
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterobimetallic Pd–K carbene complexes via one-electron reductions of palladium radical carbenes†
Peng
Cui
a,
Melissa R.
Hoffbauer
a,
Mariya
Vyushkova
b and
Vlad M.
Iluc
*a
aDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: viluc@nd.edu
bNotre Dame Radiation Laboratory, University of Notre Dame, Notre Dame, IN 46556, USA
First published on 24th March 2016
Abstract
Heterobimetallic Pd–K carbenes featuring Pd–Ccarbene–K moieties were synthesized via an unprecedented sequential substitution/reduction reaction from a radical precursor, [{PC˙(sp2)P}tBuPdI] ([PC(sp2)P]tBu = bis[2-(di-iso-propylphosphino)-4-tert-butylphenyl]methylene). Polymeric structures were observed in the solid state for the heterobimetallic compounds that can be interrupted in the presence of a donor solvent.
Introduction
The influence of Lewis acids in catalysis cannot be underestimated. While most efforts have focused on using them as the sole mediators of chemical transformations,1–5 few examples have been reported that discuss the role of Lewis acids as co-activators in homogeneous catalysis.6–9 These are related to heterogeneous processes,10,11 with a singular example discussing the role of potassium ions on the hydrogenation of dinitrogen in the Haber–Bosch process,12 and biological systems, with several examples reporting that Lewis acids are necessary cofactors that help modulate the redox properties of the oxygen-evolving complex's manganese cluster and likely its reactivity.13–17 In organic synthesis, Shibasaki's rare-earth alkali-metal heterobimetallic complexes18 are among the most enantioselective and broadly used catalysts to date,19–21 but few studies discuss the role of Lewis acids in applications of late transition metals, in general, and of carbene complexes, in particular.22–24Transition metal carbenes are of vital importance to science and have witnessed a tremendous progress in their applications in the past few decades.25–32 Late transition metal complexes with N-heterocyclic,33,34 carbocyclic,35 and heteroatom stabilized carbene ligands36–38 have been extensively studied as a consequence; however, the corresponding non-heteroatom stabilized species, [M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CRR′] (R, R′ = alkyl or H), are less explored.39–45 This situation is even more pronounced for group 10 metals, likely because these metals are too electron rich to stabilize the MC bond. Pioneering work by Hillhouse showed that a Ni(0) carbene containing a CPh2 moiety could be isolated by the thermolysis or photolysis of its diphenyldiazoalkane precursor.46,47 The isolation of the corresponding Pd and Pt carbenes is, however, more challenging than that of nickel complexes,48–54 because of the highly reactive nature of the former.55,56 On the other hand, these species are crucial intermediates in a variety of catalytic transformations,57,58 such as palladium carbene mediated cyclopropanations, cross-coupling with diazo compounds, and migratory insertion reactions.59
CRR′] (R, R′ = alkyl or H), are less explored.39–45 This situation is even more pronounced for group 10 metals, likely because these metals are too electron rich to stabilize the MC bond. Pioneering work by Hillhouse showed that a Ni(0) carbene containing a CPh2 moiety could be isolated by the thermolysis or photolysis of its diphenyldiazoalkane precursor.46,47 The isolation of the corresponding Pd and Pt carbenes is, however, more challenging than that of nickel complexes,48–54 because of the highly reactive nature of the former.55,56 On the other hand, these species are crucial intermediates in a variety of catalytic transformations,57,58 such as palladium carbene mediated cyclopropanations, cross-coupling with diazo compounds, and migratory insertion reactions.59
It is worth mentioning that only two examples of cationic Pd(II) carbene complexes are known (Chart 1, type A), synthesized via triflate or hydride abstraction, and that their reactivity has not been studied.48,49 A salt metathesis strategy was also reported for the synthesis of methanediide-based Pd(II) carbene complexes.50,51 We have recently applied dehydrohalogenation reactions60 to synthesize Pd(II) carbene complexes, [{PC(sp2)P}RPd(PMe3)] (R = H, [PC(sp2)P]H = bis[2-(di-iso-propylphosphino)phenyl]methylene); R = tBu, ([PC(sp2)P]tBu = bis[2-(di-iso-propylphosphino)-4-tert-butylphenyl]methylene).61–63 Interestingly, the Pd–Ccarbene bonds in these compounds are best described as ylide-like (Chart 1, type B), as demonstrated by their strong nucleophilic reactivity toward polar substrates (MeI, HCl, MeOH, para-toluidine),61 strong Lewis acids,62 C–H64 and Si–H65 bond activation reactions. Furthermore, our recent study on the redox-induced umpolung of palladium carbenes revealed that the radical carbene [{PC˙(sp2)P}tBuPdI] (1, Chart 1, type C) bridges cationic and anionic carbenes via reversible one-electron transfer processes.66,67
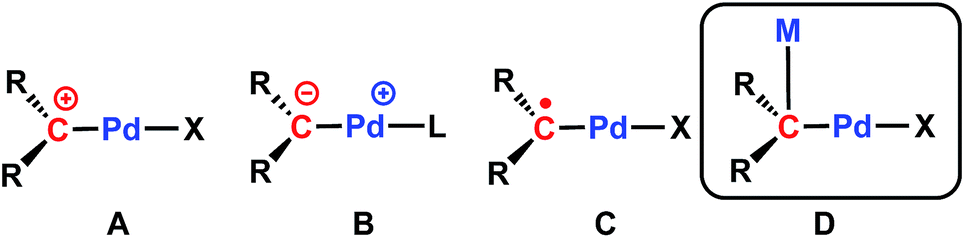 | ||
| Chart 1 Illustration of known palladium carbene complexes: (A) cationic carbene; (B) ylide-like carbene; (C) radical carbene; (D) heterobimetallic carbene discussed in this work. | ||
We reasoned that the presence of iodide as a leaving group in 1 would facilitate its substitution with various anionic nucleophiles to afford new radical carbene species, which would generate Pd(II) carbenes with functional groups when subsequent one-electron reductions are applied. Herein, we report a sequential substitution/reduction reaction of the radical carbene 1 to form heterobimetallic Pd–K carbene complexes featuring amides or benzyl ligands that represent rare examples of late transition metal complexes containing Pd–Ccarbene–M units (Chart 1, type D). Such bonding motifs were recently proposed to be instrumental in the hydroarylation of dienes catalysed by rhodium carbodicarbene complexes, but structural characterization was not available.22
Results and discussion
Synthesis and characterization of metal complexes
Treatment of 1 with potassium amides, R1R2NK, in THF afforded new radical complexes, [{PC˙(sp2)P}tBuPdNR1R2] (2: R1 = H, R2 = pTol; 3: R1 = R2 = Ph, Scheme 1), which were crystallized from n-pentane at −35 °C as dark-green crystals in high yield. As observed for 1,672 and 3 are silent by 1H and 31P NMR spectroscopy and are thermally robust. The effective magnetic moments μeff of 1.70 μB and 1.54 μB obtained using the Evans method68 indicated an S = 1/2 ground state for both compounds. EPR spectra of radicals 2 and 3 (Fig. 1) exhibit similar hyperfine patterns arising from an interaction of the unpaired electron with three pairs of magnetically equivalent protons, one nitrogen nucleus, and a 105Pd nucleus. As the natural abundance of the magnetic 105Pd isotope is 22.33%, each spectrum exhibits contributions from the non-magnetic palladium isotope (strong, well-resolved central signal) and the 105Pd isotope (broad wings). Proton hyperfine couplings were assigned to protons of the phenyl rings adjacent to the radical center. | ||
| Scheme 1 Synthesis of heterobimetallic carbene complexes 4 and 5. | ||
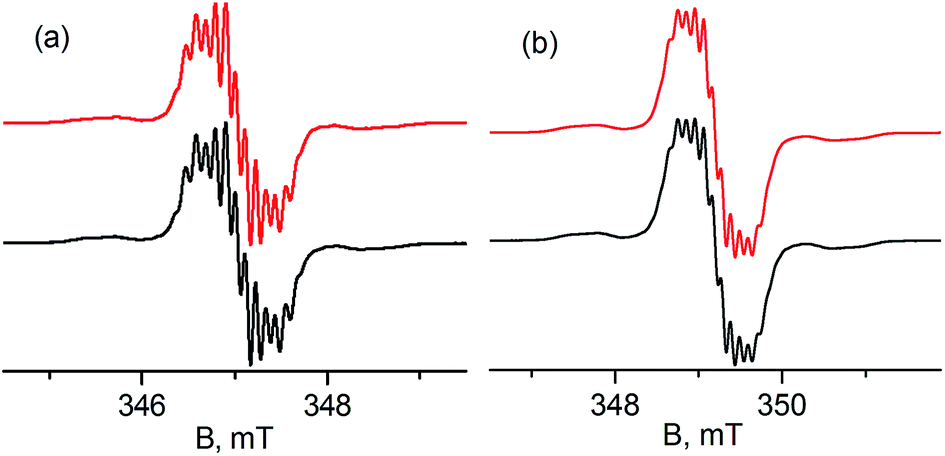 | ||
| Fig. 1 Experimental (black) and simulated (red) X-band EPR spectra of 10−4 M toluene solutions of radicals 2 (a) and 3 (b) at 298 K. Simulation parameters for 2: a1(2H) = 0.32 mT, a2(2H) = 0.13 mT, a3(2H) = 0.11 mT, a(N) = 0.09 mT, a(105Pd) = 0.47 mT, g = 2.0088; for 3: a1(2H) = 0.31 mT, a2(2H) = 0.13 mT, a3(2H) = 0.10 mT, a(N) = 0.09 mT, a(105Pd) = 0.51 mT, g = 2.0079. Contribution from the 105Pd species is 22.33% for both radicals. | ||
The solid state molecular structures of 2 and 3 are consistent with their radical carbene nature (Fig. 2): both contain square-planar palladium centers bound to sp2 hybridized backbone carbons (Σangles at Ccarbene are 359.9° for 2 and 360.0° for 3). The Pd–Ccarbene distances in 2 (2.019(2) Å) and 3 (2.024(2) Å) are comparable to each other and close to the value of 2.022(3) Å in 1.67 Trigonal planar geometries were observed for both amide nitrogen atoms in 2 and 3, with the amide planes roughly perpendicular to the planes defined by Ccarbene, C(11), and C(21) or C(11)# (80.6° for 2 and 85.8° for 3). A longer Pd–N distance of 2.149(2) Å was observed in 3 compared to 2.0787(18) Å for 2, attributed to steric reasons.
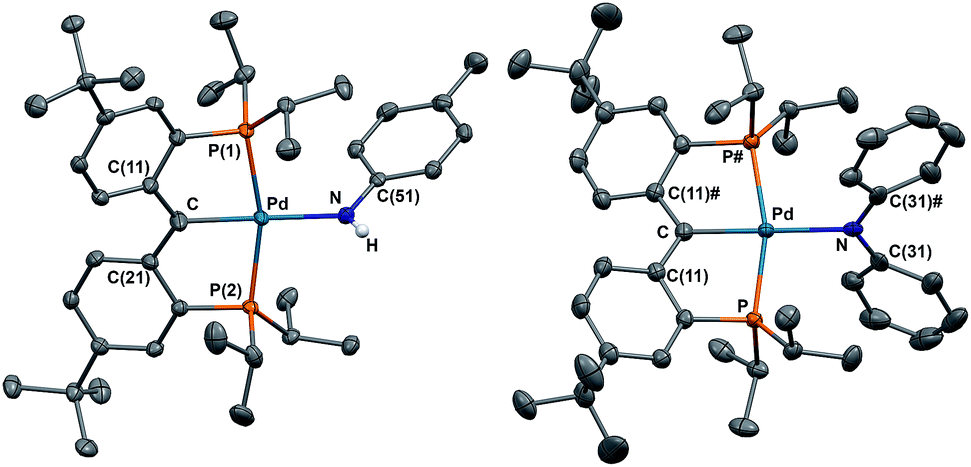 | ||
| Fig. 2 Thermal-ellipsoid (35% probability) representation of 2 (left) and 3 (right); most hydrogen atoms were removed for clarity. Selected distances [Å] and angles [°] for 2: Pd–C 2.019(2), Pd–P(1) 2.2983(5), Pd–P(2) 2.2841(5), Pd–N 2.0787(18), Pd–C–C(11) 118.57(15), Pd–C–C(21) 119.30(15), C(11)–C–(21) 121.98(19); for 3: Pd–C 2.024(2), Pd–P 2.3021(5), Pd–N 2.149(2), Pd–C–C(11) 118.97(11), C(11)–C–(11)# 122.1(2). | ||
Both radical complexes were subsequently treated with an equivalent of KC8 in benzene, and the corresponding diamagnetic heterobimetallic complexes [K(OEt2)n][{PC(sp2)P}tBuPdNR1R2] (4: R1 = H, R2 = pTol, n = 0; 5: R1 = R2 = Ph, n = 1) were obtained in high yield (Scheme 1). Both compounds are only soluble in ethereal solvents and were recrystallized by diethyl ether/n-pentane diffusion at ambient temperature. The solid state molecular structure of 5 was determined by X-ray diffraction studies (Fig. 3). Compound 5 exists as a notable polymer in which the anionic [{PC(sp2)P}tBuPdNPh2] moieties are bridged by potassium ions through the carbene units and one phenyl ring of the amide group. The average K–C distances are slightly shorter when potassium binds the phenyl ring of the amide group rather than the carbene moiety (3.08 Å vs. 3.17 Å). The coordination sphere of potassium was further completed by an additional diethyl ether molecule. The Ccarbene carbon retains its trigonal planar geometry (Σangles at Ccarbene are 359.9°) with a Pd–Ccarbene distance of 2.043(2) Å, which is shorter than the corresponding value of 2.076(3) Å in [{PC(sp2)P}tBuPd(PMe3)].62 The trigonal planar geometry at the amide nitrogen was also retained, but the dihedral angle between the amide plane and the plane defined by Ccarbene, C(11) and C(21) was much smaller than that in 3 (59.4° vs. 85.8°), probably due to the more congested environment found in the polymer.
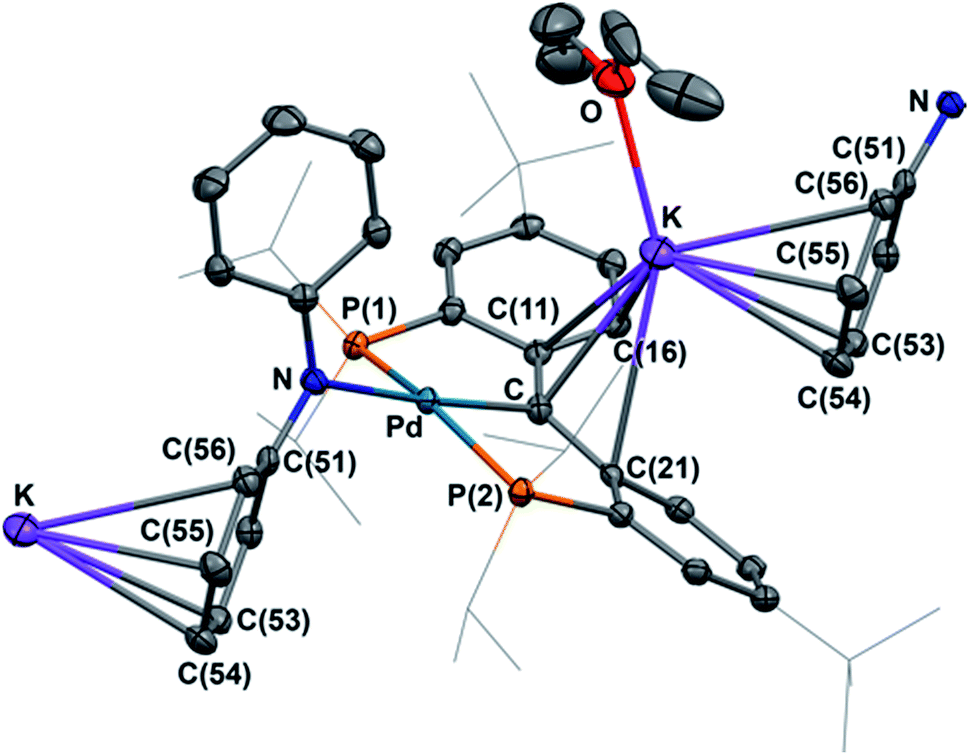 | ||
| Fig. 3 Thermal-ellipsoid (35% probability) representation of 5; most hydrogen atoms were removed for clarity. Selected distances [Å] and angles [°]: Pd–C 2.043(2), Pd–P(1) 2.2827(5), Pd–P(2) 2.3064(5), Pd–N 2.1445(16), K–C 2.939(2), K–C(11) 3.047(2), K–C(21) 3.292(2), K–C(16) 3.069(2), K–C(53) 3.240(2), K–C(54) 3.004(2), K–C(55) 3.062(2), K–C(56) 3.372(2), Pd–C–C(11) 118.45(14), Pd–C–C(21) 118.31(14), C(11)–C–(21) 123.14(18). | ||
Due to its poor solubility in non-coordinating solvents, the solution behaviour of 5 was studied in THF-d8. The 1H NMR spectrum in THF-d8 recorded at 298 K showed the peaks for the free Et2O at δ = 3.39 (q) and 1.12 (t) ppm, thus indicating its labile coordination that led to its displacement by the THF-d8 molecules. Unlike the polymeric structure observed in the solid state, 5 exhibited a C2 symmetry in the THF-d8 solution, as evidenced by only a broad peak at δ = 48.64 ppm (Δν1/2 ≈ 870 Hz) in the 31P{1H} NMR spectrum, as well as three broad peaks for the iPr (δ = 1.98 and 1.14 ppm) and tBu (δ = 1.06 ppm) groups. Therefore, the polymeric structure of 5 is disrupted in the donor solvent and even in the presence of a small amount of THF-d8 since the 1H NMR spectrum of 5 in C6D6 with 2 drops of THF-d8 also showed C2 symmetry.
Interestingly, the NPh2 group on palladium exhibited three well-resolved peaks at δ = 7.35 (d), 6.80 (t), and 6.18 (t) ppm in the 1H NMR spectrum of 5 in THF-d8, however, no resonances were observed from the phenyl rings of the supporting ligand in both 1H and 13C{1H} NMR spectra. The variable temperature NMR spectra recorded in the temperature range of 208 to 323 K in THF-d8 did not show any changes for these resonances. Only broadening of all the other peaks was observed in the 1H NMR spectrum at 208 K, while a relatively sharp (Δν1/2 ≈ 320 Hz) peak was found in the 31P{1H} NMR spectrum (see Fig. S35 and S36†). The phenyl rings of the supporting ligand are NMR silent in this whole temperature range. It is known that larger alkali metals such as potassium prefer anionic units that can delocalize the electron density, thus shifting the coordination of potassium away from the carbanion to the peripheral phenyl rings, leading to an η6-coordination mode.69,70 Therefore, the observed solution behaviour might be explained by a fast motion of the potassium ions between the two phenyl rings of the supporting ligand that cannot be frozen out at 208 K, causing a C2 symmetry on the NMR time scale. A comparable system has been reported for arene complexes of yttrium supported by a macrocyclic [P2N2] ligand ([P2N2] = [PhP(CH2SiMe2NSiMe2CH2)2PPh]).71 However, since all the signals from the phenyl rings of the supporting ligand could not be observed in the whole temperature range, a non-dynamic process caused by the formation of other polymers or oligomers containing bridging potassium ions cannot be ruled out.72
The 1H NMR spectrum of 4 in THF-d8 also exhibited a similar C2 symmetry as observed for 5. No coordination of diethyl ether was observed, even though 4 was crystallized from a diethyl ether/n-pentane mixture, indicating a slightly different bonding mode of potassium in 4, caused by the different amide substituent on palladium. Unlike those in 5, the phenyl groups from the supporting ligand in 4 exhibited three broad singlets at δ = 7.23, 6.61 and 6.54 ppm in the 1H NMR spectrum. Only one sharp singlet at δ = 46.36 ppm was observed in the 31P{1H} NMR spectrum. Therefore, a similar polymeric chain can be proposed for complex 4 (vide infra). Notably, the proton of the NH group appears as a singlet at δ = 1.33 ppm, thus the formation of a possible [PdNpTol] or [Pd–NpTol–Pd] imido species is excluded.
Our study of the solution behavior of 4 and 5 shows that not only the polymeric structure can be disrupted by donor solvents such as THF, but that the mobility of the potassium cation increases as well. These findings support the proposal by Meek and coworkers that the interaction of a rhodium catalyst for the hydroarylation of dienes with a Lewis acid has to be reversible in order to observe an increased activation of the substrate.22
Heterobimetallic carbene complexes73,74 containing K–Ccarbene–M units, in which the carbene is not stabilized by adjacent heteroatoms, are rare;23,75 to the best of our knowledge, complexes 4 and 5 represent the only known characterized heterobimetallic carbene species possessing such binding motifs. The successful synthesis of amide substituted carbene complexes 4 and 5 from the radical precursor 1 prompted us to investigate other nucleophiles, especially those featuring an alkyl group. Interestingly, the reaction of 1 with two equivalents of PhCH2K in THF led to a dark-brown diamagnetic complex, K[{PC(sp2)P}tBuPd(CH2Ph)] (6), in 64% yield, instead of the expected benzyl substituted Pd(II) radical carbene [{PC˙(sp2)P}tBuPd(CH2Ph)] (7, Scheme 2). Using one equivalent of PhCH2K also led to 6, albeit with a lower conversion.
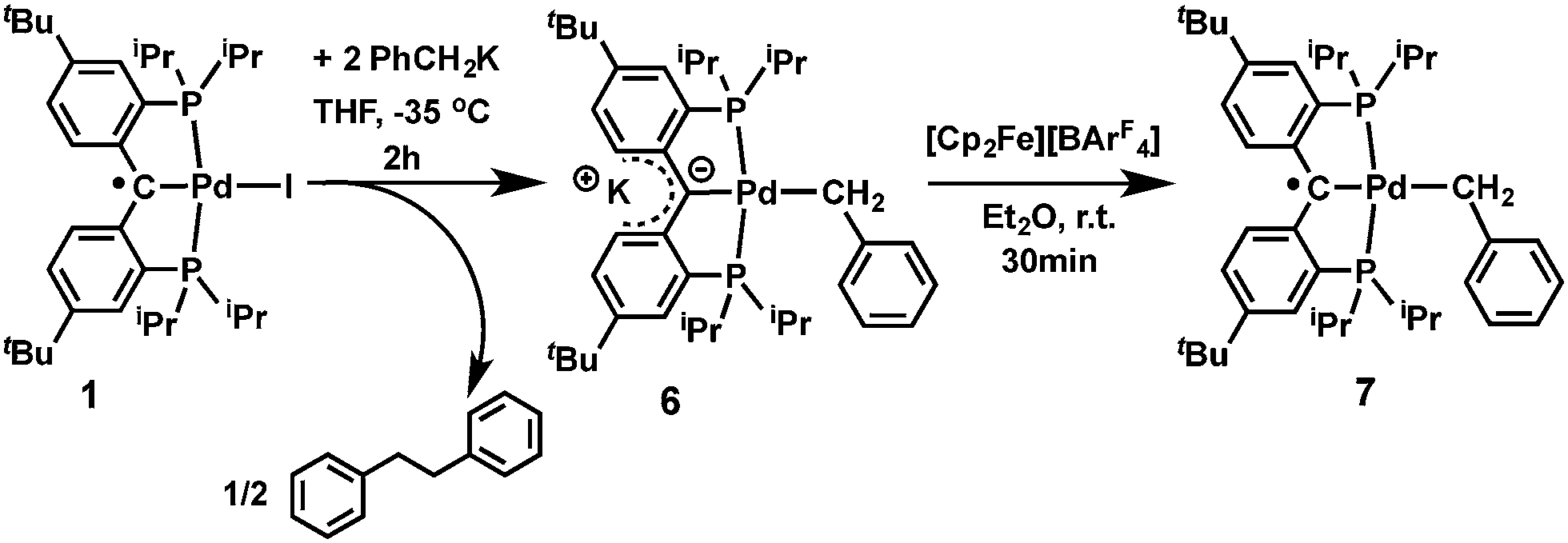 | ||
| Scheme 2 Synthesis of 6 and 7. | ||
As observed for 5, X-ray diffraction studies on single crystals of 6 grown from a diethyl ether solution also showed a polymeric structure, where the anionic carbene moieties [{PC(sp2)P}tBuPdCH2Ph] were bridged by potassium ions (Fig. 4). Although the Ccarbene carbon still retained its trigonal planar geometry (Σangles at Ccarbene are 359.8°), the Pd–Ccarbene distance of 2.095(2) Å is slightly longer than that of 2.076(3) Å in [{PC(sp2)P}tBuPd(PMe3)],62 and of 2.043(2) Å in 5. Two different coordination modes were found for potassium ions: one is sandwiched between two [{PC(sp2)P}tBuPdCH2Ph] moieties in an η4 fashion, with K–C distances ranging from 2.998(2) to 3.176(3) Å, while the other is only η1 coordinated to the carbene carbons of two anionic carbene moieties, with a K–C distance of 3.040(2) Å. The K–Ccarbene–K–Ccarbene chain is not strictly linear, with the K–Ccarbene–K and Ccarbene–K–Ccarbene angles being 163.40(8)° and 173.86(9)°, respectively. The different bridging modes observed for potassium in 5 and 6 are largely attributed to the bulkier Ph2N amide group in 5 than the benzyl group in 6.
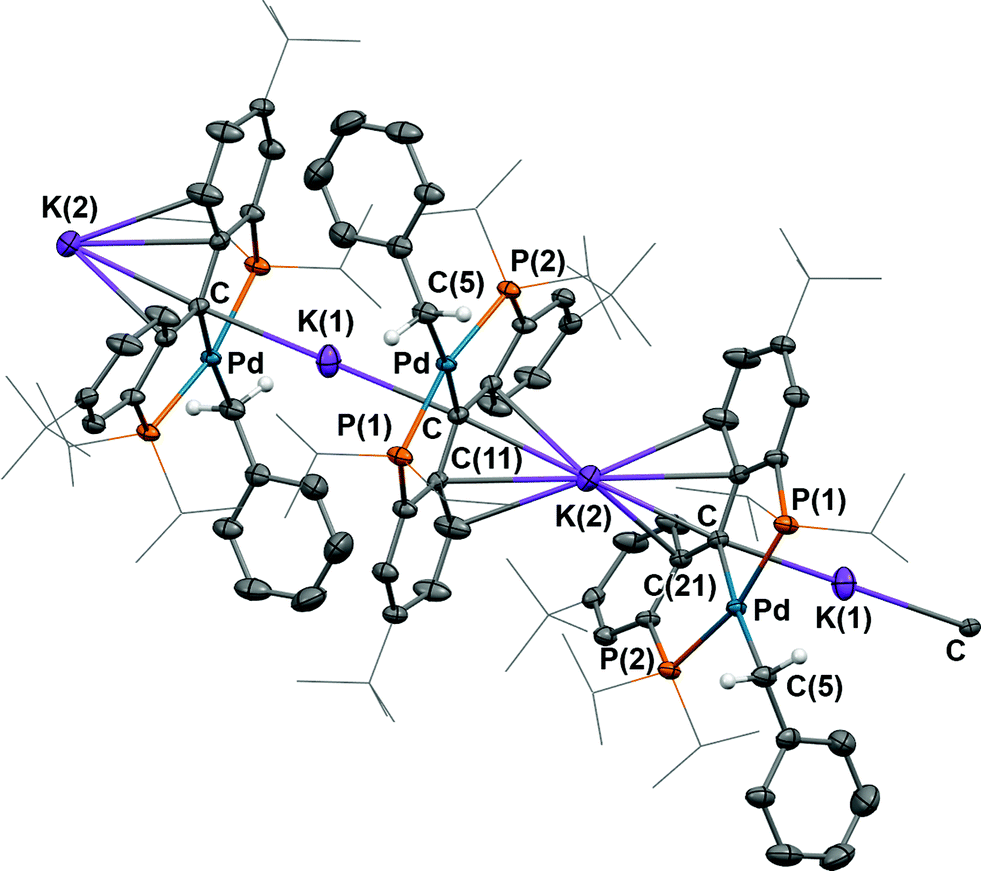 | ||
| Fig. 4 Thermal-ellipsoid (35% probability) representation of 6; most hydrogen atoms were removed for clarity. Selected distances [Å] and angles [°]: Pd–C 2.095(2), Pd–P(1) 2.2695(6), Pd–P(2) 2.2868(6), Pd–C(5) 2.163(2), K(1)–C 3.040(2), K(2)–C 3.028(2), K(2)–C(11) 3.162(2), K(2)–C(16) 3.176(3), K(2)–C(21) 2.998(2), Pd–C–C(11) 118.76(15), Pd–C–C(21) 118.06(15), C(11)–C–(21) 123.02(19). | ||
Similarly to 4 and 5, complex 6 is only soluble in ethereal solvents and its 1H NMR spectrum recorded in THF-d8 is consistent with a C2 symmetric structure in solution. The 31P {1H} NMR spectrum only shows a sharp singlet at δ = 48.52 ppm. A triplet at δ = 2.45 ppm (3JPH = 5.0 Hz) was observed for the benzyl CH2 group in the 1H NMR spectrum that correlates with a triplet at δ = 14.12 ppm (2JPC = 9.0 Hz) in the corresponding 13C{1H} NMR spectrum. The peak corresponding to the carbene carbon could not be assigned unambiguously in the 13C{1H} NMR spectrum, a situation previously observed for [{PC(sp2)P}HPdPMe3],61 as well as for other palladium carbene complexes.50
As observed for 4 and 5, a discrepancy between the solid state and THF-d8 solution structures of 6 exists that suggests a disruption of the polymeric chain by the donor solvent. In the 1H NMR spectrum, three well-resolved peaks at δ = 7.20 (dt), 6.66 (td) and 6.52 (dd) ppm were assigned to the signals from the phenyl groups of the supporting ligand, which are very close to those of 4. Considering that benzyl and NHpTol groups have similar steric profiles, complexes 4 and 6 most likely possess similar solution structures. The 1H NMR spectrum of 6 in C6D6 with 2 drops of THF-d8 also showed a C2 symmetric structure. Notably, resonances of the phenyl groups of the supporting ligand are significantly shifted upfield in THF-d8 compared to those in C6D6 with only a small amount of THF-d8 (from 7.61, 6.94, and 6.86 ppm to 7.20, 6.66, and 6.52 ppm, respectively), consistent with the generation in THF-d8 of a monomer, in which the potassium ion interacts with only one carbene moiety and not with two, as observed in the solid state or in a non-donor solvent.76
The formation of 6 is a combined substitution and reduction of radical complex 1: the benzyl group on palladium was introduced by substitution of iodide by a benzyl anion. Due to the reductive nature of the benzyl anion, the loss of an electron from it also reduced the radical species to an anionic carbene. This process is accompanied by the formation of the benzyl radical [PhCH2˙], which dimerized to form PhCH2CH2Ph. Analysis of the crude reaction mixture by 1H NMR spectroscopy indicated the formation of PhCH2CH2Ph, which was confirmed by comparison with an authentic sample. A minor palladium containing species was also observed, which showed two sets of peaks for the benzylic CH2 groups at δ = 3.81 (s) and 2.94 (3JPH = 5.5 Hz) ppm in the 1H NMR spectrum, and a sharp singlet at δ = 39.86 ppm in the 31P{1H} NMR spectrum. We tentatively assigned this species as complex [{PC(sp3)(CH2Ph)P}tBuPd(CH2Ph)], containing another benzyl group on the backbone carbon via radical coupling. Attempts to isolate this compound were hampered by its high solubility in aliphatic solvents.
Although the substitution reaction of 1 with PhCH2K led directly to the formation of the heterobimetallic carbene complex 6, the subsequent one-electron oxidation of 6 with [Cp2Fe][BArF4] afforded the radical complex 7 in quantitative yield (Scheme 2). The high solubility of 7 in aliphatic solvents prevented its separation from the byproduct, Cp2Fe, but its EPR spectrum (in the presence of Cp2Fe) in toluene at 298 K revealed a hyperfine splitting from four pairs of magnetically equivalent protons and a 105Pd nucleus (Fig. S5 and S6†). The smallest hyperfine constant was attributed to the CH2 group protons (0.08 mT), while the remaining proton hyperfine constants (0.32, 0.19 and 0.12 mT) were assigned to the six phenyl ring protons of the supporting ligand, which are close to the corresponding values for 2 and 3. Accordingly, the one-electron oxidations of the heterobimetallic carbene complexes 4 and 5 with [Cp2Fe][BArF4] also regenerated the radical complexes 2 and 3, respectively (Scheme 1), as confirmed by EPR spectroscopy (Fig. S1 and S3†).
Preliminary reactivity studies showed that the reaction of 6 with CH3CN (5 equivalents) in THF quantitatively afforded [{PC(sp3)HP}tBuPd(CH2Ph)] (8, eqn (1)), which is the product of C–H activation, similarly to [{PC(sp2)P}HPd(PMe3)].61,62 Given the presence of benzyl and amide ligands that could have been protonated by CH3CN, it is interesting to note that the former carbene carbon is more nucleophilic than these ligands, consistent with a localized negative charge on that carbon.
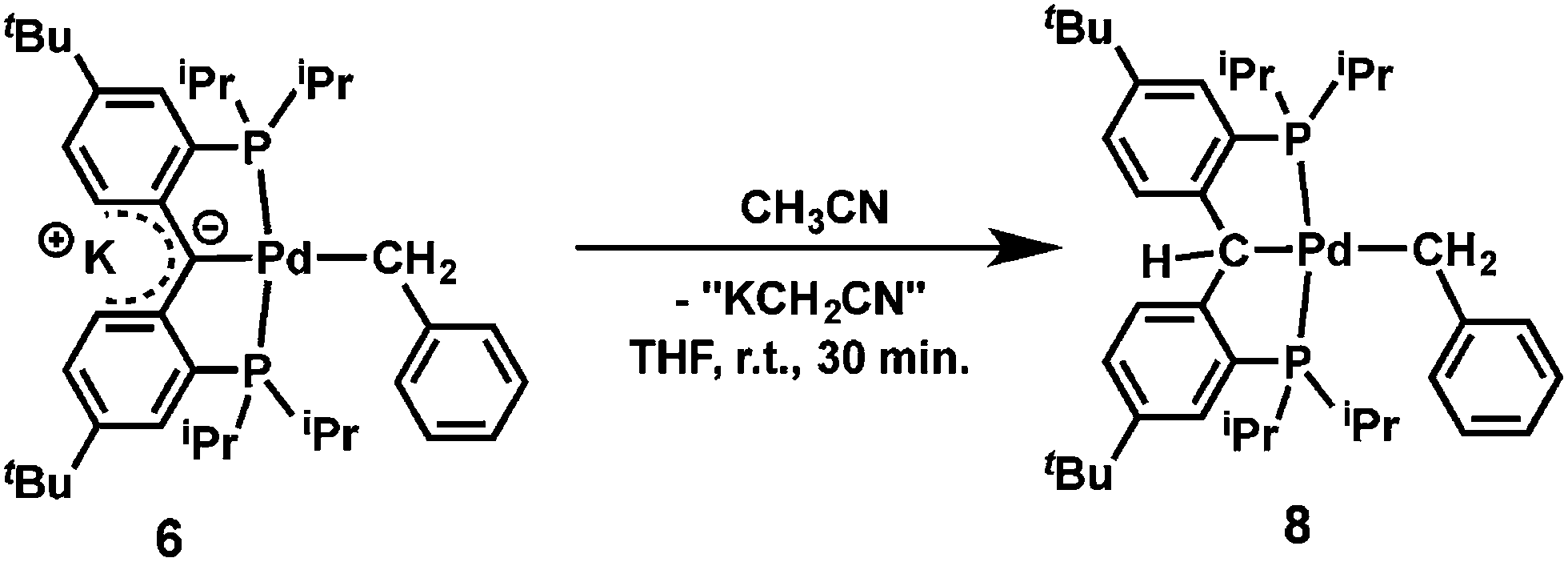 | (1) |
We also tested the direct nucleophilic substitutions of the parent carbene complex [{PC(sp2)P}tBuPd(PMe3)] with pTolNHK, Ph2NK and PhCH2K. Although the formation of the heterobimetallic complexes 4 and 6 was confirmed (pTolNHK, 65% and PhCH2K, 60% conversion) by 31P{1H} NMR spectra, some unknown species as well as unreacted starting material were also observed after 24 h. Attempts to isolate pure 4 and 6 from these mixtures met with difficulty and were inefficient. The bulky nucleophile Ph2NK does not react with [{PC(sp2)P}tBuPd(PMe3)] under similar conditions, probably due to its steric hindrance. The sluggish substitution reactions observed for these nucleophiles could also be attributed to the relatively strong coordination of PMe3 to palladium, therefore, the substitution/reduction strategy presented herein is preferable because it takes advantage of a facile redox process that provides a relatively easier access to heterobimetallic Pd–K carbene complexes.
DFT calculations
DFT calculations (B3LYP functional, LANL2DZ basis set) were performed on a model of the anion of 5, 5′, with the tBu and iPr groups replaced by H and methyl groups, respectively (Fig. 5). As was observed for [{PC(sp2)P}RPd(PMe3)],61,62 the HOMO of 5′ shows a π type interaction between the carbene carbon p orbital and the appropriate symmetry d orbital of Pd, while the LUMO shows the corresponding σ interaction, with both molecular orbitals having antibonding character. Thus the carbene moiety in these heterobimetallic carbene complexes is predicted to show some similar reactivity to that observed for [{PC(sp2)P}RPd(PMe3)], as was shown in eqn (1).62,64,66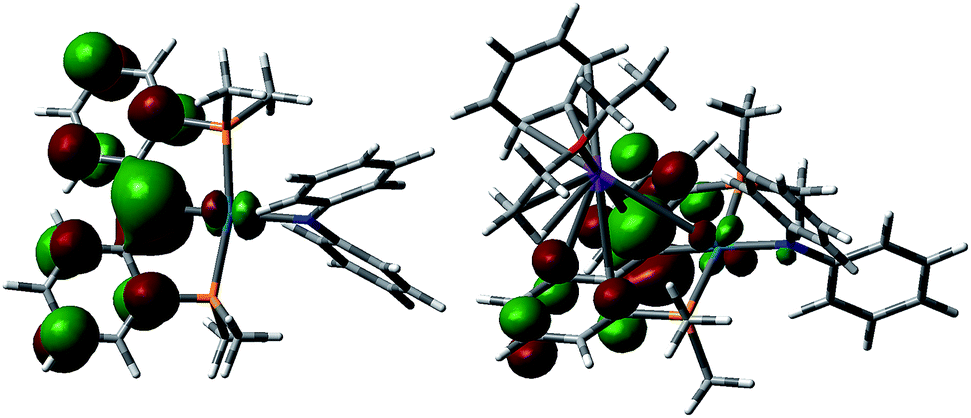 | ||
| Fig. 5 HOMO for [{PC(sp2)P}MePdNPh2]− (5′, left) and [{PC(sp2)-K(OEt2)(C6H6)P}MePdNPh2] (5′′, right). | ||
In order to study the influence of the potassium cation on the Pd–C interaction, another simplified model, 5′′, was considered, in which the interaction between potassium and a NPh2 group from a neighboring molecule was replaced with a potassium benzene interaction. The HOMO of 5′′ (Fig. 5) resembles that calculated for 5′, in agreement with an electrostatic interaction between the potassium cation and the anionic carbon, while the LUMO is localized on the benzene ring coordinated to the potassium atom. The interaction between the cation and the carbene ligand observed in both the solid state structure and the computed model involves the delocalized π orbital of the two phenyl rings attached to the carbene atom. That carbon maintains its planar geometry in both 5 and 6, similarly to other delocalized π systems coordinated to potassium.77–79
A similar situation was observed for 6. We studied the electronic structure of the free anion (6′) and a model of the heterobimetallic carbene (6′′). In both, the iso-propyl groups and the tert-butyl groups were replaced by hydrogen atoms. For 6′′, in order to simulate the coordination environments of both potassium atoms, we reduced the polymer to a dimer (Fig. S24 and S25†). Both HOMO and HOMO−1 for 6′′ are comprised of the lone pair on the carbenic carbon; LUMO and LUMO+1 are localized on the benzene rings coordinated to the terminal potassium cations. The planarity of the carbenic carbons can be attributed to the symmetric donation from the lone pair on these atoms to the two potassium atoms.
Conclusions
In conclusion, we showed that a substitution/reduction strategy employing the radical carbene [{PC˙(sp2)P}tBuPdI] (1) can be utilized to synthesize a series of heterobimetallic Pd–K carbene complexes {[{PC(sp2)P}tBuPdX]K(OEt2)n} (4: X = NHpTol, n = 0; 5: X = NPh2, n = 1; 6: X = CH2Ph, n = 0) bearing functional groups on palladium. Polymeric structures were exhibited by these heterobimetallic Pd–K carbenes in the solid state, featuring unprecedented Pd–Ccarbene–K units that are not easily accessible by other synthetic strategies. The present carbenes complete the palladium carbene series containing a cationic (Chart 1, type A), an ylide-like (type B), a radical (type C), and a heterobimetallic (type D) carbene. The isolation of these novel species not only provides a fundamental understanding of their bonding and structural features, but also sheds some light on the role of alkalis in transition metal catalysis. Notably, the solution behaviour of these complexes showed that the interaction of the potassium ion with the carbene moiety is highly influenced by the donating ability of the solvent, which led to a lower aggregation state of the complex; this may better represent species related to transition metal catalytic systems with alkali metals in polar solvents. More importantly, the flexible interaction of potassium might also be crucial in terms of substrate activation, as proposed recently in the rhodium catalyzed hydroarylation of dienes, that a reversible interaction with Lewis acids enhance the reactivity.22 Finally, the facile redox properties exhibited by these palladium carbenes also indicate their potential non-innocent ligand-based reactivity, and, therefore, future studies can lead to new and useful transformations in synthesis.Experimental
All experiments are performed under an inert atmosphere of N2 using standard glovebox techniques. Solvents hexanes, n-pentane, diethyl ether, and CH2Cl2 were dried by passing through a column of activated alumina and stored in the glovebox. THF and THF-d8 were dried over LiAlH4 followed by vacuum transfer and stored in the glovebox, while C6D6 was dried over CaH2 followed by vacuum transfer, and stored in the glovebox. Complex 1,67 [Cp2Fe][BArF4],80 and KC881 were prepared according to literature procedures. pTolNHLi, pTolNHK and Ph2NK were prepared by deprotonation of amines with nBuLi and KH, respectively. 1H, 13C{1H}, 31P{1H} NMR spectra were recorded on a Bruker DRX 500 spectrometer. All chemical shifts are reported in δ (ppm) with reference to the residual solvent resonance of deuterated solvents for proton and carbon chemical shifts, and to external H3PO4, for 31P chemical shifts, respectively. Magnetic moments were determined by the Evans method68,82,83 by using a capillary containing 1,3,5-trimethoxybenzene in C6D6 as a reference. EPR spectra were recorded on a Bruker EMXplus EPR spectrometer with a standard X-band EMXplus resonator and an EMX premium microwave bridge, at microwave power of 2 mW, modulation frequency 100 kHz and amplitude 0.01 mT. Elemental analyses were performed on a CE-440 Elemental analyzer, or by Midwest Microlab. Gaussian 03 (revision D.02) was used for all reported calculations. The B3LYP (DFT) method was used to carry out the geometry optimizations on model compounds specified in text using the LANL2DZ basis set. The validity of the true minima was checked by the absence of negative frequencies in the energy Hessian.Synthesis of [{PC˙(sp2)P}tBuPdNHpTol] (2)
p TolNHK (10.2 mg, 0.071 mmol) in 1 mL of THF was slowly added to a dark-green solution of 1 (50 mg, 0.067 mmol) in 1 mL of THF at −35 °C. The resulted greenish slurry was allowed to stir at room temperature for 1 h. All volatiles were removed under reduced pressure and the residue was extracted with 6 mL of n-pentane, and filtered to give a dark-green solution. The volume of this n-pentane solution was reduced to about 0.5 mL under reduced pressure and stored at −35 °C to give compound 2 as green crystals. Yield 42 mg (87%). For 2: [{PC˙(sp2)P}tBuPdNHpTol] (2) is paramagnetic. Magnetic moment (Evans method, 298 K): μeff = 1.70 μB; EPR: g = 2.0088. Anal. calcd for C40H60NP2Pd (723.28 g mol−1): C, 66.42; H, 8.36; N, 1.94. Found: C, 66.52; H, 8.31; N, 1.68.Synthesis of [{PC˙(sp2)P}tBuPdNPh2] (3)
Ph2NK (19.5 mg, 0.094 mmol) and 1 (70 mg, 0.094 mmol) were mixed in 5 mL of THF and heated at 60 °C for 6 days, during which time a greenish slurry was formed. All volatiles were removed under reduced pressure and the residue was extracted with n-pentane (3 × 4 mL), and filtered to give a dark-green solution. The volume of the n-pentane solution was reduced to about 1 mL under reduced pressure and stored at −35 °C to give compound 2 as green crystals. Yield 59 mg (80%). For 3: [{PC˙(sp2)P}tBuPdNPh2] (3) is paramagnetic. Magnetic moment (Evans method, 298 K): μeff = 1.54 μB; EPR: g = 2.0079. Anal. calcd for C45H62NP2Pd (785.35 g mol−1): C, 68.82; H, 7.96; N, 1.78. Found: C, 68.91; H, 7.55; N, 1.40.Synthesis of [{PC(sp2)P}tBuPdNHpTol]−K+ (4)
KC8 (10.3 mg, 0.076 mmol) and 2 (55 mg, 0.076 mmol) were mixed in 3 mL of C6H6 at room temperature. The resulted dark-brown reaction mixture was stirred at room temperature for 30 min. All volatiles were removed under reduced pressure and the residue was extracted with 4 mL of diethyl ether and filtered to give a dark-brown solution. The volume of this solution was reduced to about 1.5 mL under reduced pressure and layered with 7 mL of n-pentane at room temperature. Compound 4 crystallized from this solution as dark-brown crystals. Yield 48 mg (83%). For 4: 1H NMR (500 MHz, 25 °C, THF-d8): δ = 7.23 (br s, 2H, ArH), 6.61 (br s, 2H, ArH), 6.54 (br s, 2H, ArH), 6.35 (d, 3JHH = 7.5 Hz, 2H, ArH), 6.10 (d, 3JHH = 8.0 Hz, 2H, ArH), 2.36 (m, 4H, CH(CH3)2), 1.98 (s, 3H, pTolCH3), 1.33 (s, 1H, NH), 1.24 (dt, 3JHH = 7.5 Hz, 3JHP = 7.0 Hz, 12H, CH(CH3)2), 1.16 (dt, 3JHH = 7.5 Hz, 3JHP = 8.0 Hz, 12H, CH(CH3)2), 1.16 (s, 18H, C(CH3)3) ppm; 13C{1H} NMR (126 MHz, 25 °C, THF-d8): δ = 162.42 (s, ArC), 161.98 (m, ArC), 130.91 (br s, ArC), 129.56 (br s, ArC), 129.40 (s, ArC), 128.50 (br s, ArC), 119.08 (br s, ArC), 115.73 (s, ArC), 115.53 (br s, ArC), 112.95 (s, ArC), 110.35 (br s, ArC), 34.03 (s, C(CH3)3), 32.22 (br s, C(CH3)3), 25.85 (br s, CH(CH3)2), 20.97 (s, pTolCH3), 19.25 (s, CH(CH3)2), 18.99 (s, CH(CH3)2) ppm. The peak for the carbenic carbon cannot be assigned in the 162–110 ppm region, but the total number of carbon peaks are correct. 31P{1H} NMR (202 MHz, 25 °C, THF-d8): δ = 46.36 (s) ppm. Anal. calcd for C40H60KNP2Pd (762.38 g mol−1): C, 63.02; H, 7.93; N, 1.84. Found: C, 63.72; H, 8.42; N, 1.39.Synthesis of [{PC(sp2)P}tBuPdNPh2]−[KOEt2]+ (5)
KC8 (8.6 mg, 0.064 mmol) and 3 (50 mg, 0.064 mmol) were mixed in 2 mL of C6H6 at room temperature. The resulted greenish-brown reaction mixture was stirred at room temperature for 30 min. All volatiles were removed under reduced pressure and the residue was extracted with ether (3 × 5 mL) and filtered to give a greenish-brown solution. The volume of this solution was reduced to about 3 mL under reduced pressure and layered with 9 mL of n-pentane at room temperature. Compound 5 crystallized from this solution as greenish brown crystals. Yield 41 mg (71%). For 5: 1H NMR (500 MHz, 25 °C, THF-d8): δ = 7.35 (d, 3JHH = 7.5 Hz, 4H, ArH), 6.80 (t, 3JHH = 7.3 Hz, 4H, ArH), 6.18 (t, 3JHH = 7.0 Hz, 2H, ArH), 3.39 (q, 3JHH = 7.0 Hz, 4H, OCH2CH3), 1.98 (br s, 4H, CH(CH3)2), 1.14 (br s, 24H, CH(CH3)2), 1.12 (t, 3JHH = 7.0 Hz, 4H, OCH2CH3), 1.06 (br s, 18H, C(CH3)3) ppm; 13C{1H} NMR (126 MHz, 25 °C, THF-d8): δ = 158.82 (s, ArC), 128.14 (s, ArC), 121.73 (s, ArC), 113.63 (s, ArC), 66.47 (s, OCH2CH3), 31.85 (s, C(CH3)3), 19.04 (br s, CH(CH3)2), 18.19 (br s, CH(CH3)2), 15.85 (s, OCH2CH3) ppm. The peaks for the phenyl of the (PC(sp2)P) ligand backbone were not observed in the 1H and 13C{1H} NMR spectra (vide supra). 31P{1H} NMR (202 MHz, 25 °C, THF-d8): δ = 48.64 (s) ppm. Anal. calcd for C49H72KNOP2Pd (898.57 g mol−1): C, 65.50; H, 8.08; N, 1.56. Found: C, 65.61; H, 8.12; N, 1.44.Synthesis of [{PC(sp2)P}tBuPdCH2Ph]−K+ (6)
PhCH2K (17.5 mg, 0134 mmol) in 1 mL of THF was slowly added to 1 (50 mg, 0.067 mmol) in 1 mL of THF at −35 °C. The dark-red reaction mixture was stirred at room temperature for 2 hours. Volatiles were removed under reduced pressure and the residue was extracted with 8 mL of diethyl ether and filtered to give a dark-brown solution. The volume of this solution was reduced to about 1.5 mL under reduced pressure and layered with 7 mL of n-pentane. Compound 6 crystallized from this solution at −35 °C as dark-brown solid. Yield 32 mg (64%). For 6: 1H NMR (500 MHz, 25 °C, THF-d8): δ = 7.20 (dt, 3JHH = 9.0 Hz, 3JHP = 4.5 Hz, 2H, ArH), 6.98 (d, 3JHH = 7.0 Hz, 2H, ArH), 6.81 (t, 3JHH = 7.5 Hz, 2H, ArH); 6.66 (td, 4JHH = 2.3 Hz, 3JHP = 4.8 Hz, 2H, ArH), 6.52 (dd, 3JHH = 9.0 Hz, 4JHH = 2.0 Hz, 2H, ArH), 6.46 (t, 3JHH = 7.3 Hz, 1H, ArH), 2.45 (t, 3JHP = 5.0 Hz, 2H, CH2Ph), 2.26 (m, 4H, CH(CH3)2), 1.16 (dt, 3JHH = 7.0 Hz, 3JHP = 6.5 Hz, 12H, CH(CH3)2), 1.15 (s, 18H, C(CH3)3), 1.14 (3JHH = 6.0 Hz, 3JHP = 7.5 Hz, 12H, CH(CH3)2) ppm; 13C{1H} NMR (126 MHz, 25 °C, THF-d8): δ = 161.56 (t, JCP = 18.6 Hz, ArC), 159.81 (t, JCP = 1.3 Hz, ArC), 130.35 (t, JCP = 3.0 Hz, ArC), 129.94 (s, ArC), 128.43 (s, ArC), 128.27 (s, ArC), 127.46 (s, ArC), 121.85 (t, JCP = 19.0 Hz, ArC), 118.62 (s, ArC), 115.38 (t, JCP = 10.3 Hz, ArC), 34.00 (s, C(CH3)3), 32.27 (s, C(CH3)3), 26.07 (t, 1JCP = 10.5 Hz, CH(CH3)2), 20.26 (t, 2JCP = 3.2 Hz, CH(CH3)2), 19.04 (s, CH(CH3)2), 14.12 (t, 2JCP = 9.0 Hz, CH2Ph) ppm; 31P{1H} NMR (202 MHz, 25 °C, THF-d8): δ = 48.52 (s) ppm. Anal. calcd for C40H59KP2Pd (747.36 g mol−1): C, 64.28; H, 7.96. Found: C, 64.81; H, 8.53.Synthesis of [{PC˙(sp2)P}tBuPdCH2Ph] (7)
[Cp2Fe][BArF4] (32 mg, 0.031 mmol) in 1 mL of THF was slowly added to 6 (23 mg, 0.031 mmol) in 1 mL of THF at −35 °C. The dark-red reaction mixture was stirred at room temperature for 30 min. All volatiles were removed under reduced pressure and the residue was extracted with 4 mL of n-pentane and filtered to give a red solution. The volatiles were removed under reduced pressure to give a red oily mixture of 7 and Cp2Fe, which are not separable due to similar solubilities in common solvents. For 7: [{PC˙(sp2)P}tBuPdCH2Ph] (7) is paramagnetic. EPR: g = 2.0086.Oxidation of [{PC(sp2)P}tBuPdNHpTol]−K+ (4)
[Cp2Fe][BArF4] (6.9 mg, 0.007 mmol) in 1 mL of diethyl ether was added to [{PC(sp2)P}tBuPdNHpTol]−K+ (4) (5 mg, 0.007 mmol) in 1 mL of diethyl ether at −35 °C. The resulted brownish-green solution was allowed to stir at room temperature for 30 min. The volatiles were removed under reduced pressure and the residue was extracted with n-pentane. This solution was filtered and the volatiles were removed under reduced pressure to give a green solid. The EPR spectrum measured in toluene at 298 K is identical to that of a pure sample of 2 isolated by a different method (vide supra).Oxidation of [{PC(sp2)P}tBuPdNPh2]−[KOEt2]+ (5)
[Cp2Fe][BArF4] (5.8 mg, 0.006 mmol) in 1 mL of diethyl ether was added to 5 (5 mg, 0.006 mmol) in 1 mL of diethyl ether at −35 °C. The resulted green solution was allowed to stir at room temperature for 30 min. The volatiles were removed under reduced pressure and the residue was extracted with n-pentane and filtered. The volatiles were removed under reduced pressure to give a green solid. The EPR spectrum measured in toluene at 298 K is identical to that of a pure sample of 3 isolated by a different method (vide supra).Synthesis of [{PCHP}tBuPdCH2Ph] (8)
CH3CN (4 mg, 0.1 mmol) in 0.5 mL of THF was added to a solution of 6 (15 mg, 0.02 mmol) in 1 mL of THF at room temperature. The dark-red solution immediately turned to orange, which was stirred for another 30 min. Volatiles were removed under vacuum to give an yellow oil, which was extracted into 3 mL of pentane, filtered and removal of all volatiles under reduced pressure afforded 8 as an yellow foam. Yield 14 mg (quantitative). For 8: 1H NMR (500 MHz, 25 °C, C6D6): δ = 7.51 (d, 3JHH = 7.0 Hz, 2H, ArH); 7.46–7.44 (m, 4H, ArH); 7.26 (t, 3JHH = 7.8 Hz, 2H, ArH); 7.21 (dd, 3JHH = 8.5 Hz, 4JHH = 2.0 Hz, 2H, ArH); 6.96 (t, 3JHH = 7.3 Hz, 1H, ArH), 5.65 (s, 1H, CHbackbone), 3.00 (t, 3JHP = 5.3 Hz, 2H, CH2Ph), 2.41 (m, 2H, CH(CH3)2), 2.27 (m, 2H, CH(CH3)2), 1.28 (dt, 6H, 3JHH = 8.0 Hz, 3JHP = 7.5 Hz, CH(CH3)2), 1.27 (s, 18H, C(CH3)3), 1.17 (dt, 3JHH = 6.5 Hz, 3JHP = 6.5 Hz, 6H, CH(CH3)2), 1.06 (m, 12H, CH(CH3)2) ppm; 13C{1H} NMR (126 MHz, 25 °C, C6D6): δ = 157.67 (t, JCP = 15.0 Hz, ArC), 155.51 (s, ArC), 146.47 (s, ArC), 136.23 (t, JCP = 17.3 Hz, ArC), 128.70 (s, ArC), 128.34 (s, ArC), 127.90 (s, ArC), 127.72 (t, JCP = 8.4 Hz, ArC), 127.00 (s, ArC), 120.48 (s, ArC), 56.26 (s, CHbackbone), 34.36 (s, C(CH3)3), 31.65 (s, C(CH3)3), 25.62 (t, 1JPC = 11.2 Hz, CH(CH3)2), 25.52 (t, JPC = 9.3 Hz, CH(CH3)2), 19.87 (t, 2JPC = 3.6 Hz, CH(CH3)2), 19.32 (t, 2JPC = 2.5 Hz, CH(CH3)2), 19.17 (t, 2JPC = 2.1 Hz, CH(CH3)2), 18.00 (s, CH(CH3)2), 13.22 (t, 2JPC = 7.8 Hz, CH2Ph); 31P{1H} NMR (202 MHz, 25 °C, C6D6): δ = 45.76 (s) ppm. Anal. calcd for C40H60P2Pd (709.27 g mol−1): C, 67.74; H, 8.53. Found: C, 67.88; H, 8.62.Substitution reactions of carbene [{PC(sp2)P}tBuPd(PMe3)] with PhCH2K, pTolNHK and Ph2NK nucleophiles
PhCH2K (2.8 mg, 0.022 mmol) in 1 mL of THF was added to carbene (15 mg, 0.022 mol) in 0.5 mL of THF at −35 °C. The dark-red solution was then stirred at room temperature for 24 h. Volatiles were removed under reduced pressure and the residues were dissolved in C6D6 (with 2 drops of THF-d8) and monitored by 1H and 31P{1H} NMR spectra. Same procedures were applied for pTolNHK and Ph2NK. The reactions with PhCH2K and pTolNHK showed conversion to the heterobimetallic carbene 6 and 4 in 60% and 65% conversion, respectively, based on the 31P{1H} NMR spectra. No conversion was observed for Ph2NK.Acknowledgements
We thank Dr Allen Oliver for crystallographic assistance and the Center for Sustainable Energy at Notre Dame for a fellowship to P. C. M. V. acknowledges support from the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences under Award Number DOE DE-FC02-04ER15533. Notre Dame Radiation Laboratory document number: 5094.References
- T. Caneque, F. M. Truscott, R. Rodriguez, G. Maestri and M. Malacria, Chem. Soc. Rev., 2014, 43, 2916–2926 RSC.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- E. Dunach, Chem. Biodiversity, 2014, 11, 1752–1763 CAS.
- A. Gualandi, L. Mengozzi, C. M. Wilson and P. G. Cozzi, Chem.–Asian J., 2014, 9, 984–995 CrossRef CAS.
- R. Qiu, Y. Chen, S.-F. Yin, X. Xu and C.-T. Au, RSC Adv., 2012, 2, 10774–10793 RSC.
- D. T. Cohen and K. A. Scheidt, Chem. Sci., 2012, 3, 53–57 RSC.
- O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed.
- R. Hartmann and P. Chen, Angew. Chem., Int. Ed., 2001, 40, 3581–3585 CrossRef CAS.
- A. Bellomo, J. Zhang, N. Trongsiriwat and P. J. Walsh, Chem. Sci., 2013, 4, 849–857 RSC.
- K. Hayek, R. Kramer and Z. Paál, Appl. Catal., A, 1997, 162, 1–15 CrossRef CAS.
- P. M. Maitlis and V. Zanotti, Chem. Commun., 2009, 1619–1634 RSC.
- M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780–783 CrossRef CAS PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef PubMed.
- S. Fukuzumi, Y. Morimoto, H. Kotani, P. Naumov, Y.-M. Lee and W. Nam, Nat. Chem., 2010, 2, 756–759 CrossRef CAS PubMed.
- Y. Morimoto, H. Kotani, J. Park, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 403–405 CrossRef CAS PubMed.
- Y. J. Park, J. W. Ziller and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 9258–9261 CrossRef CAS PubMed.
- Y. J. Park, S. A. Cook, N. S. Sickerman, Y. Sano, J. W. Ziller and A. S. Borovik, Chem. Sci., 2013, 4, 717–726 RSC.
- J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723–1732 RSC.
- J. R. Robinson, J. Gu, P. J. Carroll, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2015, 137, 7135–7144 CrossRef CAS PubMed.
- S. Matsunaga and M. Shibasaki, Chem. Commun., 2014, 50, 1044–1057 RSC.
- M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117–1127 CrossRef CAS PubMed.
- C. C. Roberts, D. M. Matías, M. J. Goldfogel and S. J. Meek, J. Am. Chem. Soc., 2015, 137, 6488–6491 CrossRef CAS PubMed.
- M. Fang, N. D. Jones, R. Lukowski, J. Tjathas, M. J. Ferguson and R. G. Cavell, Angew. Chem., Int. Ed., 2006, 45, 3097–3101 CrossRef CAS PubMed.
- M. Fang, N. D. Jones, K. Friesen, G. Lin, M. J. Ferguson, R. McDonald, R. Lukowski and R. G. Cavell, Organometallics, 2009, 28, 1652–1665 CrossRef CAS.
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS PubMed.
- Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef PubMed.
- R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748–3759 CrossRef CAS PubMed.
- A. J. Arduengo and G. Bertrand, Chem. Rev., 2009, 109, 3209–3210 CrossRef CAS PubMed.
- A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef CAS PubMed.
- A. Fürstner, Science, 2013, 341, 1229713 CrossRef PubMed.
- F. Z. Dorwald, Metal carbenes in organic synthesis, Wiley-VCH, Weinheim, New York, 1999 Search PubMed.
- F. Glorius, in Topics in organometallic chemistry, Springer, Berlin, New York, 2007vol. 21 Search PubMed.
- O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- K. Öfele, E. Tosh, C. Taubmann and W. A. Herrmann, Chem. Rev., 2009, 109, 3408–3444 CrossRef PubMed.
- M. T. Whited and R. H. Grubbs, Acc. Chem. Res., 2009, 42, 1607–1616 CrossRef CAS PubMed.
- H. Werner, Angew. Chem., Int. Ed., 2010, 49, 4714–4728 CrossRef CAS PubMed.
- S. Conejero, M. Paneque, M. L. Poveda, L. L. Santos and E. Carmona, Acc. Chem. Res., 2010, 43, 572–580 CrossRef CAS PubMed.
- Y. Li, J.-S. Huang, Z.-Y. Zhou, C.-M. Che and X.-Z. You, J. Am. Chem. Soc., 2002, 124, 13185–13193 CrossRef CAS PubMed.
- S. L. Marquard, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2013, 135, 6018–6021 CrossRef CAS PubMed.
- P. Schwab, N. Mahr, J. Wolf and H. Werner, Angew. Chem., Int. Ed. Engl., 1993, 32, 1480–1482 CrossRef.
- R. J. Burford, W. E. Piers and M. Parvez, Organometallics, 2012, 31, 2949–2952 CrossRef CAS.
- L. E. Doyle, W. E. Piers and J. Borau-Garcia, J. Am. Chem. Soc., 2015, 137, 2187–2190 CrossRef CAS PubMed.
- E. A. LaPierre, W. E. Piers, D. M. Spasyuk and D. W. Bi, Chem. Commun., 2016, 52, 1361–1364 RSC.
- L. E. Doyle, W. E. Piers, J. Borau-Garcia, M. J. Sgro and D. M. Spasyuk, Chem. Sci., 2016, 7, 921–931 RSC.
- D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2002, 124, 9976–9977 CrossRef CAS PubMed.
- V. M. Iluc and G. L. Hillhouse, J. Am. Chem. Soc., 2014, 136, 6479–6488 CrossRef CAS PubMed.
- M. Broring, C. D. Brandt and S. Stellwag, Chem. Commun., 2003, 2344–2345 RSC.
- W. Weng, C.-H. Chen, B. M. Foxman and O. V. Ozerov, Organometallics, 2007, 26, 3315–3320 CrossRef CAS.
- T. Cantat, N. Mézailles, L. Ricard, Y. Jean and P. Le Floch, Angew. Chem., Int. Ed., 2004, 43, 6382–6385 CrossRef CAS PubMed.
- V. H. Gessner, F. Meier, D. Uhrich and M. Kaupp, Chem.–Eur. J., 2013, 19, 16729–16739 CrossRef CAS PubMed.
- J. Campos, R. Peloso and E. Carmona, Angew. Chem., Int. Ed., 2012, 51, 8255–8258 CrossRef CAS PubMed.
- N. D. Jones, G. Lin, R. A. Gossage, R. McDonald and R. G. Cavell, Organometallics, 2003, 22, 2832–2841 CrossRef CAS.
- G. Lin, N. D. Jones, R. A. Gossage, R. McDonald and R. G. Cavell, Angew. Chem., Int. Ed., 2003, 42, 4054–4057 CrossRef CAS PubMed.
- K. K. Irikura and W. A. Goddard, J. Am. Chem. Soc., 1994, 116, 8733–8740 CrossRef CAS.
- C. Heinemann, R. H. Hertwig, R. Wesendrup, W. Koch and H. Schwarz, J. Am. Chem. Soc., 1995, 117, 495–500 CrossRef CAS.
- A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed.
- E. Soriano and J. Marco-Contelles, Acc. Chem. Res., 2009, 42, 1026–1036 CrossRef CAS PubMed.
- Y. Zhang and J. Wang, Eur. J. Org. Chem., 2011, 1015–1026 CrossRef.
- D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS PubMed.
- C. C. Comanescu and V. M. Iluc, Organometallics, 2014, 33, 6059–6064 CrossRef CAS.
- P. Cui, C. C. Comanescu and V. M. Iluc, Chem. Commun., 2015, 51, 6206–6209 RSC.
- C. C. Comanescu and V. M. Iluc, Inorg. Chem., 2014, 53, 8517–8528 CrossRef CAS PubMed.
- C. C. Comanescu and V. M. Iluc, Organometallics, 2015, 34, 4684–4692 CrossRef CAS.
- C. C. Comanescu and V. M. Iluc, Chem. Commun., 2016 10.1039/C5CC09468B.
- C. C. Comanescu, M. Vyushkova and V. Iluc, Chem. Sci., 2015, 6, 4570–4579 RSC.
- P. Cui and V. M. Iluc, Chem. Sci., 2015, 6, 7343–7356 RSC.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- H. Schumann, D. M. M. Freckmann and S. Dechert, Organometallics, 2006, 25, 2696–2699 CrossRef CAS.
- H. Viebrock, T. Panther, U. Behrens and E. Weiss, J. Organomet. Chem., 1995, 491, 19–25 CrossRef CAS.
- M. D. Fryzuk, J. B. Love and S. J. Rettig, J. Am. Chem. Soc., 1997, 119, 9071–9072 CrossRef CAS.
- S. Harder, Coord. Chem. Rev., 1998, 176, 17–66 CrossRef CAS.
- P. L. Arnold and S. T. Liddle, Organometallics, 2006, 25, 1485–1491 CrossRef CAS.
- R. A. Musgrave, R. S. P. Turbervill, M. Irwin and J. M. Goicoechea, Angew. Chem., Int. Ed., 2012, 51, 10832–10835 CrossRef CAS PubMed.
- J. A. R. Schmidt, S. A. Chmura and J. Arnold, Organometallics, 2001, 20, 1062–1064 CrossRef CAS.
- H. Schumann, S. Schutte, H.-J. Kroth and D. Lentz, Angew. Chem., Int. Ed., 2004, 43, 6208–6211 CrossRef CAS PubMed.
- H. Bock, Z. Havlas, K. Gharagozloo-Hubmann, S. Holl and M. Sievert, Angew. Chem., Int. Ed., 2003, 42, 4385–4389 CrossRef CAS PubMed.
- F. Feil and S. Harder, Organometallics, 2000, 19, 5010–5015 CrossRef CAS.
- C. Unkelbach, H. S. Rosenbaum and C. Strohmann, Chem. Commun., 2012, 48, 10612–10614 RSC.
- I. Chávez, A. Alvarez-Carena, E. Molins, A. Roig, W. Maniukiewicz, A. Arancibia, V. Arancibia, H. Brand and J. Manuel Manríquez, J. Organomet. Chem., 2000, 601, 126–132 CrossRef.
- I. S. Weitz and M. Rabinovitz, J. Chem. Soc., Perkin Trans. 1, 1993, 117–120 RSC.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- S. K. Sur, J. Magn. Reson., 1989, 82, 169–173 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for all new compounds, computational results, single crystal X-ray structure analysis of complexes 2, 3, 5 and 6. CCDC 1436940–1436943. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00948d |
| This journal is © The Royal Society of Chemistry 2016 |