interfaces (“blocking” hypothesis).
In this work, we describe the self-assembly of a set of CIIIs and CIVs into supercomplexes using our coarse grain molecular dynamics (CGMD) simulation approach.30,31 This technique allows the issues of system size and slow kinetics pertaining to such systems to be alleviated.32,33 The role and impact of CL on SC architecture is evaluated from the comparison of a CL:POPC mixture with a pure POPC bilayer. The simulations reveal the mechanism by which CL glues the complexes together and suggest how it drives them towards the correct heterodimer. They also reveal the non-additive forces underlying such specific behavior. As a pioneering simulation of multiple proteins in a heterogeneous membrane system, this work will inspire more studies in crowded and complex mixed membrane environments34–36 to unravel biophysical principles of lipid–protein interplay.
Methods
All molecular dynamics (MD) simulations were performed using the GROMACS simulation package version 4.x37 and the Martini-2.0 CG force field for biomolecules38 and its extension 2.1 to protein39 together with the ElNeDyn approach.40 This CGMD approach is well suited to study a large variety of membrane related processes,32 including membrane protein self-assembly30,31 and lipid binding.23,24 See Extended methods in the ESI† for the setup details.The main system studied contains 9 CIII dimers and 27 CIVs (Fig. 1) embedded in a 1:15 CL:POPC molar ratio lipid bilayer used to mimic mitochondrial lipid composition.41,42 While PE lipid head groups are also present in native systems, we show in the ESI (Fig. S9†) that they do not affect lipid binding in a significant manner and were thus not included in the main simulations. The models were built as described previously23,24 and in the ESI.†
 | ||
Fig. 1 Molecular system. (A) Bovine cytochrome bc1 (CIII) and cytochrome c oxidase (CIV). The models of CIII and CIV are identical to the ones used in our previous studies.23,24 Briefly, CIII’s dimer was built from a combination of four experimental structures (PDB entries: 1 l0l,531sqb/1sqq54 and 2a06 (ref. 55)), excluding the six hemes and two iron-sulfur clusters. CIV's monomer was built from the PDB entries 1occ and 2occ,56 also excluding two deeply buried hemes. Details are given in the ESI† Methods section. (B) Matrix view of the membrane-exposed CL binding sites on both complexes.23,24 The detail of the CIII and CIV subunits, their nomenclature (Fig. S1†) and the comparison of predicted CL binding sites to experimental data have been described previously.23,24 The location of the non-CL-binding surface on CIV is indicated by a star. In the simulated system the complexes are embedded in a POPC bilayer containing CL at a 1:15 CL:POPC molar ratio; side (C) and top (D) views. The system shown contains 9 CIII dimers, 27 CIV, 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 462 POPCs, 1175 CLs (∼32000 beads) and the aqueous phase (∼1116000 water beads and ∼2600 sodium ions), thus a total of slightly less than 1400000 CG beads. To ease visualization, the aqueous phase is omitted and to emphasize the relative orientation of the complexes two subunits of each complex are highlighted (A and B in red for CIII and D and G in yellow for CIV). CL topology and parameters were taken from the work of Dahlberg et al.57 (E) View of the CL-containing system after 20 μs of self-assembly simulation. 462 POPCs, 1175 CLs (∼32000 beads) and the aqueous phase (∼1116000 water beads and ∼2600 sodium ions), thus a total of slightly less than 1400000 CG beads. To ease visualization, the aqueous phase is omitted and to emphasize the relative orientation of the complexes two subunits of each complex are highlighted (A and B in red for CIII and D and G in yellow for CIV). CL topology and parameters were taken from the work of Dahlberg et al.57 (E) View of the CL-containing system after 20 μs of self-assembly simulation. | ||
Supercomplex characterization is described in detail in the ESI.† Briefly we used (a) protein contact surfaces based on buried protein surfaces, aij, (b) lipid content at the surface of the individual complexes or at their interface in supercomplexes, a contact being counted when <1.0 nm, (c) the architecture of a SC was defined by the relative orientation of the two complexes following virtual bond analysis43 as we did previously in the case of rhodopsin.31Φ1 and Φ3 define the orientation of CIII and CIV relative to CIV and CIII, respectively, in the plane of the bilayer (Fig. S14A†). Alternatively the relative orientation of the complexes in a SC can be monitored from the location of the COM of the protein interfaces on both complexes defining γ1 and γ3 (Fig. S14B†). The two definitions provide similar results (see Fig. S14C and D†) although slightly different information. γ1 and γ3 are used in Fig. 2; (d) a CL binding site contact map was built considering two sites in contact or shared between the two complexes when their COM distance was less than 3.0 nm, and (e) lipid binding strengths were quantified from the potentials of mean force (PMFs) of lipid molecules to the surfaces of CIV. PMFs were calculated using an umbrella sampling technique (6 μs per umbrella simulation) with the distance between the center of mass of the lipid head group and protein surface as a reaction coordinate covering from the lipid-bound situation to free in the bulk membrane (up to 3 nm spaced by 0.1 nm). The weighted histogram analysis method (WHAM)44 was used to combine and unbias the simulations, and to produce free energy profiles. The Bayesian bootstrapping method on independent umbrella windows was used to estimate the statistical error of the calculations.
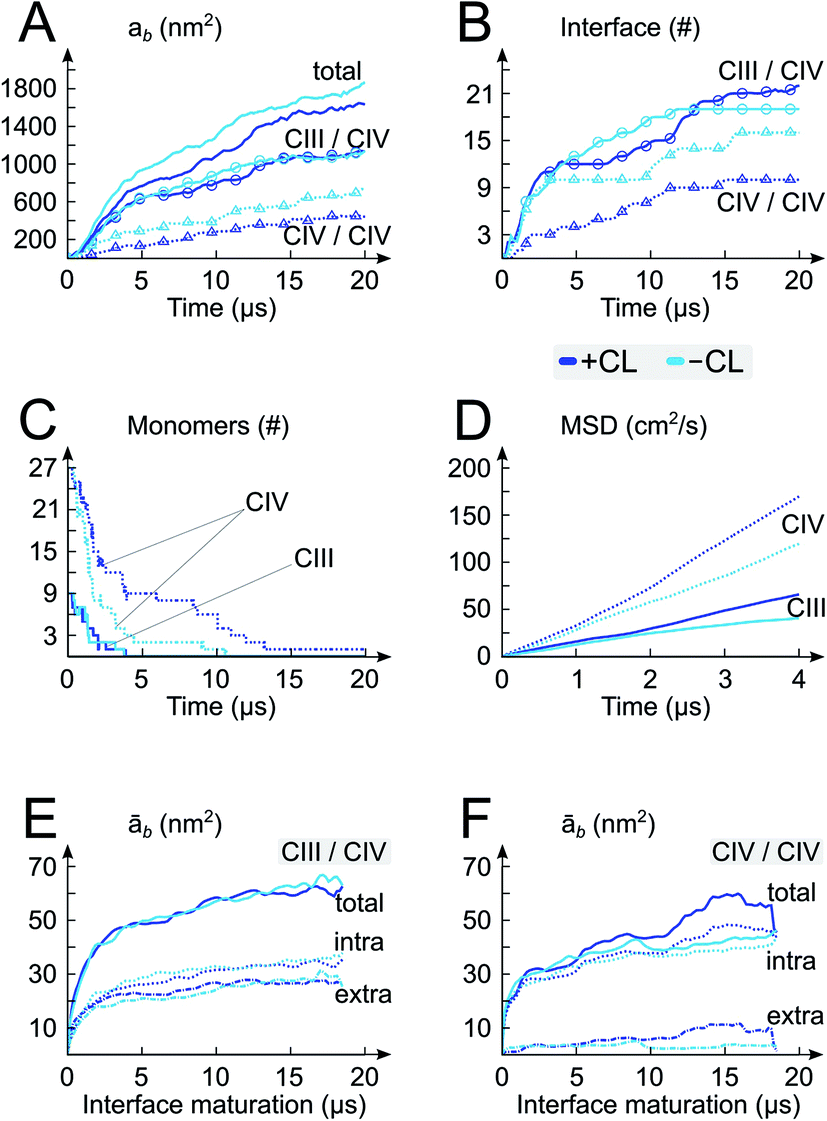 | ||
| Fig. 2 Self-assembly process and architecture of respiratory chain supercomplexes. The time evolution of properties of the system with (+CL; blue) and without (-CL; cyan) cardiolipins are reported: (A) protein burial, ab, with contributions from the CIII/CIV and CIV/CIV interfaces, (B) numbers of interfaces, (C) numbers of CIII and CIV monomers, (D) the mean square displacement (MSD) of the CIII and CIV monomers in isolation in both membrane environments (+CL and -CL), and (E and F) average protein burial during CIII/CIV and CIV/CIV interface maturation with intra- and extra-membraneous contributions. | ||
Results
We performed a 20 μs CGMD simulation of the self-assembly of a set of 9 cytochrome bc1 dimers (CIII) and 27 cytochrome c oxidase monomers (CIV) into supercomplexes (SCs) in a mitochondrial membrane modelled by a POPC lipid bilayer containing ∼13% CLs (phosphorus content, Fig. 1). A bilayer with the same phosphorus content but composed solely of POPC lipids, was used as a control. Starting from a crowded initial distribution of the complexes designed to favor the formation of CIII/CIV interfaces (alternating CIII and CIV on a grid, see Fig. 1 and S2†), SCs formed in both membrane environments involving most proteins in a SC (Fig. 1E and 2A–C). Although we performed only two simulations, abundant CIII/CIV and CIV/CIV interfaces are observed. One CIII/CIII formed but was discarded from the analysis presented.CL disfavor interactions between CIVs
A quantitative analysis of the progressive oligomerization of the complexes points towards a specific effect of CL on CIV interactions (Fig. 2 and 3). This observation is possible by monitoring the protein buried surface area or protein burial, ab, which corresponds to the amount of protein surface area involved in contacts with other proteins and thus not available for contacts with lipids or water molecules. The presence of CL decreases the protein burial associated with CIV/CIV contact but not of CIII/CIV (Fig. 2A), in line with the reduced number of interfaces in which CIV is involved with itself while those with CIII are barely affected (Fig. 2B) and with the reduced involvement of CIV monomers into protein contacts (Fig. 2C). It is also interesting that the rate of formation of CIV/CIV contacts is significantly decreased in presence of CL (Fig. 2B) despite the fact that CL increases the translational self-diffusion of both CIV and CIII (Fig. 2D). | ||
| Fig. 3 Supercomplex architectures: (A) location of CIII/CIV interfaces on CIII (top panels) and CIV (bottom panels) in +CL and -CL membranes. Each stick represents the projection of an interface onto a circle surrounding the protein. The location of an interface, γ1 and γ3, is determined by the position of the centers of mass of the residues contributing to it for each partner (see Fig. S14 and the ESI† Methods for details). The length of a stick reports the protein burial, ab, corresponding to the interface. Similar analysis for CIV/CIV interfaces is shown in Fig. S5.† The favored interfaces found with CL (see panels B and E) are shown by a larger head stick. A triangle points towards the denser zone of contacts on CIII and CIV in the system with and without CL. (B) Contact map of CL binding sites in CIII/CIV SCs from the simulations +CL and -CL. Sites are assumed to form a contact when distant by <3.0 nm (see Methods in the ESI†). Similar analysis for CIV/CIV SCs is shown in Fig. S5.† Sites on the inter-membrane space side of the bilayers (VICIV, VIIaCIV and VIIbCIV) are not reported. In all cases, the C2 symmetry axis of the CIII dimer is used to average over both monomers. (C and E) Simulated vs. experimental and plasticity of CIII/CIV interfaces. Experimental models were derived: for bovine heart mitochondria (panel C) from Althoff et al.4 or Dudkina et al.,3 (panel E) from Schäfer et al.2 and for yeast (panel D) Heinemeyer et al.7 or Mileykovskaya et al.8 The CL binding sites are colored when in contact (<3.0 nm) following the color code defined in Fig. 1 or left grey otherwise. The yeast model was provided by Heinemeyer et al. Althoff's bovine model was taken from the PDB entry 2ybb. Schäfer's model was built by visual fitting of CIII and CIV onto Fig. 2 and 3 from ref. 2 and thus should be considered qualitatively. The surface SCs are projected onto the membrane plane from the matrix with the orientation of CIII conserved. The distance between cytochrome c, dCytc, binding sites on CIII and CIV are given in (C)–(E) and detailed in Fig. S11.† In (E), the compatibility of the configuration with its association with complex I is indicated by a green (possible) or red (not possible) circle, see Fig. S12† for details. | ||
Maturation of supercomplexes and their interfaces
Overall the formation and maturation of the interfaces corroborate the generic features of membrane protein assembly30,31 and proteins in general.45–47 After the formation of a diffusional encounter complex, a subsequent re-orientation of the proteins maximizing the protein complementarity (burial) is associated with delipidation of the protein interfaces (maturation phase, Fig. 2–4, S4 and S5†).We determined the protein burial for each SC interface individually and analyzed them collectively as a function of the protein interface maturation (Fig. 2E and F and S3†). We found that all interfaces form ∼75% of the final protein burial within 4 μs at similar rates. The main structural reorientation of the protein complexes occurs concomitantly with the initial protein contact followed by a slow maturation of the interfaces. It is important to stress the large number and relative variety of complexation events observed in our simulations. The spread in our data is important with maturation times ranging from 4 μs to still evolving after 15 μs (Fig. 4 and S4†) and individual interfacial lipid content (see below) varying by a factor up to three (Fig. 4 and S5†). This variety is illustrated by two examples of interface maturation of the CIII/CIV SC (Fig. 4), the full set is shown in Fig. S3.†
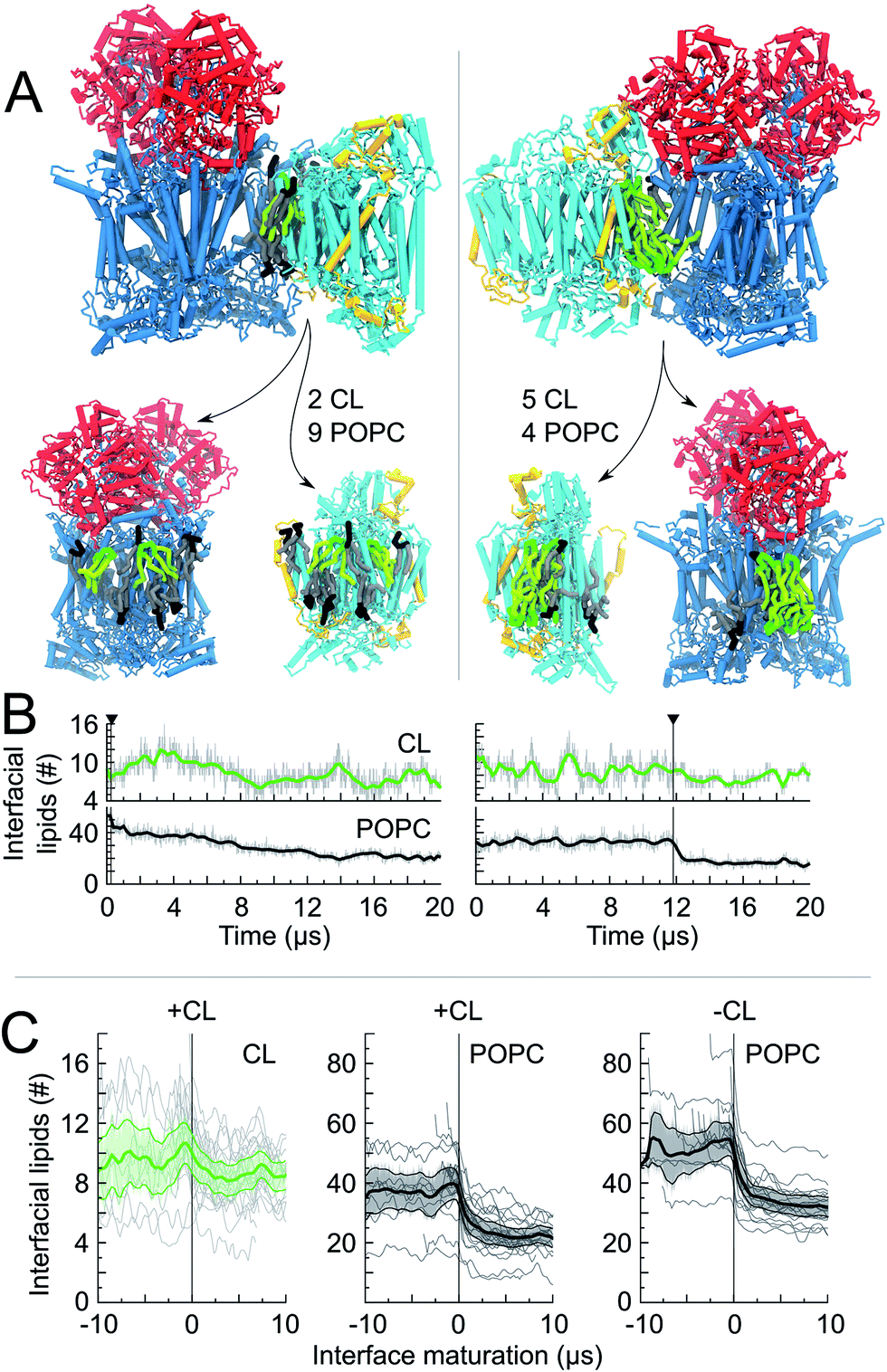 | ||
| Fig. 4 Lipid content of CIII/CIV interfaces. For two CIII/CIV SCs we show: (A) a snapshot of the interfaces, t = 20 μs, which is cut open to show the lipid content (CL and POPC) at the protein surfaces. The number of shared lipids is indicated. In one case (left) the interface is formed early in the simulation and is followed by a long maturation of the interface with POPC being removed at timescales up to 14 μs. In the second case (right) the interface is formed late and POPC molecules are expelled quickly; (B) the time evolution of the number of interfacial lipids for the interface depicted in the snapshot. The vertical line denotes the formation of the interface; (C) the average number of interfacial lipids (CL and POPC) over all interfaces formed in the simulations; time relates here to interface maturation: t = 0 corresponds to the time of formation of the first contact of an interface. | ||
Analysis of the relative contributions of extra-membraneous versus intra-membraneous protein burials to the total protein burial (Fig. 4B and S4†) demonstrates a significant amount of extra-membraneous contacts between CIII–CIV, reflecting the contribution from the large periplasmic domain of CIII (Fig. 1). The effect of CL on the relative contribution of intra- and extra-membraneous interfaces appears limited.
Preferential CIII/CIV contact in presence of CL involves CL binding sites
Analysis of the location of the protein contacts on CIII and CIV in CIII/CIV SCs reveals that both proteins show a dispersed but non-uniform distribution of their interaction sites (Fig. 3A). Notably, interfaces appear denser on a specific region for both CIII and CIV (see the arrows in Fig. 3A). This region on CIII is not affected by CL, but in the case of CIV, CL induces a shift of the densest contact zone from one side of CIV to the other side. Interestingly, for both CIII and CIV, large regions of the protein surface remain devoid of contacts and are therefore not involved in SC formation. For CIII, the non-interacting region corresponds to the side where subunit K is located; the dynamic gate keeper of CIII's inner cavity23 is thus operational (Fig. S10†). No effect of CL was observed for CIV/CIV contacts and the same regions were excluded from contacts (Fig. S6 and S7†).Projection maps of the relative orientations of CIII and CIV in SCs reflect the large variety of SC organization found in our simulations (Fig. 3A and B, S3 and S6–S8†), and also reveal that a few SCs combine the favored interfaces from both complexes (encircled in Fig. S8A†) and do so in a CL-dependent manner. These features become more pronounced when analyzing the proximity of CL binding sites in the SCs (Fig. 3B). In ∼30% (6 vs. 21) of the CIII/CIV SCs formed in the presence of CL, CIII and CIV assemble in such way that sites III and IVa on CIII and site Va on CIV are in close proximity (Fig. 3B). Similar behavior is not observed in the absence of CL, nor in the case of CIV/CIV SCs (Fig. S6†). Interestingly, the CL binding site contacts in CIII/CIV SCs seem an intrinsic property since the presence of CL only slightly increases their number per CIII/CIV interface from 1.7 to 2.0.
Experimental SC models may be compared to the ones formed in our simulations (Fig. 3C–E). Among the CIII/CIV SC configurations formed in the simulations we found configurations that strongly resemble the SC models derived from EM experiments on bovine heart2–4 and yeast7,8 mitochondria. SC interfaces connecting similar CL binding sites on both complexes (see previous paragraph) present a certain plasticity, which is illustrated by a collection of CIII/CIV SCs interfaces experiencing small differences in the relative orientation of the proteins (Fig. 3E).
CL enrichment at the supercomplex interfaces precedes their assembly
The quantification of the lipid content of the complexes' solvation shell shows that the CL:POPC at the surface of the proteins is already drastically higher than the overall 1:15 ratio of the bulk membrane. This increase reflects the occupation of surface-exposed CL binding sites we reported earlier (Fig. 1), but also transient contacts.23,24 Furthermore, this analysis shows a strong CL enrichment of the overall solvation shell of the complexes during SC assembly, and most interestingly of the protein surfaces engaging in SC interfaces (Fig. 4C and Table 1, S1†). These increases mainly result from a large depletion of POPC, while CL mainly remains at the protein surfaces (Fig. 4, S5A and Table 1, S1†).
|
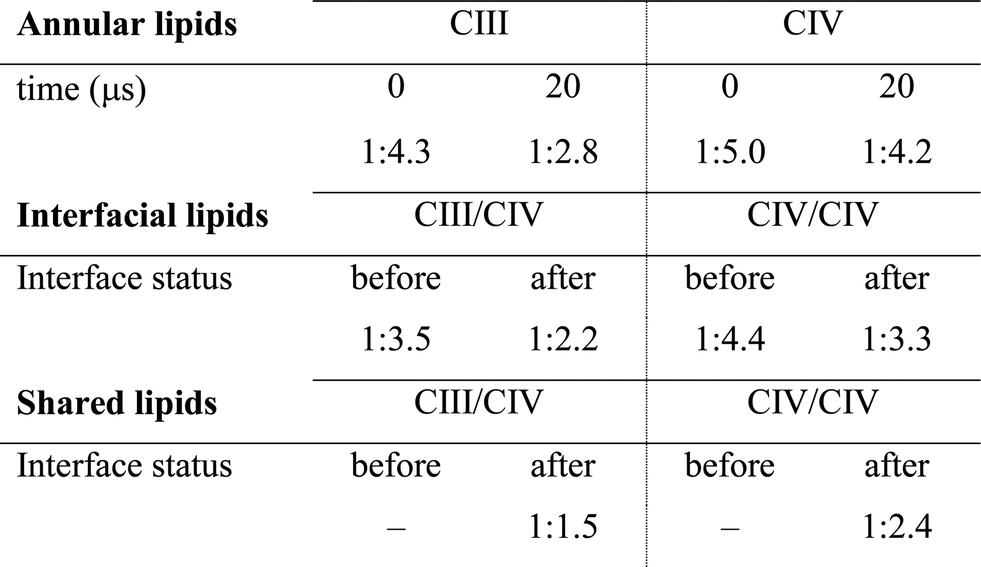
Note the higher CL:POPC molar ratios of the protein surfaces engaging in SC interfaces prior to their involvement in a contact compared to the ratios averaged over the entire protein surface: 1:3.5 and 1:4.4 vs. 1:4.3 and 1:5.0 for CIII/CIV and CIV/CIV, respectively (Table 1). This difference unambiguously shows that the protein surfaces involved in contact are enriched in CL prior to their assembly, demonstrating that supercomplexes associate where CL is denser on the complexes prior to assembly. CL thus determines SC interfaces.
The interfacial lipid content takes a few μs of interface maturation to reach stable values (Fig. 4C, S3, S5A, B and Table S1†). On average, in a CIII/CIV interface the complexes share 6 POPC and 4 CL molecules in the mixed membrane, and 10 POPC in the membrane devoid of CL. In the case of a CIV/CIV interface, we found 6 POPC and 2.5 CL molecules, and 7 POPC for systems with and without CL, respectively. The CL:POPC molar ratios of these shared lipids, 1:1.5 and 1:2.4 for CIII/CIV and CIV/CIV, respectively (Table 1), show a further increase of CL enrichment of the shared section of SCs interfaces.
Non-additive electrostatic and van der Waals forces make CL a stronger glue
The enrichment in CL over POPC at the protein surface is in line with our previous estimate of the binding free energy,24 showing CL to be a stronger binder than POPC. We have extended this analysis to find that CL is actually a stronger binder than all lipid types present in the inner mitochondrial membrane: POPG, POPE and POPC (Fig. 5A). Interestingly, this differentiation in binding strength is specific to CL's binding sites. It is not observed on a non-binding region of the protein where most lipids tested do not show any binding affinity (Fig. 5B). This data suggests that the conservation of CL at the interface of the complexes is predominately due to their binding to specific sites, not to the full protein surface.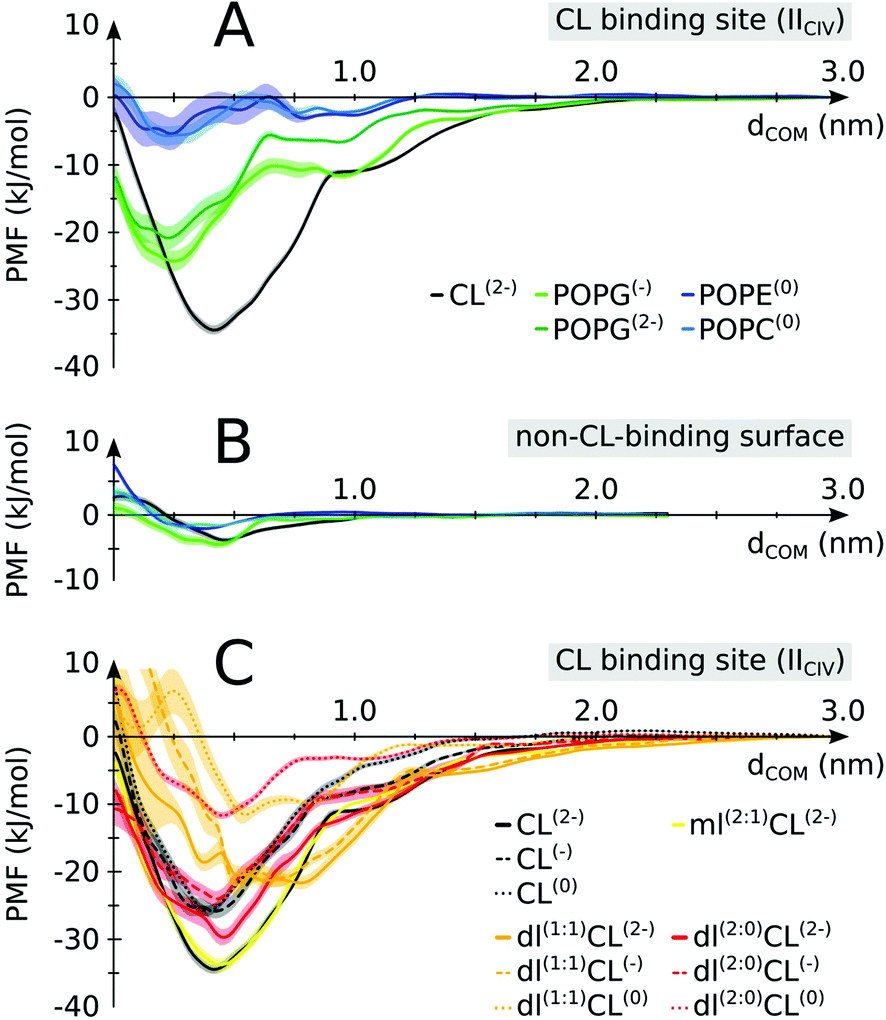 | ||
| Fig. 5 Lipid binding affinities. Potentials of mean force (PMFs) were determined for (A) CL vs. other lipids on a CIV CL binding site (IICIV); (B) CL vs. other lipids on a non-CL-binding region (indicated in Fig. 1 and defined in S1,† color coding as in panel A). (C) Native vs. non-native CL: full tailed CL is compared to monolyso-CL (mlCL) and dilyso-CL (dlCL) variants with different charged head groups. Shaded areas indicate the error bars. For mlCL and dlCL, the number of aliphatic tails carried by each glycerol moiety are indicated within brackets. E.g.: dl(1:1)CL(2−) is a dilyso-CL with one acyl chain on each head glycerol moiety and a −2e charge. See Table S3† for a detailed account of the respective values. | ||
To elucidate the specificity of CL in gluing complexes into SCs we determined the PMF of binding for modified CLs. We altered the two features that make CL unique: the double negative charge carried by its head group and the four acyl chains that make it very bulky (Fig. 1). The progressive decrease in the head group charge and the number of tails reduces the binding strength of CL (Fig. 5C). Expectedly, the data show that the strength of CL binding results from a delicate balance of Coulomb and van der Waals forces. Most striking is that these forces are not strictly additive, but cooperative, i.e. removing the 2e− charge or two tails from the native CL has much less an effect than if CL already has tails or its charge missing (Fig. 5C and Table S3†). This might be a natural protective mechanism to preserve CL specific function and avoid interferences from other lipids, either bulky or carrying a charge but not both simultaneously as CL. Also of note is that our calculation predicts monolyso-CL would be as efficient as CL to stabilize SCs when this particular CL binding site is involved. The stability of dlCL2− compared to an artificial POPG2− (Fig. 5A and C) indicates that the distribution of the charge over two sites is an important feature of CL binding strength. The distribution of the tails in dlCL also has a significant effect on its binding strength. dlCL binds stronger with the two tails attached to the same lipid head (Fig. 5C).
Discussion
Biological relevance of an alternative CIII/CIV interface
It is particularly exciting that a similar CIII/CIV interface showed up in ∼30% of the SCs formed in the membrane containing CL (Fig. 3B). Comparison to the CIII/CIV interface modelled from cryo-EM experiments on bovine heart mitochondria3,4 (comparable to potato)6 shows that the interface is similar on CIII but CIV is rotated by ∼180° (Fig. 3C and E). But it is identical to an earlier model also built from EM data on bovine heart mitochondria,2,48 later dismissed as resulting from a low resolution of the EM data. Dudkina/Althoff's bovine heart model and Heinemeyer/Mileykovskaya yeast model of CIII/CIV interfaces are also found in the simulation (Fig. 3C and D).It is important to recall at this point that there is absolutely no direct evidence for a biological function of supercomplexes and even less for a specific CIII/CIV interface besides their possible extraction from in vivo and in vitro systems. In contrast, recent work has suggested the heterogeneity of respirasomes.11,12 In line with these observations, it is interesting to stress the relative diversity in the way the complexes are organized into supercomplexes, although denser zones of contact are visible on each complex. In particular, the apparent flexibility of the favored interface (Fig. 3E) allows the integration of a few of these CIII/CIV supercomplex configurations to the CI/CIII/IV bovine supercomplex4 (Fig. 3E and S12†).
Of potential biological relevance to the alternative CIII/CIV supercomplex model found in the simulation is the reduced distance between the cytochrome c binding sites on CIII and CIV, dCytc, compared to previous experimental models (Fig. 3C–E). Cytochrome c uses these binding sites to transport an electron from CIII to CIV. The distance is systematically smaller than in the models by Schäfer et al.2 and by Dudkina et al.3 or Althoff et al.4 for bovine heart (Fig. 3C and E and S11†). In all conformations of the alternative model, dCytc would allow direct channelling of cytochrome c between the two complexes. Moreover, in these conformations the distance remains in a small range (4.5–6.1 nm) (Fig. 3E) as in previous bovine (8–10 nm) and yeast (5.4 nm) experimental models (Fig. S11,†3,4). Also notable is the systematic reduction of dCytc in the models obtained through the simulations compared to the experimental ones in the bovine system. In the case of yeast the CG model has a larger dCytc but the difference might not be significant. However, dCytc might be highly subjective to the method of estimation.16 We provide here a coherent set of values.
The formation of the various models in the simulations with and/or without CL shows that they are accessible to the complexes, and suggests that structural features of the proteins allow only a restricted set of interfaces. The membrane composition and other environmental or experimental conditions might intervene to favor a particular interface. These observations would be in line with a flexible model with a set of non-functional weak interfaces.17 In the simulations despite the unprecedented level of realism of the complexity and crowdedness of membrane matrix we can not exclude that the lack of complex I,4 PE headed lipids and Rcf1 (ref. 49–51) might affect the CIII/CIV complexation that we observe (see discussion in the ESI†).
Bridging or blocking mechanism for CL gluing
The main hypothesis tested in this work is how CL may act to “glue the respiratory chain together”.20 Since it was first established that CL glues respiratory complexes, it was proposed that CLs from the membrane bulk (as opposed to the tightly bound ones found in the crystal) are actually involved in the mechanism. The presence of CL in SCs, filling the gap at the complex interface, was first suggested by data showing that 50 CLs associate to the CIII–CIV SC,8 in large excess to the number of CLs found on individual complexes.52 Our simulations confirm a CL excess in the SCs and more specifically at their interfaces. We found that the CL:POPC molar ratio increases from 1:15 in the bulk to 1:5 in the annular shell of the isolated proteins and 1:2–3 considering the contact zones (Tables S1 and S2†). Close to 50% of the CLs at the protein interface are shared between the two complexes.Furthermore, we have previously shown that bulk CL binds to the individual complexes at specific locations defining CL binding sites on the surface of the complexes,23,24 suggesting that CL binding sites may act by either bridging or blocking complexes' surfaces. We found here that, on average, 2 contacts between CL binding sites are formed per CIII/CIV interface with an average of 4 shared CLs, thus pointing to a bridging mechanism for CL in gluing the respiratory chain complexes together through CL binding sites. The comparison of CLs’ binding strengths to those of other lipids (Fig. 5A) clearly indicates that CL is a significantly stronger binding moiety and thus a stronger glue. This model of the role of CL in SC formation is summarized by the schematic in Fig. 6.
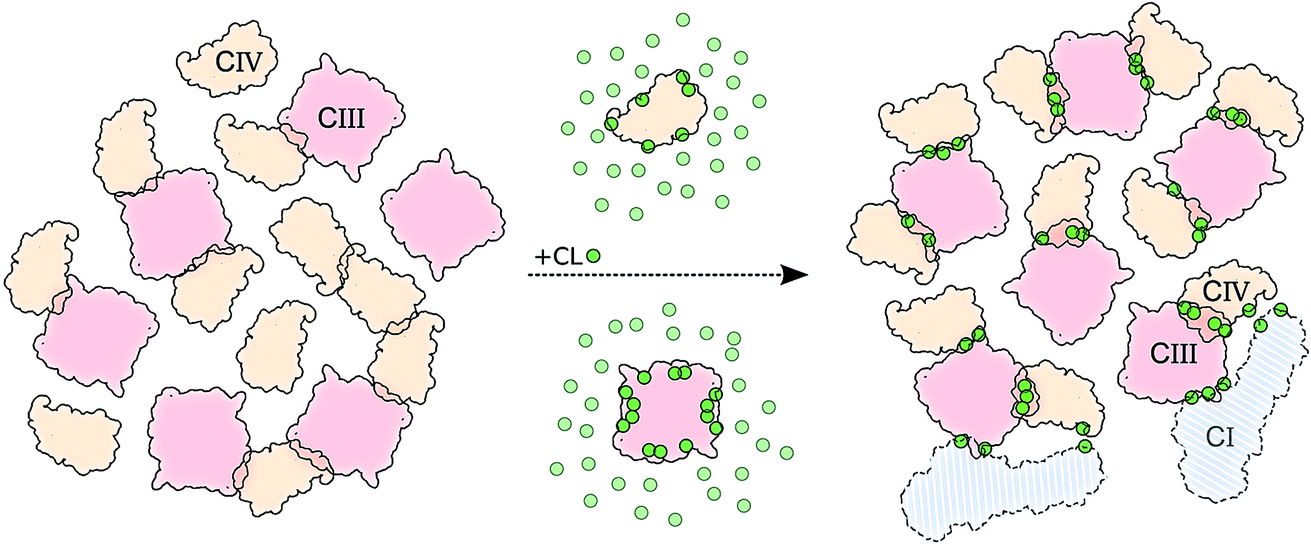 | ||
| Fig. 6 Schematic model of CL implication in the formations of supercomplexes. The model shows how in the presence (right side) of CL (green dots) we observed: (i) an increased number of CIII/CIV interfaces but not of CIV/CIV, and (ii) stronger and (iii) more specific interfaces. CIII is depicted in light red and CIV in light orange. Two copies of CI are shown to illustrate its possible integration to the CIII/CIV SC formed in the simulations (Fig. S12†). | ||
It is tempting to further hypothesize that the variable binding strength and occupation of CL for different binding sites24 could be a strategy used to favor certain interfaces, steering the complexes. Steering is also supported by the observation that protein surfaces densely populated in CL prior SCs assembly. Further experimental and computational studies are needed to explore this idea.
In summary, our simulations have revealed important features of supercomplex formation of the respiratory chain complexes embedded in a mitochondrial membrane model. Most notable is the mechanism by which CL glues and steers the complexes into SCs using its binding sites at the surface of both complexes. We also elucidate CL specificity. On a more general note, our study shows that the mechanisms underlying supercomplex formation are accessible to contemporary cutting-edge computer simulations, opening the way for simulating lipid-mediated protein–protein association in realistic membrane environments.34
Acknowledgements
The authors would like to express their gratitude to W. Dowhan, E. Mileykovskaya and T. Huber for critical reading of the manuscript and insightful discussions. This work was supported by the Netherlands Organisation for Scientific Research (NWO) (to X.P and SJM through an ECHO and a Dutch Computational Challenge Grant).References
- H. Schägger and K. Pfeiffer, EMBO J., 2000, 19, 1777–1783 CrossRef PubMed.
- E. Schäfer, H. Seelert, N. H. Reifschneider, F. Krause, N. A. Dencher and J. Vonck, J. Biol. Chem., 2006, 281, 15370–15375 CrossRef PubMed.
- N. V. Dudkina, M. Kudryashev, H. Stahlberg and E. J. Boekema, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15196–15200 CrossRef CAS PubMed.
- T. Althoff, D. J. Mills, J.-L. Popot and W. Kühlbrandt, EMBO J., 2011, 30, 4652–4664 CrossRef CAS PubMed.
- H. Eubel, J. Heinemeyer and H. P. Braun, Plant Physiol., 2004, 134, 1450–1459 CrossRef CAS PubMed.
- J. B. Bultema, H. P. Braun, E. J. Boekema and R. Kouřil, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 60–67 CrossRef CAS PubMed.
- J. Heinemeyer, H. P. Braun, E. J. Boekema and R. Kouřil, J. Biol. Chem., 2007, 282, 12240–12248 CrossRef CAS PubMed.
- E. Mileykovskaya, P. A. Penczek, J. Fang, V. K. P. S. Mallampalli, G. C. Sparagna and W. Dowhan, J. Biol. Chem., 2012, 287, 23095–23103 CrossRef CAS PubMed.
- Y. Chaban, E. J. Boekema and N. V. Dudkina, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 418–426 CrossRef CAS.
- C. R. Hackenbrock, B. Chazotte and S. S. Gupte, J. Bioenerg. Biomembr., 1986, 18, 331–368 CrossRef CAS PubMed.
- R. Acín-Pérez, M. P. Bayona-Bafaluy, P. Fernández-Silva, R. Moreno-Loshuertos, A. Perez-Martos, C. Bruno, C. T. Moraes and J. A. Enriquez, Mol. Cell, 2004, 13, 805–815 CrossRef.
- R. Acín-Pérez, P. Fernández-Silva, M. L. Peleato, A. Pérez-Martos and J. A. Enriquez, Mol. Cell, 2008, 32, 529–539 CrossRef PubMed.
- R. Acín-Pérez and J. A. Enriquez, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 444–450 CrossRef PubMed.
- C. A. Porras and Y. Bai, Front. Biosci., 2015, 20, 621–634 CrossRef.
- J. F. Allen, Physiol. Plant., 1995, 93, 196–205 CAS.
- M. L. Genova and G. Lenaz, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 427–443 CrossRef CAS PubMed.
- J. N. Blaza, R. Serreli, A. J. Y. Jones, K. Mohammed and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15735–15740 CrossRef CAS PubMed.
- S. R. Shaikh, E. M. Sullivan, R. J. Alleman, D. A. Brown and T. N. Zeczycki, Biochemistry, 2014, 53, 5589–5591 CrossRef CAS PubMed.
- G. Paradies, V. Paradies, V. De Benedictis, F. M. Ruggiero and G. Petrosillo, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 408–417 CrossRef CAS PubMed.
- M. Zhang, E. Mileykovskaya and W. Dowhan, J. Biol. Chem., 2002, 277, 43553–43556 CrossRef CAS PubMed.
- K. Pfeiffer, V. Gohil, R. A. Stuart, C. Hunte, U. Brandt, M. L. Greenberg and H. Schägger, J. Biol. Chem., 2003, 278, 52873–52880 CrossRef CAS PubMed.
- E. Mileykovskaya and W. Dowhan, Chem. Phys. Lipids, 2014, 179, 42–48 CrossRef CAS PubMed.
- C. Arnarez, J.-P. Mazat, J. Elezgaray, S.-J. Marrink and X. Periole, J. Am. Chem. Soc., 2013, 135, 3112–3120 CrossRef CAS PubMed.
- C. Arnarez, S.-J. Marrink and X. Periole, Sci. Rep., 2013, 3, 1263 CAS.
- T. Wenz, R. Hielscher, P. Hellwig, H. Schägger, S. Richers and C. Hunte, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 609–616 CrossRef CAS PubMed.
- C. Lange, J. H. Nett, B. L. Trumpower and C. Hunte, EMBO J., 2001, 20, 6591–6600 CrossRef CAS PubMed.
- S. Pöyry, O. Cramariuc, P. A. Postila, K. Kaszuba, M. Sarewicz, A. Osyczka, I. Vattulainen and T. Róg, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 769–778 CrossRef PubMed.
- L. Qin, C. Hiser, A. Mulichak, R. M. Garavito and S. Ferguson-Miller, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16117–16122 CrossRef CAS PubMed.
- K. Shinzawa-Itoh, H. Aoyama, K. Muramoto, H. Terada, T. Kurauchi, Y. Tadehara, A. Yamasaki, T. Sugimura, S. Kurono, K. Tsujimoto, T. Mizushima, E. Yamashita, T. Tsukihara and S. Yoshikawa, EMBO J., 2007, 26, 1713–1725 CrossRef CAS PubMed.
- X. Periole, T. Huber, S.-J. Marrink and T. P. Sakmar, J. Am. Chem. Soc., 2007, 129, 10126–10132 CrossRef CAS PubMed.
- X. Periole, A. M. Knepp, T. P. Sakmar, S.-J. Marrink and T. Huber, J. Am. Chem. Soc., 2012, 134, 10959–10965 CrossRef CAS PubMed.
- M. Baaden and S.-J. Marrink, Curr. Opin. Struct. Biol., 2013, 23, 878–886 CrossRef CAS PubMed.
- H. I. Ingólfsson, C. A. López, J. J. Uusitalo, D. H. de Jong, S. M. Gopal, X. Periole and S.-J. Marrink, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 225–248 CrossRef PubMed.
- H. I. Ingólfsson, M. N. Melo, F. J. van Eerden, C. Arnarez, C. A. López, T. A. Wassenaar, X. Periole, A. H. de Vries, D. P. Tieleman and S.-J. Marrink, J. Am. Chem. Soc., 2014, 136, 14554–14559 CrossRef PubMed.
- T. Reddy, D. Shorthouse, D. L. Parton, E. Jefferys, P. W. Fowler, M. Chavent, M. Baaden and M. S. P. Sansom, Structure, 2015, 23, 584–597 CrossRef CAS PubMed.
- H. Koldsø and M. S. P. Sansom, J. Am. Chem. Soc., 2015, 137, 14694–14704 CrossRef PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- S.-J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- L. Monticelli, S. K. Kandasamy, X. Periole, R. G. Larson, D. P. Tieleman and S.-J. Marrink, J. Chem. Theory Comput., 2008, 4, 819–834 CrossRef CAS PubMed.
- X. Periole, M. Cavalli, S.-J. Marrink and M. A. Ceruso, J. Chem. Theory Comput., 2009, 5, 2531–2543 CrossRef CAS PubMed.
- G. Daum, Biochim. Biophys. Acta, Rev. Biomembr., 1985, 822, 1–42 CrossRef CAS.
- G. van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS PubMed.
- S. Boresch, F. Tettinger, M. Leitgeb and M. Karplus, J. Phys. Chem. B, 2003, 107, 9535–9551 CrossRef CAS.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- R. R. Gabdoulline and R. C. Wade, Curr. Opin. Struct. Biol., 2002, 12, 204–213 CrossRef CAS PubMed.
- R. Grünberg, J. Leckner and M. Nilges, Structure, 2004, 12, 2125–2136 CrossRef PubMed.
- C. Tang, J. Iwahara and G. M. Clore, Nature, 2006, 444, 383–386 CrossRef CAS PubMed.
- E. Schäfer, N. A. Dencher, J. Vonck and D. N. Parcej, Biochemistry, 2007, 46, 12579–12585 CrossRef PubMed.
- M. Vukotic, S. Oeljeklaus, S. Wiese, F. N. Voegtle, C. Meisinger, H. E. Meyer, A. Zieseniss, D. M. Katschinski, D. C. Jans, S. Jakobs, B. Warscheid, P. Rehling and M. Deckers, Cell Metab., 2012, 15, 336–347 CrossRef CAS PubMed.
- Y.-C. Chen, E. B. Taylor, N. Dephoure, J.-M. Heo, A. Tonhato, I. Papandreou, N. Nath, N. C. Denko, S. P. Gygi and J. Rutter, Cell Metab., 2012, 15, 348–360 CrossRef CAS PubMed.
- V. Strogolova, A. Furness, M. Robb-McGrath, J. Garlich and R. A. Stuart, Mol. Cell. Biol., 2012, 32, 1363–1373 CrossRef CAS PubMed.
- S. Bazan, E. Mileykovskaya, V. K. P. S. Mallampalli, P. Heacock, G. C. Sparagna and W. Dowhan, J. Biol. Chem., 2013, 288, 401–411 CrossRef CAS PubMed.
- X. Gao, X. Wen, C.-A. Yu, L. Esser, S. Tsao, B. Quinn, L. Zhang, L. Yu and D. Xia, Biochemistry, 2002, 41, 11692–11702 CrossRef CAS PubMed.
- L. Esser, B. Quinn, Y.-F. Li, M. Zhang, M. Elberry, L. Yu, C.-A. Yu and D. Xia, J. Mol. Biol., 2004, 341, 281–302 CrossRef CAS PubMed.
- L.-S. Huang, D. Cobessi, E. Y. Tung and E. A. Berry, J. Mol. Biol., 2005, 351, 573–597 CrossRef CAS PubMed.
- T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono and S. Yoshikawa, Science, 1996, 272, 1136–1144 CAS.
- M. Dahlberg, J. Phys. Chem. B, 2007, 111, 7194–7200 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04664e |
| ‡ Current address: Department of Physics and Astronomy, University of Delaware, 258 Sharp Lab, 19712 Newark, Delaware, United States of America. |
| This journal is © The Royal Society of Chemistry 2016 |