
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrostatic binding of polyanions using self-assembled multivalent (SAMul) ligand displays – structure–activity effects on DNA/heparin binding†
Loryn E.
Fechner
a,
Buthaina
Albanyan
a,
Vânia M. P.
Vieira
a,
Erik
Laurini
b,
Paola
Posocco
 b,
Sabrina
Pricl
*b and
David K.
Smith
*a
b,
Sabrina
Pricl
*b and
David K.
Smith
*a
aDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: david.smith@york.ac.uk
bSimulation Engineering (MOSE) Laboratory, Department of Engineering and Architectures (DEA), University of Trieste, Trieste, 34127, Italy. E-mail: sabrina.pricl@di3.units.it
First published on 18th April 2016
Abstract
This paper reports that modifying the ligands in self-assembled multivalent (SAMul) displays has an impact on apparent binding selectivity towards two nanoscale biological polyanions – heparin and DNA. For the nanostructures assayed here, spermidine ligands are optimal for heparin binding but spermine ligands are preferred for DNA. Probing subtle differences in such nanoscale binding interfaces is a significant challenge, and as such, several experimental binding assays – competition assays and isothermal calorimetry – are employed to confirm differences in affinity and provide thermodynamic insights. Given the dynamic nature and hierarchical binding processes involved in SAMul systems, we employed multiscale modelling to propose reasons for the origins of polyanion selectivity differences. The modelling results, when expressed in thermodynamic terms and compared with the experimental data, suggest that DNA is a shape-persistent polyanion, and selectivity originates only from ligand preferences, whereas heparin is more flexible and adaptive, and as such, actively reinforces ligand preferences. As such, this study suggests that inherent differences between polyanions may underpin subtle binding selectivity differences, and that even simple electrostatic interfaces such as these can have a degree of tunability, which has implications for biological control and regulation on the nanoscale.
Introduction
Biology is dominated by polyanions, such as nucleic acids, glycosaminoglycans, proteoglycans, micro-tubules and membranes – subtle discrimination between these nanoscale species is important to regulate and control this ‘polyanion world’.1 However, discrimination between polyanions is a highly challenging target given their similarities in terms of charge density, their charge providing the primary mechanism by which they can be bound. As such, in terms of supramolecular and nanoscale chemistry, polyanions are interesting, but difficult targets for selective binding. As a result, most studies focus on a specific anion with a defined application in mind, e.g., DNA binding for gene delivery,2 or heparin binding for coagulation control.3 The development of active agents in these two areas is of considerable clinical relevance and has made these polyanions of great interest. Considering these two specific anions, DNA is a negatively charged as a result of phosphate links in the sugar backbone, while the polysaccharide backbone of heparin is appended with anionic sulfates and carboxylates. Clearly there are some inherent differences between these polyanions, but surprisingly, there has been relatively little interest in probing binding selectivity. Obviously, if we could understand the factors which allow receptors to preferentially bind to one polyanion over another, as well as addressing a genuine challenge in supramolecular design, we would also be able to develop systems that are able to intervene much more precisely in processes of biomedical relevance and are better optimised for specific clinical applications.2,3Nanoscale targets such as polyanions are also a significant challenge for supramolecular chemistry because of their relatively large, solvent-exposed surfaces.4 Effective binding is best achieved using multivalency, i.e., employing ligands with many points of interaction.5 Given the relative difficulty of using synthetic chemistry to construct covalent multivalent arrays, there has recently been increasing interest in developing self-assembled multivalent (SAMul) ligand displays, in which multiple ligands non-covalently assemble to generate a nanoscale display which interacts with a binding partner.6 This hierarchical approach to nanoscale recognition has been used to target (e.g.) sugar binding proteins,7 integrins,8 nucleic acids9 and heparin.10 SAMul is a tunable strategy because it only requires the synthesis of small molecules – it is therefore easy to introduce structural variation and explore structure–activity relationships. To bind polyanions such as DNA or heparin requires cationic ligands,9,10 which bind via multiple electrostatic ion–ion interactions. We recently reported that ligand chirality could influence apparent heparin/DNA binding selectivity,11 but selective polyanion binding, for the reasons outlined above, remains a rarely explored, challenging target.
There has been considerable interest in colloid science in investigating the interaction of self-assembled simple cationic lipids with specific polyanions in both practical and theoretical terms.12 In the widely accepted model, charge density plays the dominant role in binding – it is noted that other interactions can then influence selectivity, but there are relatively few specific experimental examples of this.13 Synthetic polyanions have been studied, and it has been shown, for example, that hydrophobic interactions between the polymer chain and the hydrophobic unit in the lipid can play an important role in moderating charge–charge binding effects.14 In this paper, we determine the impact of ligand choice in our SAMul displays and report an experimental example in which we uncover apparent selectivities of naturally occurring biopolyanions, DNA and heparin, for different nanoscale assemblies – we use multiscale modelling methods to provide further insight into the complex, interdependent, hierarchical self-assembly and nanoscale binding processes.
Results and discussion
For the purposes of this structure–activity effect study, amphiphilic molecules with different ligands were synthesised (Fig. 1) each of which could, in principle, self-assemble into micelles displaying cationic ligand surfaces. As hydrophobe we selected palmitic acid (C16), with different amines as ligands, connected using TBTU-mediated peptide coupling with an appropriate protecting group strategy (see ESI†). This yielded C16-DAP, C16-DAPMA, C16-SPD and C16-SPM, with nominal ligand charges of +1, +2, +2 and +3 respectively at physiological pH (7.4). Singly-charged C16-DAP was largely insoluble in water/buffer – its +1 charge is insufficient to counterbalance the hydrophobicity, and it was not studied further. Compound C16-DAPMA (+2) had good aqueous solubility, spermidine-based C16-SPD (+2) was slightly less soluble, and spermine-derived C16-SPM (+3) was more difficult to dissolve. We reason the +3 charge of C16-SPM hinders assembly, and hence solubility, because cation–cation repulsions on the micellar surface are not fully offset by the hydrophobic driving force for assembly.15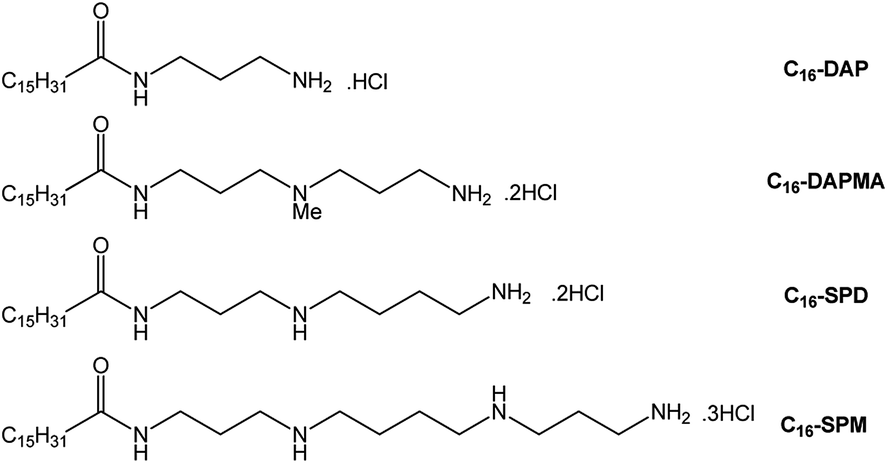 | ||
| Fig. 1 Compounds investigated in this paper. | ||
We initially quantified self-assembly using a Nile Red assay16 in 150 mM NaCl. All studies in this paper were performed at this salt concentration; ionic strength can have a major impact on electrostatic binding and it is important to keep it constant. The resulting critical micelle concentrations (CMCs) supported the macroscopic solubility observations, with C16-DAPMA having the lowest CMC and C16-SPM the highest, with C16-SPM requiring heating to encourage solubility – in agreement with entropically-driven hydrophobic self-assembly. Further analysis of CMC data in different buffer/salt conditions is provided in the ESI.† For validation, we also used isothermal titration calorimetry (ITC) and the resulting CMCs were in very good agreement with those from the Nile Red assay (Table 1). Importantly, treatment of the ITC data also provided thermodynamic parameters for self-assembly (ΔHmic, TΔSmic and ΔGmic) which support the proposal that C16-SPM had the least favourable self-assembly, primarily as a result of the enthalpic term. Dynamic light scattering (DLS, Fig. S1–S3,†Table 1) indicated, based on the volume contribution, that all three compounds formed similar-sized assemblies, as may be expected given the relatively similar molecular sizes of the three compounds. Perhaps surprisingly, the most highly-charged C16-SPM actually formed the assembly with the lowest zeta potential – significantly lower than that observed for C16-DAPMA, which may reflect the difficulty of bringing these more highly charged ligands into close proximity on the nanoscale surface. We also used transmission electron microscopy (TEM) to visualise the self-assembled nanostructures formed by these ligands (Fig. S4–S6†). On drying aqueous samples on a TEM grid, we observed that in each case, self-assembled spherical nanostructures could be visualised, in good general agreement with the DLS data.
| C16-DAPMA (+2) | C16-SPD (+2) | C16-SPMa (+3) | |
|---|---|---|---|
| a Heating was required to encourage solubility under these conditions. | |||
| CMCNR/μM | 40 ± 1 | 51 ± 2 | 65 ± 20 |
| CMCITC/μM | 34 | 52 | 71 |
| ΔHmic/kJ mol−1 | −10.81 | −8.61 | −8.41 |
| TΔSmic/kJ mol−1 | 14.72 | 15.86 | 15.29 |
| ΔGmic/kJ mol−1 | −25.52 | −24.47 | −23.70 |
| Diameter/nm | 6.2 ± 1.3 | 6.6 ± 0.2 | 6.2 ± 0.1 |
| Zeta potential/mV | +51.9 ± 2.6 | +44.0 ± 1.7 | +40.5 ± 0.9 |
To further understand self-assembly, we used multiscale molecular simulation in 150 mM aqueous NaCl (see ESI†).17 Spherical micelles were obtained in all cases (e.g., Fig. 2). Interestingly, simulation indicated that the compounds formed micelles with different packing densities. Specifically, the aggregation number (Nagg, Table 2) suggests that C16-DAPMA forms more tightly packed micelles than C16-SPD, which in turn is more densely packed than C16-SPM. As suggested from experiment, the hydrophobic C16 chain struggles to bring together the more highly charged SPM ligands. As a result of the decrease in Nagg for C16-SPM, the electrostatic potential ψs also decreases, leading to simulated zeta potentials (ζ, Table 2) in good agreement with the experimental data (Table 1), with C16-DAPMA > C16-SPD > C16-SPM.
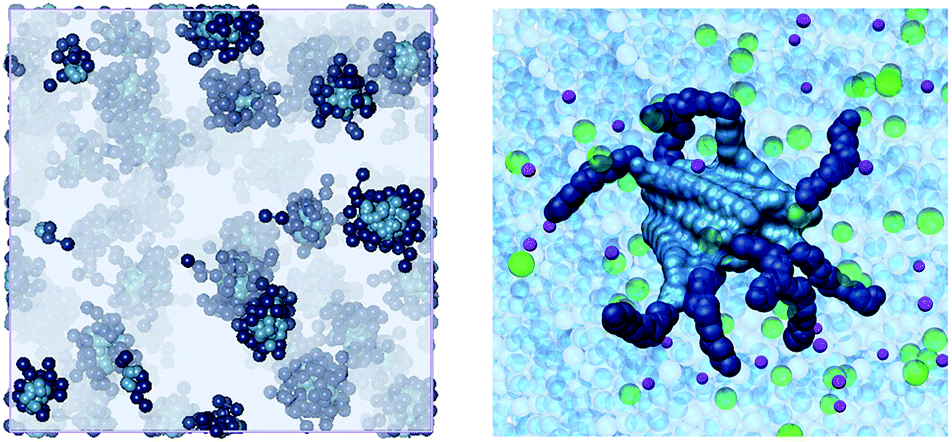 | ||
| Fig. 2 Mesoscopic (left) and atomistic (right) simulations of C16-SPM self-assembling into micelles. The C16 hydrophobic portion is shown as steel blue spheres whereas the SPM residues are portrayed as navy blue spheres. In the left panel, water, ions and counterions are shown as light grey field; in the right panel, water molecules are depicted as transparent light blue spheres, some Na+ and Cl− ions are shown as purple and green spheres, respectively. | ||
| Compound | N agg | D m (nm) | ψ s (mV) | ζ (mV) |
|---|---|---|---|---|
| C16-DAPMA | 16 ± 2 | 6.0 ± 0.3 | 172.4 | 50.2 |
| C16-SPD | 13 ± 1 | 6.3 ± 0.1 | 153.3 | 45.1 |
| C16-SPM | 10 ± 1 | 5.8 ± 0.2 | 144.6 | 41.8 |
The ability of SAMul nanostructures to bind polyanions was then tested experimentally. DNA binding was assessed using an ethidium bromide (EthBr) displacement assay18 in which the compounds displace EthBr from calf thymus DNA, as monitored by fluorimetry. Heparin binding was monitored by UV-Vis spectroscopy using our recently introduced Mallard Blue (MalB) dye in a competition assay.19 All assays were performed in triplicate. These simple, rapid approaches allowed us to determine CE50 values (cation![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anion charge excess at which 50% of dye is displaced). These can be converted into EC50 values (effective concentration of binder at the same point). Under assay conditions, all compounds had good solubility – interactions between cationic ligands and the polyanionic binding partner decrease cation–cation repulsion at the micellar surface (see below for further discussion).
:anion charge excess at which 50% of dye is displaced). These can be converted into EC50 values (effective concentration of binder at the same point). Under assay conditions, all compounds had good solubility – interactions between cationic ligands and the polyanionic binding partner decrease cation–cation repulsion at the micellar surface (see below for further discussion).
From these rapid assays, it was found (Table 3) that the more highly charged C16-SPM ligand appears to be the optimal DNA binder with low CE50 and EC50 values, whereas C16-SPD and C16-DAPMA are less effective. Interestingly, although C16-DAPMA and C16-SPD have the same ligand charge (+2), C16-DAPMA is a slightly better DNA binder. In contrast, for heparin binding, C16-SPD is the most charge-efficient binder as measured by its CE50 value, significantly outperforming C16-DAPMA, even though the latter compound was the slightly better DNA binder and both systems have the same nominal charge. C16-SPD even performs better than more highly charged C16-SPM in terms of its CE50 value. Although these differences are relatively small, they were reproducible and outside of error range – as such, they provide some hint that heparin and DNA behave differently when faced with these SAMul nanostructures as binding partners.
:anion charge excess at which 50% of indicator dye is displaced from its complex) and EC50 (effective concentration at which 50% of dye is displaced)
| C16-DAPMA (+2) | C16-SPD (+2) | C16-SPM (+3) | ||
|---|---|---|---|---|
| CE50 | DNA | 5.0 ± 0.1 | 6.0 ± 0.3 | 4.3 ± 0.1 |
| Heparin | 0.69 ± 0.05 | 0.34 ± 0.05 | 0.49 ± 0.01 | |
| EC50/μM | DNA | 10.1 ± 0.1 | 11.9 ± 0.5 | 5.7 ± 0.1 |
| Heparin | 37 ± 3 | 19 ± 3 | 17.5 ± 0.3 |
All of the reported EC50 values (Table 3) were below the CMC values of these compounds (Table 1). Polyanions can assist self-assembly by limiting electrostatic repulsion on the cationic SAMul surface.20 This agrees with the observation that all compounds showed excellent solubility in polyanion-binding assays, unlike in their absence. It should also be noted that the lineshapes for these binding assays are sigmoidal (Fig. 3) and can be divided into three regions: (i) non-assembled binder initially struggles to displace polyanion-binding dye, (ii) at a critical concentration, binding occurs and dye displacement is activated (iii) the system saturates and no further dye is displaced. The onset of region (ii) allows us to estimate apparent critical aggregation concentration (CAC) values in the presence of the polyanions – see ESI† for full data. These observations are consistent with a system that only self-assembles and binds polyanions at a critical concentration. It is also clear that while self-assembly evidently assists multivalent binding, the converse is also true, and multivalent binding can considerably assist self-assembly – reflecting the dynamic nature of these SAMul nanostructures.
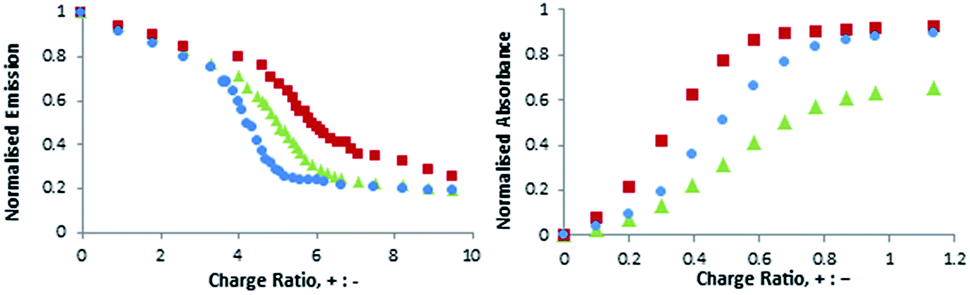 | ||
| Fig. 3 Titration curves for EthBr (left, DNA binding) and MalB (right, heparin binding) displacement. For DNA binding: [DNA] = 4 μM (per base), [EthBr] = 5.07 μM, buffer: 2 mM HEPES, 0.05 mM EDTA, 150 mM NaCl. For heparin binding: [heparin] = 27 μM (per disaccharide unit), [MalB] = 25 μM, buffer: Tris HCl 10 mM, 150 mM NaCl. Blue = C16-SPM, red = C16-SPD, green = C16-DAPMA. | ||
We were somewhat surprised by the DNA/heparin selectivity differences reported above, as we had anticipated that the polyanions would bind to the most highly charged systems best, and that both polyanions would exhibit similar orders of preference. As such, we therefore used ITC to further validate the data. These experiments are complex, as there are many near-simultaneous processes which are difficult to deconvolute: (i) non-assembled ligand binding to polyanion, (ii) ligand self-assembly, (iii) self-assembled ligands binding to polyanion, and (iv) further nanoscale assembly. Nonetheless, we reasoned that we could obtain useful thermodynamic data for the overall binding process which would still be informative (Table 4) and would provide insight into the impact of the presence of the polyanion on the self-assembly of the ligands. In particular, we reasoned that comparing the data with that obtained for the self-assembly of the ligands in the absence of polyanion (Table 1) should allow us to isolate the thermodynamic contributions which are a direct result of ligand–polyanion interactions.
| C16-DAPMA (+2) | C16-SPD (+2) | C16-SPM (+3) | ||
|---|---|---|---|---|
| CAC (with polyanion)/μM | DNA | 6.1 | 9.8 | 3.4 |
| Heparin | 13.6 | 7.2 | 9.8 | |
| ΔHagg/kJ mol−1 | DNA | −12.2 | −12.5 | −15.0 |
| Heparin | −12.8 | −13.5 | −11.0 | |
| TΔSagg/kJ mol−1 | DNA | 17.6 | 16.2 | 16.0 |
| Heparin | 15.0 | 15.9 | 17.6 | |
| ΔG agg /kJ mol −1 | DNA | −29.8 | −28.7 | −31.0 |
| Heparin | −27.8 | −29.4 | −28.6 | |
| ΔHbind/kJ mol−1 | DNA | −1.4 | −3.9 | −6.6 |
| Heparin | −1.9 | −4.9 | −2.6 | |
| TΔSbind/kJ mol−1 | DNA | 2.9 | 0.4 | 0.7 |
| Heparin | 0.3 | 0.0 | 2.3 | |
| ΔG bind /kJ mol −1 | DNA | −4.3 | −4.2 | −7.3 |
| Heparin | −2.2 | −4.9 | −4.9 |
The method was based on titrating the ligand into the polyanion, and therefore allowed us to determine polyanion-modified critical aggregation concentrations (CACs). Interestingly, these modified CAC values had good agreement with those determined from the dye displacement assay (see ESI†), which would suggest good comparability between our different binding assay approaches. Furthermore, ITC allowed us to elucidate the thermodynamic parameters for SAMul self-assembly in the presence of polyanion. By comparing these thermodynamics of aggregation in the presence (ΔHagg, TΔSagg and ΔGagg, Table 4) and absence (ΔHmic, TΔSmic and ΔGmic, Table 1) of polyanion, we estimate the difference to represent the effective binding between the SAMul nanostructures and the polyanion (e.g. ΔHbind = ΔHagg − ΔHmic) (Table 4). This simple approach allows us to extract and quantify the effective change in solution thermodynamics induced by the polyanion.
Pleasingly, the ITC data in terms of apparent binding affinity (ΔGbind, bold, Table 4) were in broad agreement with the trends obtained from the dye displacement assays. Once again, C16-SPM was the most effective DNA binder (ΔGbind), and C16-SPD was the most effective heparin binder, especially if the ΔGbind values are normalised per charge (C16-SPD, −2.45 kJ mol−1; C16-SPM, −1.63 kJ mol−1). Furthermore, as in the dye displacement assays, for DNA binding C16-DAPMA > C16-SPD, whereas for heparin binding C16-SPD > C16-DAPMA. Although the thermodynamic differences are relatively small, they are in agreement with the results of the dye displacement assay, supporting the view that specific binding preferences are able to influence the simple ion–ion interactions (which provide the bulk of the binding affinity) in order to generate different selectivity from different polyanions. As such, the ITC data support the view that ligand choice directs polyanion selectivity in this system.
Considering the data in more detail, it is evident that much of the difference in binding appears to be caused by the ability of these polyanions to influence the aggregation of the SAMul ligand displays (ΔGagg, italics, Table 4). In the presence of DNA, the magnitude of ΔGagg for C16-SPM is greater than for the other two ligands, whereas in the presence of heparin, it is C16-SPD which has the largest ΔGagg value – this is also reflected in the apparent CACs in the presence of each of these polyanions. As such, we reason the specifics of the ligand–polyanion interaction can directly influence the ability of the nanostructures to assemble and hence, as a result, exhibit high-affinity SAMul binding.
In order to provide greater insight into the binding interface between the self-assembled nanostructure and the polyanions we once again turned to multiscale simulations. We brought together the SAMul nanostructures as optimised in the absence of polyanion with the polyanions themselves in order to probe the binding interface. Clearly this assumes that the nanostructures do not significantly change in the presence of the polyanion, which is a limitation on the method, but it provides the only tractable approach for simulating the interactions between these SAMul displays and polyanions in order to provide some insight into the thermodynamics of the nanoscale binding interface.
We note that in reality, multiple DNA helices or heparin chains will contact a single micelle. Indeed, this is supported by DLS data (Table S3†) recorded on the complexes formed between these cationic micelles and either heparin or DNA. These DLS data clearly indicate the formation of larger ill-defined aggregates (ca. 100–300 nm in diameter) on binding. The DLS data also provided further support for the binding preferences reported above, with zeta potentials suggesting that C16-SPM was most effective at neutralising the negative charge of DNA, while heparin binding reduced the cationic charge of C16-SPD more than either of the other ligands.
It is important to consider whether the cationic micelles actually remain intact during the polyanion binding process, as there is a possibility that significant reorganisation could occur. To probe this experimentally, we employed transmission electron microscopy (TEM) to visualise the morphologies formed when the self-assembled nanostructures bind to polyanionic heparin (Fig. 4). In agreement with similar literature studies on related systems10b,11 the micelles remained remarkably intact, and were organised into hierarchical nanoscale arrays. We suggest this is a result of close packing interactions between the spherical micellar polycations and the linear polyanions. As such, we are confident that the micelles do indeed remain intact on binding, and this supports our suggested methodological approach for computational modelling in which the pre-formed micelle is brought into contact with a polyanion chain in order to determine the fundamental binding interactions.
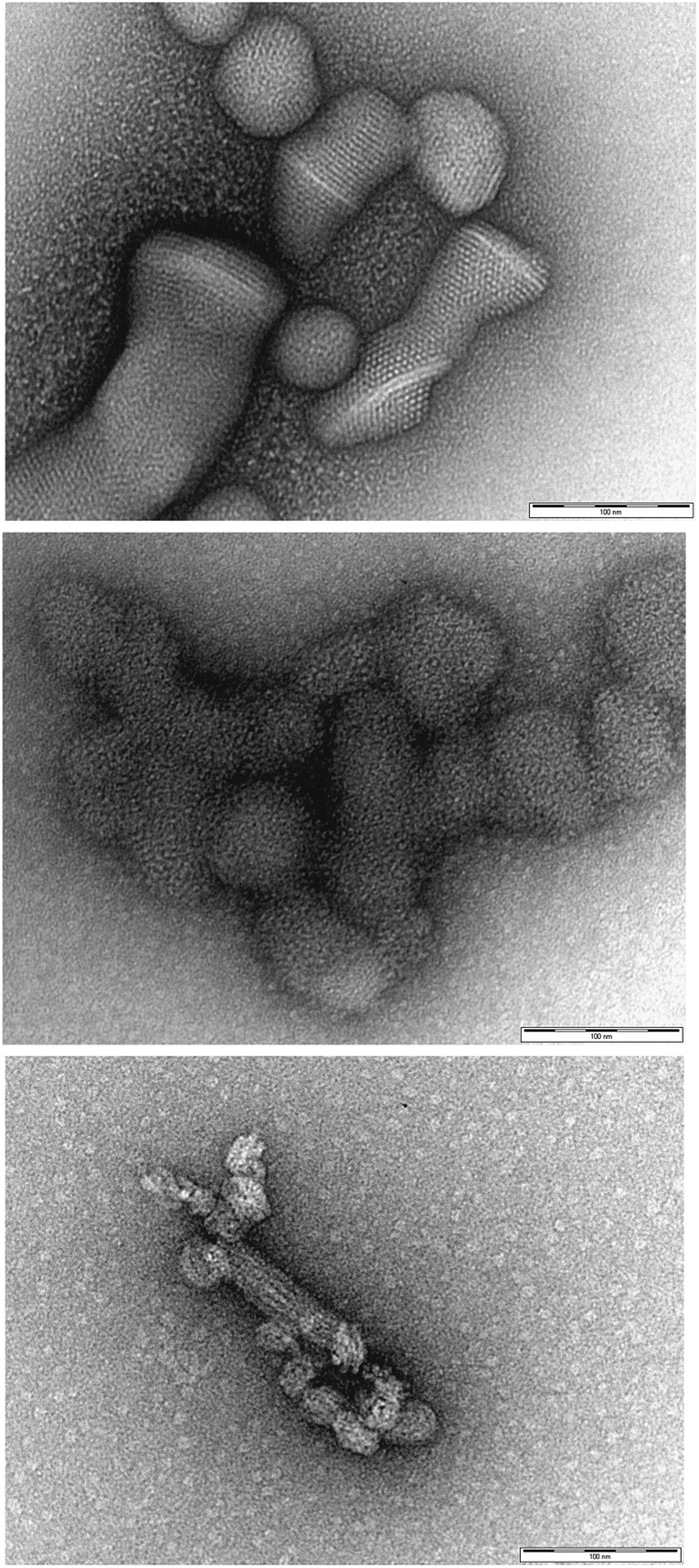 | ||
| Fig. 4 TEM images of self-assembled micellar nanostructures binding to heparin to yield a hierarchically organised self-assembled nanoscale aggregate and supporting the view that self-assembled micelles remain stable under polyanion binding conditions: (top) C16-DAPMA, (middle) C16-SPD, (bottom) C16-SPM. All scale bars are 100 nm. | ||
We therefore reason, with this experimental support from TEM, that our modelling approach which contacts a single micelle with a single polyanion is a valid methodology for gaining insight into the fundamental forces responsible for the primary binding event. In this way, we were interested to determine whether the thermodynamic insights obtained would show broad agreement with our experimental observations of binding. Obviously, understanding the further hierarchical assembly event is more complex, and was beyond the scope of this study, which was instead focussed on the differences induced by different ligands at the initial polyanion binding interface.
For DNA binding (Fig. 5, top), the C16-SPM micelles contain 10 SPM residues, 9 of which effectively contact DNA (a parameter we define as Neff), resulting in a charge-normalized binding free energy (per-effective-residue) ΔG* of −14.32 kJ mol−1. Conversely, C16-SPD and C16-DAPMA nanostructures only use 7 and 8 (out of 13 and 16) SPM residues, respectively, to bind DNA. For C16-SPD and C16-DAPMA, the per-effective-residue interactions were lower, with ΔG* values of −9.76 and −10.80 kJ mol−1, respectively. The simulated ΔG* values therefore follow the same trend as the experimental CE50 values and ITC data: C16-SPM > C16-DAPMA > C16-SPD.
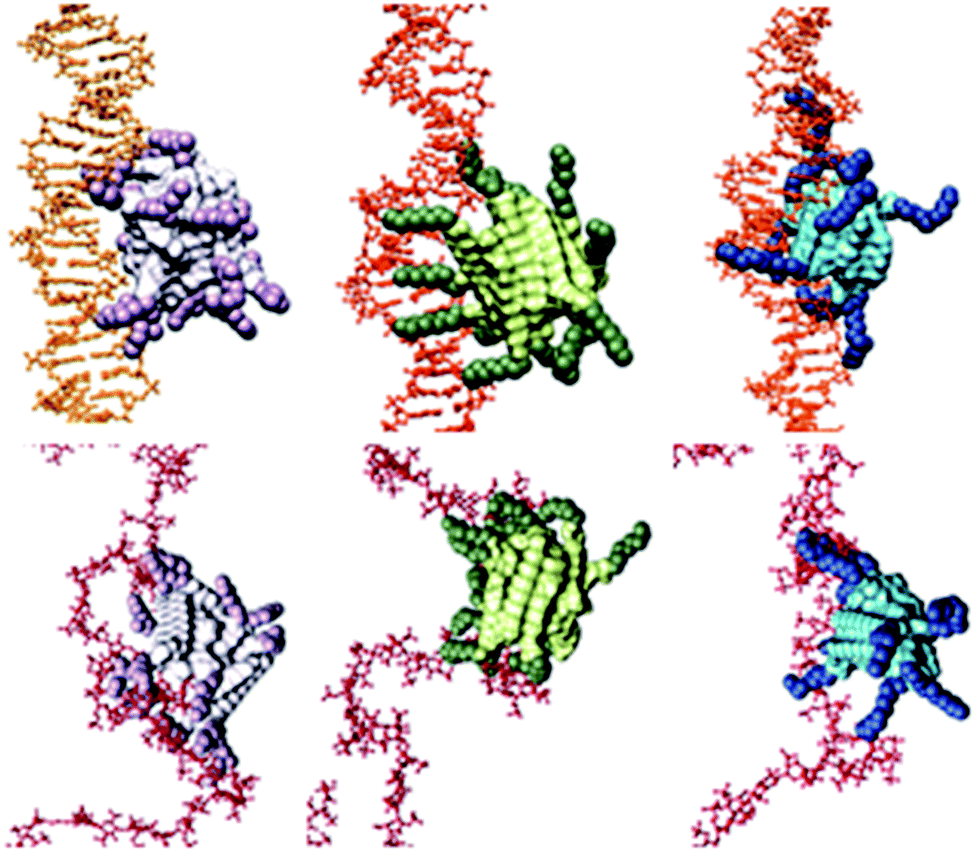 | ||
| Fig. 5 Equilibrated atomistic molecular dynamics (MD) simulation snapshots of SAMul micelles binding DNA (upper panel, orange) and heparin (lower panel, firebrick). In both panels, from left to right: C16-DAPMA (light grey (C16) and plum (DAPMA)), C16-SPD (lime green (C16) and forest green (SPD)), and C16-SPM (steel blue (C16) and navy blue (SPM)). Hydrogen atoms, water molecules, ions and counterions are not shown for clarity. | ||
For heparin binding (Fig. 5, bottom), the micelles formed by C16-SPD engage 12 out of 13 available ligands in productive binding, resulting in charge-normalized ΔG* of −14.98 kJ mol−1. However, C16-DAPMA and C16-SPM assemblies only exploit 9/16 and 6/10 ligands, giving ΔG* values of −8.65 and −11.97 kJ mol−1, respectively. The predicted ΔG* values are thus in agreement with the trend of experimental data: C16-SPD > C16-SPM > C16-DAPMA.
To understand why polyanions appear to have different selectivities towards SAMul nanostructures, we then deconvoluted these overall ΔG* values into enthalpic (ΔH*) and entropic (−TΔS*) components (Fig. 6 and ESI†).
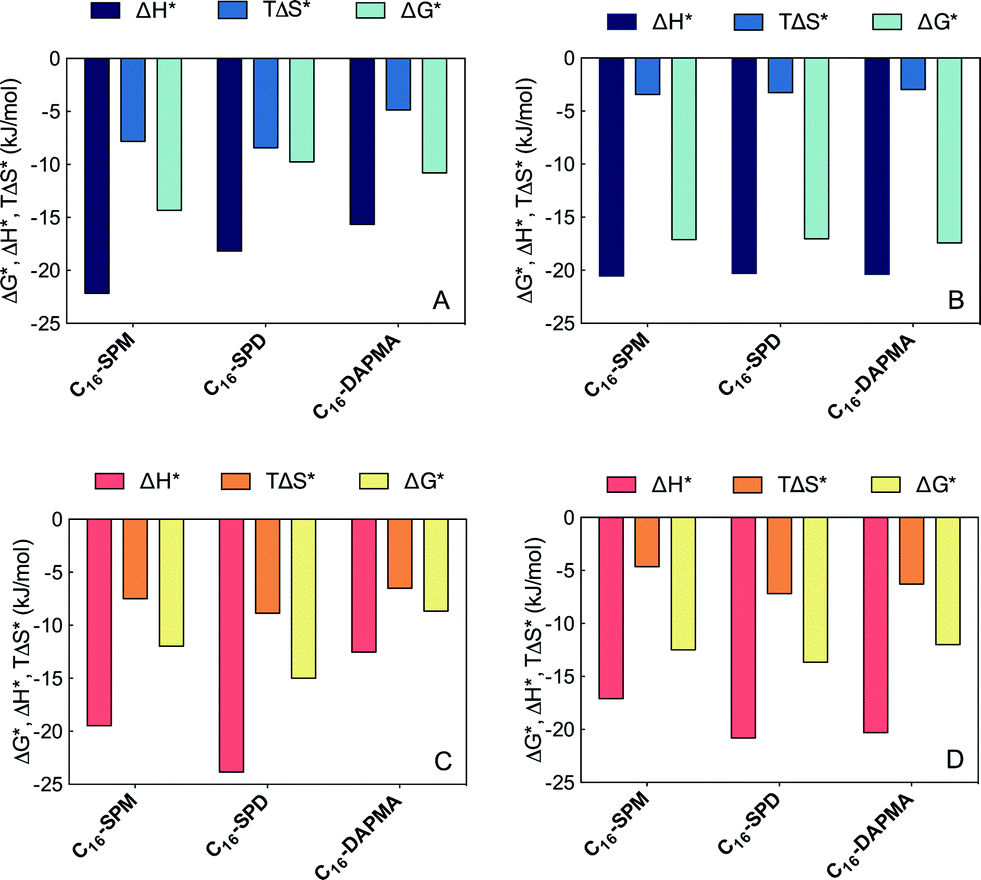 | ||
| Fig. 6 Charge-normalized per-residue effective free energy of binding (ΔG*), and enthalpic (ΔH*) and entropic (TΔS*) components for: (A) each SAMul micelle ligand-type complexed with DNA; (B) DNA-bases complexed with each of the SAMul micelles; (C) each SAMul micelle ligand-type complexed with heparin; (D) heparin sugars complexed with each of the SAMul micelles. | ||
For DNA binding (Fig. 5, upper panel) considered from the viewpoint of each effective SAMul cationic charge (Fig. 6A), the flexible C16-SPM (3+) ligands can enthalpically overcompensate the significant entropic cost associated with their organisation on binding DNA. In part, this is due to the greater reduction in cation–cation repulsions at the SAMul surface of C16-SPM on DNA binding, which in turn will assist self-assembly. The other two less-charged ligands have less enthalpic gain and bind less well – in full agreement with the experimental ITC data (Table 4). The slightly more rigid C16-DAPMA suffers less entropic penalty than C16-SPD on binding, as it does not reorganise, slightly favouring its DNA binding over C16-SPD. Once again this is in agreement with ITC data (Table 4).
Applying the same analysis, but from the viewpoint of each anionic DNA charge involved at the binding interface (Fig. 6B), ΔG*, ΔH* and TΔS* are practically independent of the choice of ligand – i.e., from the perspective of DNA, all interactions are equally good. The selectivity of the SAMul micelles towards DNA can thus be ascribed only to the optimization of the ligands – as such DNA appears to be a shape-persistent21 polyanion which will simply bind to, and organise the SAMul display with which it is presented.
For heparin binding (Fig. 6, lower panel), considered from the viewpoint of each effective SAMul cationic charge (Fig. 5C), the enthalpic gain when C16-SPD reorganises to optimise its interactions is greater than for C16-DAPMA and also greater than for C16-SPM – in broad agreement with the ITC data (ΔHbind, Table 4). It appears that C16-SPM is less effective at binding the more open surface of heparin in terms of enthalpic gain than it was for DNA. Although the entropic penalty of binding C16-DAPMA is, as for DNA binding, less than that of the more flexible C16-SPD, it is in this case outweighed by the enthalpic term. As such, C16-SPD emerges as the optimal system for heparin binding, in agreement with ITC (ΔGbind, Table 4).
Considered from the viewpoint of each heparin sugar (−2), we also observe different behaviour depending on the ligand. Each heparin residue offsets the entropic cost of binding SPD with a greater enthalpic gain of its own (Fig. 6D). This is in contrast to DNA, where each anion behaved identically, irrespective of the ligand. As such, the more effective binding of C16-SPD induces more effective binding from each residue of the heparin chain via an enthalpy/entropy optimisation, mediated through polyanion structural adaptation – i.e. heparin is an adaptive21 polyanion, which not only binds to the SAMul display, but importantly, is also able to adapt itself in response.
We believe that these insights, which correlate experimental and simulation data for the challenging problem of polyanion binding hint at fundamental differences through which some discrimination between polyanions may be achieved. Interestingly, even biology struggles to achieve selectivity between DNA and heparin within its proteins – for example many (but not all) DNA/RNA binding proteins also bind to heparin.22 Clearly understanding the factors which can lead to even small degrees of selectivity between these polyanions, and developing synthetic nanosystems with this capacity, is therefore useful. In this regard, it is worth noting that flexibility has recently been identified as a key factor in heparin binding proteins – which would be supported by the view, expressed here, of this polyanion being an adaptive binding target.23
Conclusions
In summary, this paper demonstrates that ligand choice in SAMul displays can have an influence on apparent binding selectivity. As such, electrostatic ion–ion binding depends on structural detail, not only charge density – as confirmed by the complementary experimental methods of competition binding assays and isothermal calorimetry. Of the compounds studied here, C16-SPM is optimal for DNA binding, while C16-SPD is optimal for heparin binding. We note that the polyanions play a role in assisting self-assembly and hence switching on the multivalent binding effect, and suggest that specifics of ligand–polyanion interactions help mediate subtle differences in this overall process. Molecular simulation studies lead us to propose that the shape-persistence (DNA), or adaptability (heparin) of the polyanionic targets help mediate the selectivity of interaction with different ligands. These results provide intriguing insight into molecular recognition processes at nanoscale surfaces and suggest that SAMul can deliver some selectivity in addressing the challenging problem of the ‘polyanion world’.Acknowledgements
This work was supported by a PROMOS scholarship from Freie Universität Berlin to LEF, the Saudi Arabian Government (Ministry of Education) via a PhD scholarship to BA and Marie Curie ITN ‘SMART-NET’ interdisciplinary training network funding to VMPV.Notes and references
- L. S. Jones, B. Yazzie and C. R. Middaugh, Mol. Cell. Proteomics, 2004, 3, 746–769 CAS.
- R. Srinivas, S. Samanta and A. Chaudhuri, Chem. Soc. Rev., 2009, 38, 3326–3338 RSC.
- S. M. Bromfield, E. Wilde and D. K. Smith, Chem. Soc. Rev., 2013, 42, 9184–9185 RSC.
- (a) K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef CAS PubMed; (b) D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817–2826 RSC.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- (a) A. Barnard and D. K. Smith, Angew. Chem., Int. Ed., 2012, 51, 6572–6581 CrossRef CAS PubMed; (b) K. Petkau-Milroy and L. Brunsveld, Org. Biomol. Chem., 2013, 11, 219–232 RSC.
- (a) J. E. Kingery-Wood, K. W. Williams, G. B. Sigal and G. M. Whitesides, J. Am. Chem. Soc., 1992, 114, 7303–7305 CrossRef CAS; (b) B. S. Kim, D. J. Hong, J. Bae and M. Lee, J. Am. Chem. Soc., 2005, 127, 16333–16337 CrossRef CAS PubMed; (c) M. K. Müller and L. Brunsveld, Angew. Chem., Int. Ed., 2009, 48, 2921–2924 CrossRef PubMed; (d) E. L. Dane, A. E. Ballok, G. A. O'Toole and M. W. Grinstaff, Chem. Sci., 2014, 5, 551–557 RSC.
- (a) D. J. Welsh and D. K. Smith, Org. Biomol. Chem., 2011, 9, 4795–4801 RSC; (b) D. J. Welsh, P. Posocco, S. Pricl and D. K. Smith, Org. Biomol. Chem., 2013, 11, 3177–3186 RSC.
- (a) D. Joester, M. Losson, R. Pugin, H. Heinzelmann, E. Walter, H. P. Merkle and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1486–1490 CrossRef CAS PubMed; (b) S. P. Jones, N. P. Gabrielson, D. W. Pack and D. K. Smith, Chem. Commun., 2008, 4700–4702 RSC; (c) S. P. Jones, N. P. Gabrielson, C.-H. Wong, H.-F. Chow, D. W. Pack, P. Posocco, M. Fermeglia, S. Pricl and D. K. Smith, Mol. Pharmaceutics, 2011, 8, 416–429 CrossRef CAS PubMed; (d) A. Barnard, P. Posocco, S. Pricl, M. Calderon, R. Haag, M. E. Hwang, V. W. T. Shum, D. W. Pack and D. K. Smith, J. Am. Chem. Soc., 2011, 133, 20288–20300 CrossRef CAS PubMed; (e) S. K. M. Nalluri, J. Voskuhl, J. B. Bultema, E. J. Boekema and B. J. Ravoo, Angew. Chem., Int. Ed., 2011, 50, 9747–9751 CrossRef CAS PubMed; (f) A. Tschiche, A. M. Staedtler, S. Malhotra, H. Bauer, C. Böttcher, S. Sharbati, M. Calderon, M. Koch, T. M. Zollner, A. Barnard, D. K. Smith, R. Einspanier, N. Schmidt and R. Haag, J. Mater. Chem. B, 2014, 2, 2153–2167 RSC; (g) X. Liu, J. Zhou, T. Yu, C. Chen, Q. Cheng, K. Sengupta, Y. Huang, H. Li, C. Liu, Y. Wang, P. Posocco, M. Wang, Q. Cui, S. Giorgio, M. Fermeglia, F. Qu, S. Pricl, Y. Shi, Z. Liang, P. Rocchi, J. J. Rossi and L. Peng, Angew. Chem., Int. Ed., 2014, 53, 11822–11827 CrossRef CAS PubMed.
- (a) K. Rajangam, H. A. Behanna, M. J. Hui, X. Han, J. F. Hulvat, J. W. Lomasney and S. I. Stupp, Nano Lett., 2006, 6, 2086–2090 CrossRef CAS PubMed; (b) A. C. Rodrigo, A. Barnard, J. Cooper and D. K. Smith, Angew. Chem., Int. Ed., 2011, 50, 4675–4679 CrossRef CAS PubMed; (c) S. M. Bromfield, P. Posocco, C. W. Chan, M. Calderon, S. E. Guimond, J. E. Turnbull, S. Pricl and D. K. Smith, Chem. Sci., 2014, 5, 1484–1492 RSC; (d) G. L. Montalvo, Y. Zhang, T. M. Young, M. J. Costanzo, K. B. Freeman, J. Wang, D. J. Clements, E. Magavern, R. W. Kavash, R. W. Scott, D. H. Liu and W. F. DeGrado, ACS Chem. Biol., 2014, 9, 967–975 CrossRef CAS PubMed.
- S. M. Bromfield and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 10056–10059 CrossRef CAS PubMed.
- L. Chiappisi, I. Hoffmann and M. Gradzielski, Soft Matter, 2013, 9, 3896–3909 RSC.
- (a) A. Perico and A. Ciferri, Chem.–Eur. J., 2009, 15, 6312–6320 CrossRef CAS PubMed; (b) D. Li and N. J. Wagner, J. Am. Chem. Soc., 2013, 135, 17547–17555 CrossRef CAS PubMed; (c) M. S. Sulatha and U. Natarajan, J. Phys. Chem. B, 2015, 119, 12526–12539 CrossRef CAS PubMed.
- For a review see: K. Kogej, Adv. Colloid Interface Sci., 2010, 158, 68–83 CrossRef CAS PubMed.
- K. Esumi and M. Ueno, Structure-Performance Relationships in Surfactants, Marcel Dekker, New York, 2003 Search PubMed.
- M. C. A. Stuart, J. C. van de Pas and J. B. F. N. Engberts, J. Phys. Org. Chem., 2005, 18, 929–934 CrossRef CAS.
- P. Posocco, E. Laurini, V. Dal Col, D. Marson, K. Karatasos, M. Fermeglia and S. Pricl, Curr. Med. Chem., 2012, 19, 5062–5087 CrossRef CAS PubMed.
- (a) B. F. Cain, B. C. Baguley and W. A. Denny, J. Med. Chem., 1978, 21, 658–668 CrossRef CAS PubMed; (b) D. L. Boger, B. E. Fink, S. R. Brunette, W. C. Tse and M. P. Hedrick, J. Am. Chem. Soc., 2001, 123, 5878–5891 CrossRef CAS PubMed.
- (a) S. M. Bromfield, A. Barnard, P. Posocco, M. Fermeglia, S. Pricl and D. K. Smith, J. Am. Chem. Soc., 2013, 135, 2911–2914 CrossRef CAS PubMed; (b) S. M. Bromfield, P. Posocco, M. Fermeglia, S. Pricl, J. Rodríguez-López and D. K. Smith, Chem. Commun., 2013, 49, 4830–4832 RSC.
- (a) A. J. Konop and R. H. Colby, Langmuir, 1999, 15, 58–65 CrossRef CAS; (b) H. Schiessel, M. D. Correa-Rodriguez, S. Rudiuk, D. Baigl and K. Yoshikawa, Soft Matter, 2012, 8, 9406–9411 RSC.
- S. M. Bromfield, P. Posocco, M. Fermeglia, J. Tolosa, A. Herreros-López, S. Pricl, J. Rodriguez-López and D. K. Smith, Chem.–Eur. J., 2014, 20, 9666–9674 CrossRef CAS PubMed.
- For a range of examples see: (a) K. Ishii, S. Futaki, H. Uchiyama, K. Nagasawa and T. Andoh, Biochem. J., 1987, 241, 111–119 CrossRef CAS PubMed; (b) J. Dudas, G. Ramadori, T. Knittel, K. Neubauer, D. Raddatz, K. Egedy and I. Kovalszky, Biochem. J., 2000, 350, 245–251 CrossRef CAS PubMed; (c) H. Ma, V. Leveque, A. De Witte, W. X. Li, T. Hendricks, S. M. Clausen, N. Cammack and K. Klumpp, Virology, 2005, 332, 8–15 CrossRef CAS PubMed.
- (a) J. A. Huntington, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824, 246–252 CrossRef CAS PubMed; (b) K.-W. Jeong, M.-C. Jeong, B. Jin and Y. Kim, Biochemistry, 2013, 52, 8823–8832 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic methods and characterisation data, assay and modelling methodologies, further experimental data from binding assays, DLS and TEM. See DOI: 10.1039/c5sc04801j |
| This journal is © The Royal Society of Chemistry 2016 |