
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot synthesis of a [c2]daisy-chain-containing hetero[4]rotaxane via a self-sorting strategy†
Xin
Fu‡
,
Qi
Zhang‡
,
Si-Jia
Rao
,
Da-Hui
Qu
* and
He
Tian
Key Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, China. E-mail: dahui_qu@ecust.edu.cn
First published on 12th January 2016
Abstract
The construction and efficient synthesis of hetero[n]rotaxanes with high structural complexity are always attractive challenges. Herein, we demonstrate a facile one-pot preparation of a hetero[4]rotaxane, by employing a self-sorting strategy, which contains an interpenetrated dibenzo-24-crown-8 (DB24C8) based [c2]daisy chain structure and is ended with a benzo-21-crown-7 (B21C7) based rotaxane at each side. The key to the design involved encoding the selective threading using a steric hindrance-related “language”, where highly selective self-assemblies occurred in a three-component self-sorting process, which included the threading of a benzylalkylammonium into a B21C7 and interpenetrated dimerized formation of a DB24C8 based [c2]daisy chain simultaneously; the precise pre-assembled system resulted in the efficient synthesis of hetero[4]rotaxane with a high-level of structural complexity under the “CuAAC” reaction.
Introduction
In the last twenty years, a variety of mechanically interlocked molecules (MIMs), especially rotaxanes and catenanes, have been delicately designed and constructed.1 The unique structural features of MIMs are utilized by scientists to birth elegant functional molecules,2 indicating the great potential of MIMs in functional materials. As a modification platform for functional groups, the structural complexity of MIMs significantly provides infinite possibilities in designing novel functional molecular machines.3 Hence, the construction and efficient synthesis of MIMs with high structural complexity have been greatly attractive but challenging.4 Recently, much attention has been focused on the topological,3a interpenetrating,5 knotted6 structures of MIMs with high structural complexity. In this intriguing family, [c2] daisy chain molecules,5 a class of rotaxanes constructed from the dimerization of AB-type linear monomers with two self-complementary units, A (host) and B (guest), have shown their talent in mimicking the contraction and extension movements of natural biological machines.5l Various elegant [c2] daisy chain molecules have been reported based on different host–guest systems including versatile macrocycles, such as crown ethers,5b,e,h cucurbit[n]uril,5i cyclodextrins,5a,c,d pillarenes,5g,m,p and cyclophanes.5oA variety of hetero[n]rotaxanes, especially those comprising two or more different types of ring moiety, have been designed and constructed,7 some even in a one-step strategy.7b,d This aroused our interest to integrate muscle-like daisy chain structures with hetero[n]rotaxanes to breed a hetero[n]rotaxane with a novel structural topology. However, it is difficult to synthesize this novel hetero[n]rotaxane based on traditional strategies for rotaxane synthesis,8 which are efficient in the preparation of typical [2]rotaxane but complicated when there is more than one type of ring moiety, due to them being “non-selective”. The key challenge is the dramatically increased complexity of the interpenetration process in the formation of a [c2]daisy chain when additional recognition sites and rings are incorporated into the multi-component self-assembling system. Hence, a programming language for highly selective self-assembly is desperately needed to integrate hetero[n]rotaxane with the daisy chain structure.
Self-sorting strategies have the unique capability of selective self-assembly in complex supramolecular systems.9 Schalley and co-workers10 have developed an efficient integrative self-sorting strategy for selective self-assembly to simultaneously incorporate two kinds of polyether macrocycles into a single axle molecule comprising two kinds of secondary ammoniums which, as a result, form a cascade-stoppered hetero[3]rotaxane. This self-sorting strategy has been proved to be very efficient in constructing many hetero[n]rotaxanes with increased structural complexity.11 However, to the best of our knowledge, this self-sorting strategy has not been proved to be appropriate for a system simultaneously involving both threading and interpenetration, which are known for the formation of pseudorotaxanes and daisy chain structures, respectively.
Herein, a facile one-pot synthesis of hetero[4]rotaxane 5 is successfully realized via a three-component (1, 2 and B21C7 in Scheme 1) self-sorting strategy. In principle, these initial components (1, 2 and B21C7 in Scheme 1) could assemble into several self-assembly species, while only two kinds of precursors, [c2]daisy chain 3 and [2]semi-rotaxane 4, were successfully formed through the highly selective self-assembly process as determined by 1H NMR studies. In this pre-assembly process, both the self-interpenetration of compound 1 to form [c2]daisy chain 3 and the threading of 2 into the cavity of B21C7 to form [2]semi-rotaxane 4 occurred simultaneously. Then the highly symmetrical hetero[4]rotaxane 5 containing [c2]daisy chain could be obtained by the following facile one-pot CuAAC click reaction.8d We believe that the facile one-pot synthesis of hetero[4]rotaxane 5 would inspire the successful construction of MIMs with fascinating structures and potential functions.
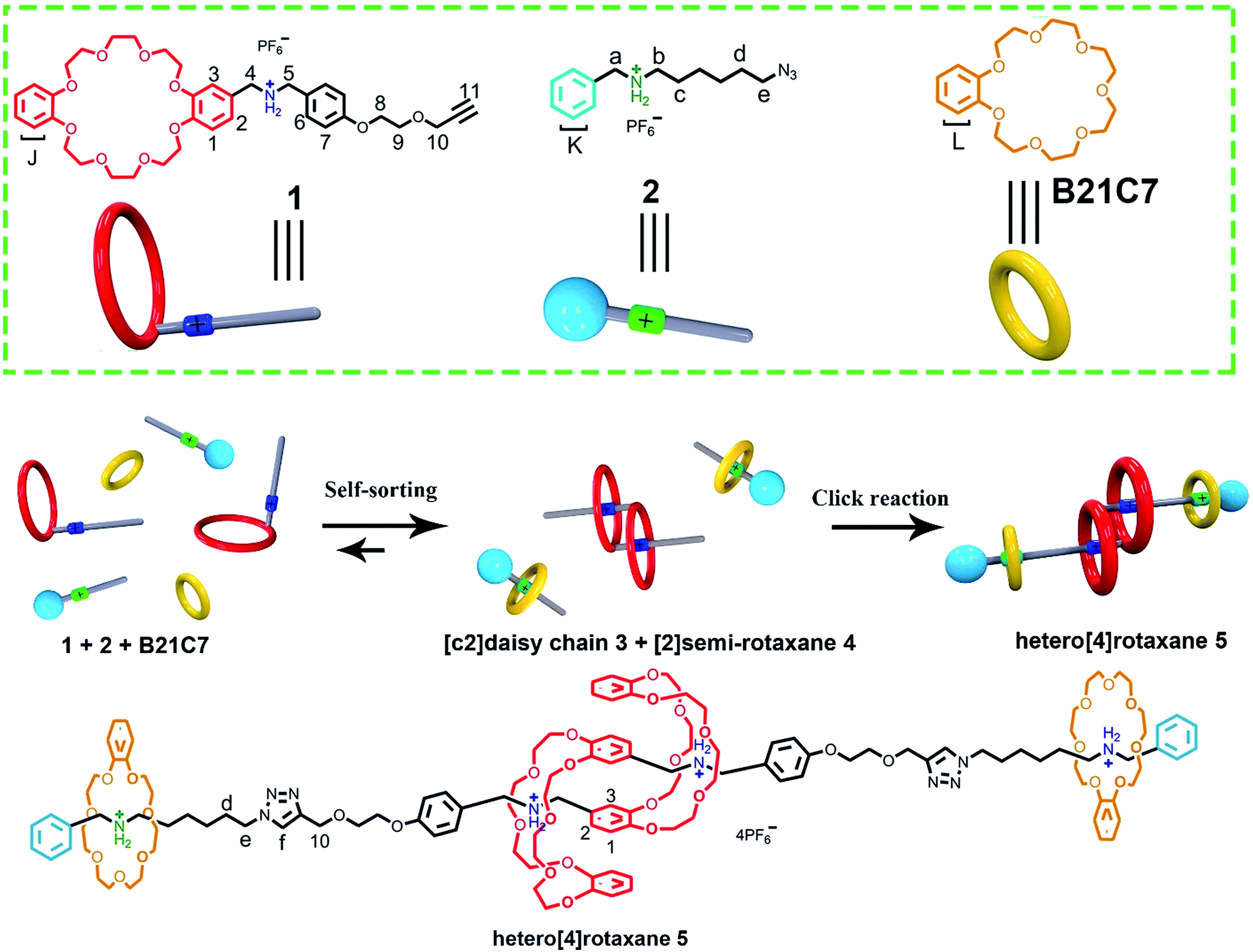 | ||
| Scheme 1 Schematic representation of the preparation of hetero[4]rotaxane 5 using a three-component modularized self-sorting system of 1, 2 and B21C7. Only one main stereoisomer of hetero[4]rotaxane 5, which is derived from the three possible [c2]daisy chain stereoisomers,5e is shown here. | ||
Results and discussion
The rational design of block building is a prerequisite to realizing the precise construction of a multi-component system. As shown in Scheme 1, initial substrates include compound 1 (the synthesis information from compounds 7 and 8 can be found in the ESI†), which contains a DB24C8 macrocycle modified with a secondary dibenzylammonium branched chain; a secondary benzylalkylammonium 2; and B21C7. The key part of the design involves the dibenzylammonium recognition site in compound 1, where the phenyl group encodes the secondary ammonium as a selective site that only can be included by the larger macrocyclic DB24C8, but not by B21C7.7b,10,12 Therefore, even in one system, B21C7 would not affect the formation of the [c2]daisy chain 3. Meanwhile, this phenyl group also makes the secondary ammonium in compound 1 a preferred recognition site for DB24C8, over that in compound 2.10 Considering the precise 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 proportion of DB24C8 and the secondary ammonium in compound 1, compound 2 would not affect the formation of [c2]daisy chain 3 from monomer molecule 1, which has been proved by the 1H NMR measurements (Fig. S1†). As a result, the self-assembly of the initial molecules was precisely encoded, where the self-sorting process included simultaneous threading and interpenetration, and then [2]semi-rotaxane 4 and [c2]daisy chain 3 were generated, respectively. Finally, in the presence of Cu(MeCN)4PF6, the subsequent CuAAC click reaction between [c2]daisy chain 3 and [2]semi-rotaxane 4 afforded the formation of hetero[4]rotaxane 5 in a one-pot strategy. In the obtained hetero[4]rotaxane 5, two B21C7 rings can be stopped by the outer phenyl groups, while the central [c2]daisy chain structures are cascade-stopped by the B21C7 rings.
:1 proportion of DB24C8 and the secondary ammonium in compound 1, compound 2 would not affect the formation of [c2]daisy chain 3 from monomer molecule 1, which has been proved by the 1H NMR measurements (Fig. S1†). As a result, the self-assembly of the initial molecules was precisely encoded, where the self-sorting process included simultaneous threading and interpenetration, and then [2]semi-rotaxane 4 and [c2]daisy chain 3 were generated, respectively. Finally, in the presence of Cu(MeCN)4PF6, the subsequent CuAAC click reaction between [c2]daisy chain 3 and [2]semi-rotaxane 4 afforded the formation of hetero[4]rotaxane 5 in a one-pot strategy. In the obtained hetero[4]rotaxane 5, two B21C7 rings can be stopped by the outer phenyl groups, while the central [c2]daisy chain structures are cascade-stopped by the B21C7 rings.
We performed 1H NMR experiments to confirm the self-sorting behaviours in this three-component system, as shown in Fig. 1. The 1H NMR spectra revealed that 1 existed as a monomer in polar solvents, such as [D6]DMSO (Fig. 1a), with normal and simple 1H NMR signals. While in less polar solvents, such as [D3]acetonitrile (Fig. 1b), [c2]daisy chain 3 became the predominant species. The 1H NMR signals of the NH2+ protons and crown ether moiety shifted and split, becoming much broader and more complicated. As shown in Fig. 1b, two sets of signals for the NH2+ protons were observed at δ = 7.0–7.4 ppm, attributed to each ammonium facing the two non-symmetrical ends of the axle. According to the previous studies about the analogous [c2]daisy chains,13,14 there are three dimeric interlocked stereoisomers arising from the unsymmetrical substitution of the DB24C8 ring, which were also observed in the 1H NMR spectrum of [c2]daisy chain 3 (Fig. 1b). As shown in Fig. 1, the aromatic protons of DB24C8 also become split, especially protons H1 and H3. H1 splits into two sets of double peaks (H1a, 6.4 ppm and H1b, 6.9 ppm), and H3 splits into two sets of single peaks (H3a, 6.8 ppm and H3b, 6.2 ppm) with the ratio of 5:1, indicating the major supramolecular stereoisomer of the “meso” type, shown in Scheme 1, which is consistent with the previous report.5e Meanwhile, the formation of [2]semi-rotaxane 4 from compound 2 and B21C7 has been detected using the 1H NMR spectra shown in Fig. 2. After mixing compound 2 and B21C7 in a 1:1 molar ratio, the signals of the NH2+ protons shifted upfield with a Δδ of −0.97 ppm, and the signals of Ha, Hb and He shifted downfield with a Δδ of 0.26, 0.24 and 0.07 ppm, respectively, suggesting the formation of [2]semi-rotaxane 4.12 Then, the three compounds 1, 2 and B21C7 were mixed in [D3]acetonitrile in a molar ratio of 1:1:1. 1H NMR spectra (Fig. 3) showed that the signals of H1a, H3b, H4 and H5 in [c2]daisy chain 3, as well as those of Ha, Hb and He in [2]semi-rotaxane 4, remain constant. These observations indicate that the two pseudorotaxanes still exist as the predominant species in this three-component self-sorting system.
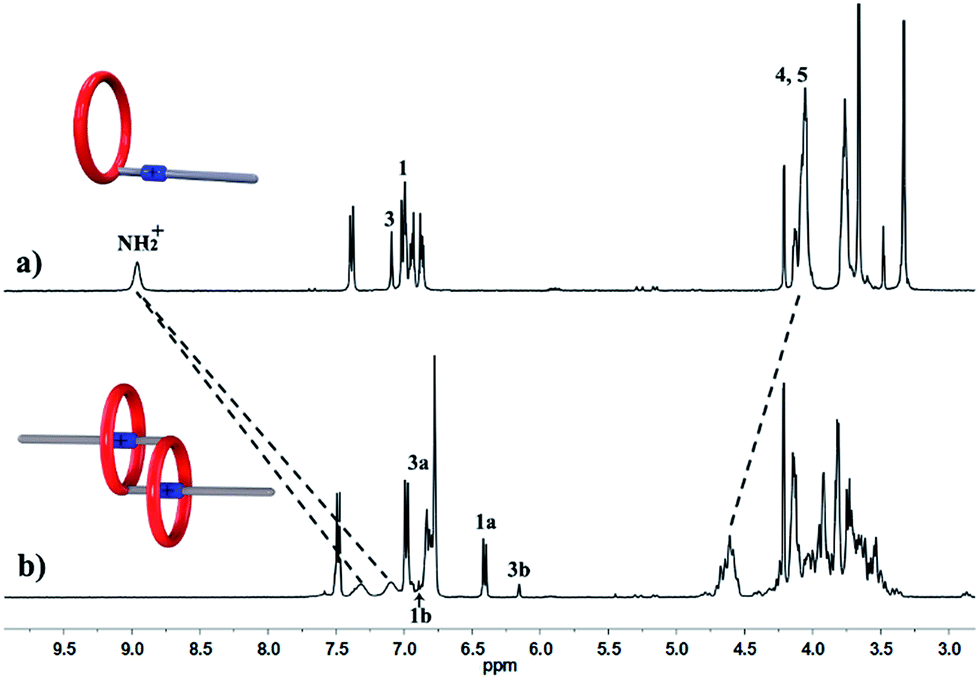 | ||
| Fig. 1 Partial 1H NMR spectra (400 MHz, 298 K) of compound 1 in (a) [D6]DMSO, and (b) [D3]acetonitrile. | ||
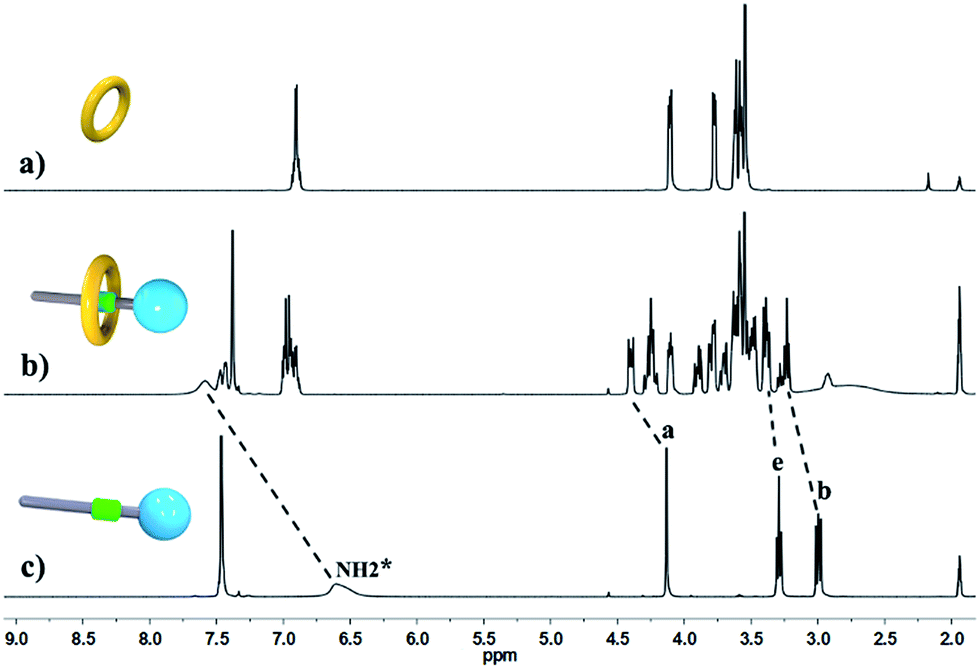 | ||
| Fig. 2 Partial 1H NMR spectra (400 MHz, 298 K, [D3]acetonitrile) of (a) B21C7, (b) a 1:1 mixture of 2 and B21C7, and (c) compound 2. | ||
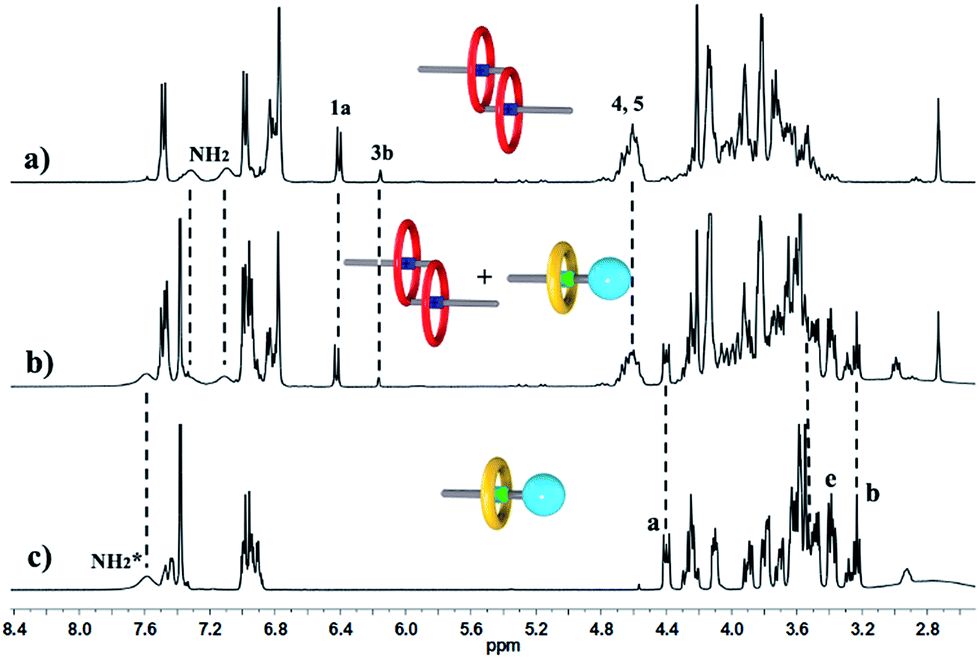 | ||
| Fig. 3 Partial 1H NMR spectra (400 MHz, 298 K, [D3]acetonitrile) of (a) compound 1, (b) a 1:1:1 mixture of 1, 2 and B21C7, and (c) a 1:1 mixture of 2 and B21C7. | ||
The synthetic route for hetero[4]rotaxane 5 is outlined in Scheme 1. At room temperature, the CuAAC reaction between [c2]daisy chain 3 and [2]semi-rotaxane 4 successfully afforded the formation of a white solid, hetero[4]rotaxane 5, with a yield exceeding 50% in the presence of Cu(MeCN)4PF6. As shown in Fig. 4a and b, the terminal alkynyl proton H11 of 3 disappeared after the CuAAC click reaction, indicating the successful formation of the triazole. The resonance for the triazole proton Hf appeared at δ = 7.72 ppm and the adjacent protons H10, Hb, Hd and He shifted downfield with a Δδ of 0.38, 0.22, 0.12 and 0.89 ppm, respectively. All of these changes demonstrated the importance of the CuAAC click reaction for the formation of hetero[4]rotaxane 5, as illustrated in Scheme 1.15 The high resolution mass spectrum (HRMS) of hetero[4]rotaxane 5 showed major signals at m/z 628.0955, 885.7819 and 1401.1543, which correspond to the product after the loss of 4, 3 and 2PF6− ions, i.e. [M − 4PF6]4+, [M − 3PF6]3+ and, [M − 2PF6]2+, respectively. The molecular ion peaks of [M − 4PF6]4+ and [M − 3PF6]3+, as well as the results of 1H NMR, give enough evidence to show the formation of hetero[4]rotaxane 5.15
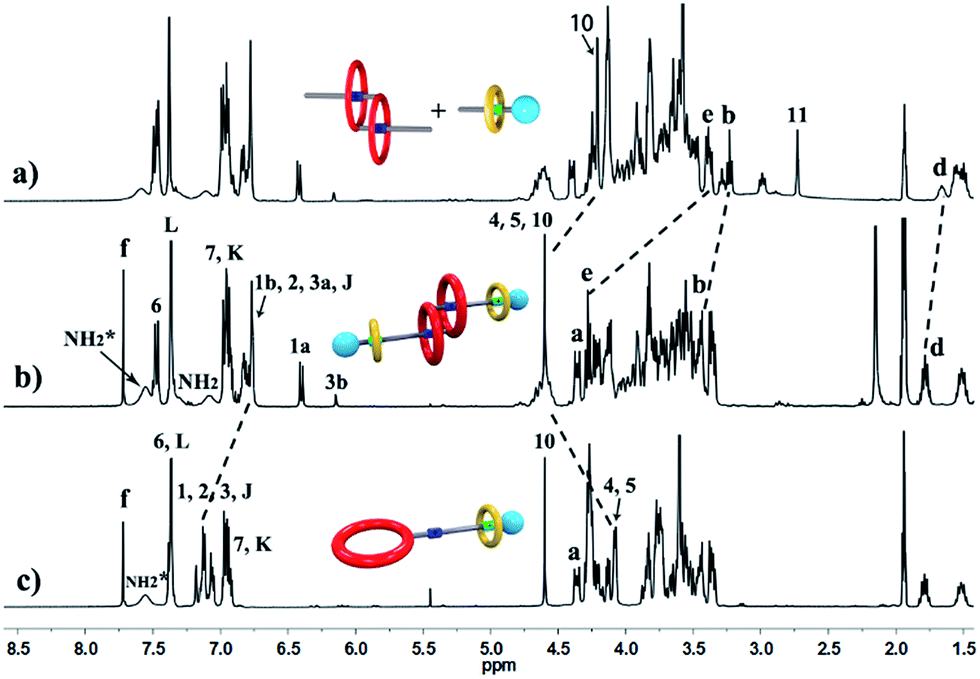 | ||
| Fig. 4 Partial 1H NMR spectra (400 MHz, 298 K, [D3]acetonitrile) of (a) a 1:1:1 mixture of 1, 2 and B21C7, (b) hetero[4]rotaxane 5, and (c) [2]rotaxane 6. | ||
For further understanding of the self-sorting process and confirming the chemical structure of hetero[4]rotaxane 5, the dibenzylammonium site in compound 1 was protected with a BOC group to yield compound 9 (Scheme 2), leading to a recognition failure between macrocyclic DB24C8 and the DBA site. As a result, there was only one type of secondary ammonium recognition site in the mixed systems of compound 9, compound 2 and B21C7, meaning that a non-self-sorting process would occur among these building blocks. Then, a CuAAC click reaction of compound 9 and [2]semi-rotaxane 4 successfully afforded [2]rotaxane 10 in the presence of Cu(MeCN)4PF6 in a 1:1 volume ratio of dichloromethane/acetonitrile at room temperature. After being treated with trifluoroacetic acid, the reference compound [2]rotaxane 6 with an unoccupied DB24C8 ring was obtained. The HRMS spectrum of [2]rotaxane 6 shows major signals at m/z 1400.6526, corresponding to the product after a loss of the PF6− ion, confirming the interlocked structure. Furthermore, we compared all of the resonances between hetero[4]rotaxane 5 and [2]rotaxane 6 by analysing their 1H NMR spectra (Fig. 4b and c). As shown in the 1H NMR spectra of 5 and 6 in [D3] acetonitrile (Fig. 4b and c), the protons H1, H2, H3 and HJ of [2]rotaxane 6 shifted downfield with a Δδ of about 0.2 ppm, and the stereoisomer signals of H1a and H3b disappeared, which was attributed to the fact that the DBA site was not surrounded by the DB24C8 macrocycle to give a simple chemical shift environment. It is notable that the DBA protons in [2]rotaxane 6 cannot be detected because there are no hydrogen bonding interactions between the crown ether and the DBA hydrogen atoms.16 Compared with the 1H NMR spectrum of hetero[4]rotaxane 5, H4 and H5 which are adjacent to the DBA site were observed to shift upfield with a Δδ of −0.53 ppm in the 1H NMR spectrum of [2]rotaxane 6, which confirmed that the DBA sites were not bound by the host macrocycle DB24C8. Therefore, the phenyl-stopped B21C7 played an important role in inhibiting the dethreading of the daisy chain structure in the middle, this is also called a cascade-stopped strategy.10,7b Importantly, this inhibiting behaviour also caused the stable structure of hetero[4]rotaxane 5 instead of a metastable pseudorotaxane.
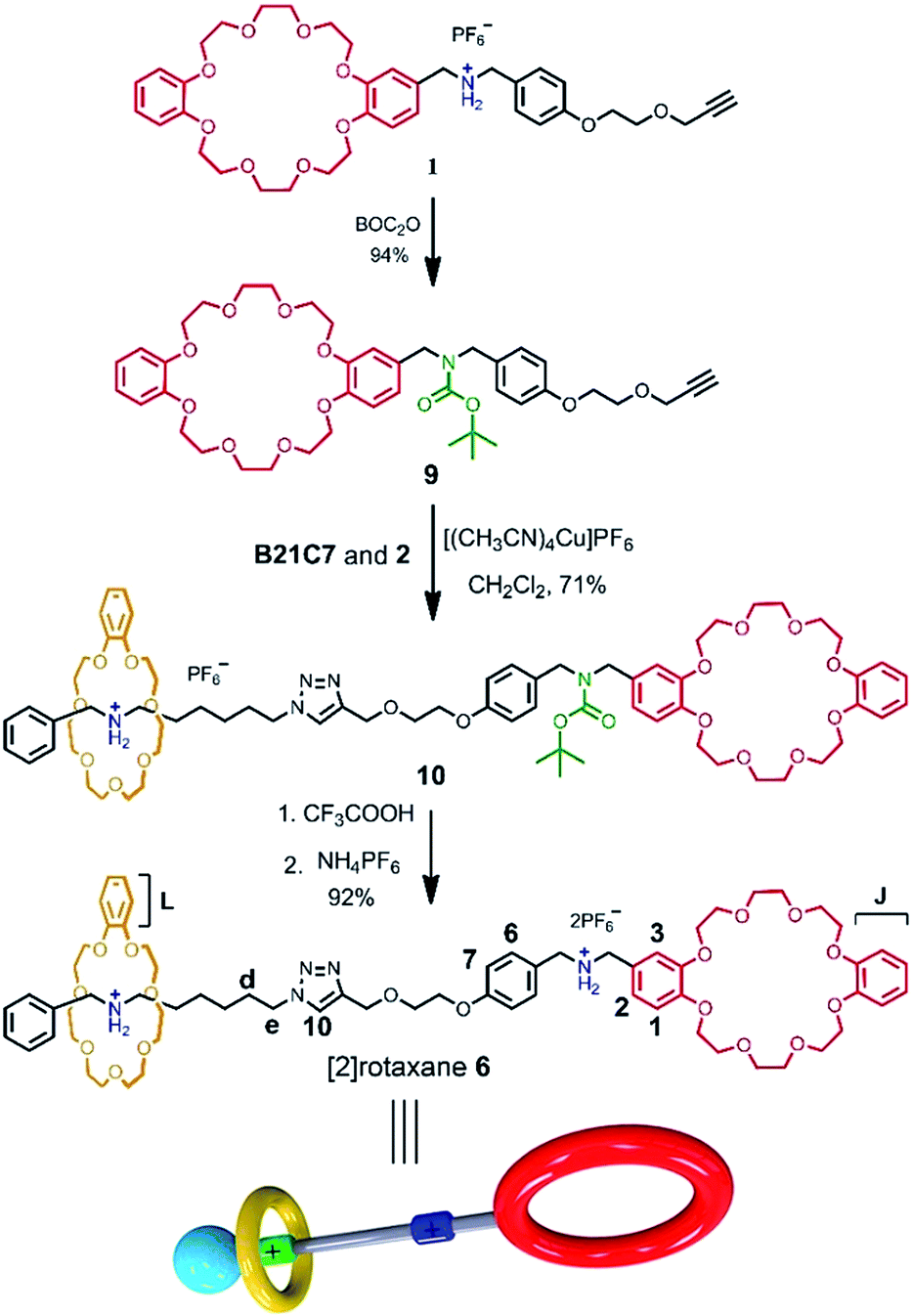 | ||
| Scheme 2 Synthetic route for [2]rotaxane 6. | ||
Meanwhile, 2D NOESY spectra of hetero[4]rotaxane 5 and [2]rotaxane 6 validated their chemical structures. The NOESY spectrum of 6 in [D3]acetonitrile (Fig. S1†) shows two cross-peaks (peak F and G) between the phenyl protons HL and ethylene protons of B21C7, thus clearly indicating that B21C7 is located on the outer ammonium site which is far away from the DB24C8 ring. In the meantime, no cross-peaks between DB24C8 and the inner DBA site were found. Nevertheless, in the NOESY spectrum of 5 (Fig. S2†), we found not only cross-peaks (peaks P, Q, R and S) between the phenyl protons HL and ethylene protons of B21C7, but also cross-peaks (peak N and O) between the phenyl protons H7 and ethylene protons of DB24C8, and cross-peaks (peak T and U) between the phenyl protons H6 and ethylene protons of DB24C8, thus clearly indicating the existence of a [c2]daisy chain structure in 5.
Conclusion
In conclusion, a novel hetero[4]rotaxane containing a [c2]daisy chain can be facilely synthesized in one pot by employing a multi-component self-sorting strategy followed by the well-known CuAAC stoppering reaction. The highly selective self-assembling process that occurred among the three kinds of building blocks provides the possibility for the formation of only two kinds of rotaxane precursors, by employing a steric hindrance-related “language”. This work serves as one of few successful examples of the construction of hetero[n]rotaxane with a [c2]daisy chain cored structure directed by a self-sorting strategy. Significantly, the integration of hetero[n]rotaxane and a [c2]daisy chain structure enlarges the family of mechanically interlocked molecules and this synthetic methodology can be utilized in the preparation of mechanically interlocked compounds with increasingly complicated structures and functions.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21421004, 20272073, 21190033), the Fundamental Research Funds for the Central Universities, Shanghai Education Development Foundation and Shanghai Municipal Education Commission.Notes and references
- (a) S. J. Loeb, Chem. Soc. Rev., 2007, 36, 226–235 RSC; (b) C. F. Lee, D. A. Leigh, R. G. Pritchard, D. Schultz, S. J. Teat, G. A. Timco and R. E. Winpenny, Nature, 2009, 458, 314–318 CrossRef CAS PubMed; (c) J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 11102–11104 CrossRef CAS PubMed; (d) K. Zhu, C. A. O'Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514–519 CrossRef CAS PubMed; (e) G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70–75 CrossRef CAS PubMed; (f) C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed; (g) G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef PubMed; (h) M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed; (i) T. Heinrich, C. H. H. Traulsen, M. Holzweber, S. Richter, V. Kunz, S. K. Kastner, S. O. Krabbenborg, J. Huskens, W. E. S. Unger and C. A. Schalley, J. Am. Chem. Soc., 2015, 137, 4382–4390 CrossRef CAS PubMed; (j) S. H. Li, H. Y. Zhang, X. Xu and Y. Liu, Nat. Commun., 2015, 6, 7590 CrossRef PubMed; (k) E. R. Kay and D. A. Leigh, Angew. Chem., Int. Ed., 2015, 54, 10080–10088 CrossRef CAS PubMed; (l) J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS PubMed; (m) S. Erbas-Cakmak, D. A. Leigh, C. T. Mcternan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- (a) B. Lewandowski, G. D. Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed; (b) J. N. Zhang, H. Li, W. Zhou, S. L. Yu, D. H. Qu and H. Tian, Chem.–Eur. J., 2013, 19, 17192–17200 CrossRef CAS PubMed; (c) H. Li, X. Li, Z. Q. Cao, D. H. Qu, H. Ågren and H. Tian, ACS Appl. Mater. Interfaces, 2014, 6, 18921–18929 CrossRef CAS PubMed; (d) M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935–1949 CrossRef CAS PubMed; (e) Z. Q. Cao, Q. Miao, Q. Zhang, H. Li, D. H. Qu and H. Tian, Chem. Commun., 2015, 51, 4973–4976 RSC.
- (a) W. Wang, L. J. Chen, X. Q. Wang, B. Sun, X. Li, Y. Zhang, J. Shi, Y. Yu, L. Zhang, M. Liu and H. B. Yang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5597–5601 CrossRef CAS PubMed; (b) S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC; (c) R. J. Bordoli and S. M. Goldup, J. Am. Chem. Soc., 2014, 136, 4817–4820 CrossRef CAS PubMed; (d) D. A. Leigh, R. G. Pritchard and A. J. Stephens, Nat. Chem., 2014, 6, 978–982 CrossRef CAS PubMed; (e) Z. Meng, Y. Han, L. N. Wang, J. F. Xiang, S. G. He and C. F. Chen, J. Am. Chem. Soc., 2015, 137, 9739–9745 CrossRef CAS PubMed.
- (a) J. D. Badjić, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef PubMed; (b) H. Li, X. Li, H. Ågren and D. H. Qu, Org. Lett., 2014, 16, 4940–4943 CrossRef CAS PubMed; (c) P. Waelès, C. Clavel, K. Fournel-Marotte and F. Coutrot, Chem. Sci., 2015, 6, 4828–4836 RSC; (d) M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897–1901 RSC.
- (a) T. Fujimoto, Y. Sakata and T. Kaneda, Chem. Commun., 2000, 2143–2144 RSC; (b) J. Wu, K. C. F. Leung, D. Benitez, J. Y. Han, S. J. Cantrill, L. Fang and J. F. Stoddart, Angew. Chem., Int. Ed., 2008, 47, 7470–7474 CrossRef CAS PubMed; (c) Y. Liu, Z.-X. Yang and Y. Chen, J. Org. Chem., 2008, 73, 5298–5304 CrossRef CAS PubMed; (d) R. E. Dawson, S. F. Lincoln and C. J. Easton, Chem. Commun., 2008, 34, 3980–3982 RSC; (e) F. Coutrot, C. Romuald and E. Busseron, Org. Lett., 2008, 10, 3741–3744 CrossRef CAS PubMed; (f) P. G. Clark, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 13631–13633 CrossRef CAS PubMed; (g) Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397–1401 CrossRef CAS PubMed; (h) G. Du, E. Moulin, N. Jouault, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2012, 51, 12504–12508 CrossRef CAS PubMed; (i) L. Cao and L. Isaacs, Org. Lett., 2012, 14, 3072–3075 CrossRef CAS PubMed; (j) Z. Zhang, C. Han, G. Yu and F. Huang, Chem. Sci., 2012, 3, 3026–3031 RSC; (k) J. Rotzler and M. Mayor, Chem. Soc. Rev., 2013, 42, 44–62 RSC; (l) C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef CAS PubMed; (m) Q. Zhang, C. H. Zhang, J. H. Yang, P. Y. Xin, X. P. Xuan, J. G. Wang, N. N. Ma, H. M. Guo and G. R. Qu, Chem. Commun., 2015, 51, 15253–15256 RSC; (n) A. Coujon, G. Du, E. Moulin, G. Fuks, M. Maaloum, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2016, 55, 703–707 CrossRef PubMed; (o) C. J. Bruns, M. Frasconi, J. Iehl, K. J. Hartlieb, S. T. Schneebeli, C. Cheng, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 4714–4723 CrossRef CAS PubMed; (p) L. Gao, Z. Zhang, B. Zheng and F. Huang, Polym. Chem., 2014, 5, 5734–5739 RSC.
- (a) J. F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Chem. Soc. Rev., 2013, 42, 1700–1712 RSC; (b) J. F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed; (c) J. F. Ayme, J. E. Beves, C. J. Campbell, G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, J. Am. Chem. Soc., 2015, 137, 9812–9815 CrossRef CAS PubMed.
- (a) E. J. F. Klotz, T. D. W. Claridge and H. L. Anderson, J. Am. Chem. Soc., 2006, 128, 15374–15375 CrossRef CAS PubMed; (b) Z. J. Zhang, H. Y. Zhang, H. Wang and Y. Liu, Angew. Chem., Int. Ed., 2011, 50, 10834–10838 CrossRef CAS PubMed; (c) C. Ke, R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 381–387 CrossRef CAS PubMed; (d) X. Hou, C. Ke, C. Cheng, N. Song, A. K. Blackburn, A. A. Sarjeant, Y. Y. Botros, Y. W. Yang and J. F. Stoddart, Chem. Commun., 2014, 50, 6196–6199 RSC; (e) P. N. Chen, C. C. Lai and S. H. Chiu, Org. Lett., 2011, 13, 4660–4663 CrossRef CAS PubMed; (f) G. Celtek, M. Artar, O. A. Scherman and D. Tuncel, Chem.–Eur. J., 2009, 15, 10360–10363 CrossRef CAS PubMed; (g) C. Ke, R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 381–387 CrossRef CAS PubMed; (h) Q. F. Luo, L. Zhu, S. J. Rao, H. Li, Q. Miao and D. H. Qu, J. Org. Chem., 2015, 80, 4704–4709 CrossRef CAS PubMed.
- (a) M. C. Fyfe and J. F. Stoddart, Acc. Chem. Res., 1997, 30, 393–401 CrossRef CAS; (b) S. J. Rowan, S. J. Cantrill and J. F. Stoddart, Org. Lett., 1999, 1, 129–132 CrossRef CAS; (c) S. J. Rowan and J. F. Stoddart, J. Am. Chem. Soc., 2000, 122, 164–165 CrossRef CAS; (d) K. D. Hänni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC.
- (a) A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed; (b) F. Wang, C. Han, C. He, Q. Zhou, J. Zhang, C. Wang, N. Li and F. Huang, J. Am. Chem. Soc., 2008, 130, 11254–11255 CrossRef CAS PubMed; (c) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed; (d) W. Wang, Y. Zhang, B. Sun, L. Chen, X. D. Xu, M. Wang, X. Li, Y. Yu, W. Jiang and H. B. Yang, Chem. Sci., 2014, 5, 4554–4560 RSC; (e) D. Račkauskaitė, R. Gegevičius, Y. Matsuo, K. Wärnmark and E. Orentas, Angew. Chem., Int. Ed., 2016, 55, 208–212 CrossRef PubMed.
- W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852–13853 CrossRef CAS PubMed.
- (a) W. Jiang and C. A. Schalley, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10425–10429 CrossRef CAS PubMed; (b) W. Jiang, A. Schäfer, P. C. Mohr and C. A. Schalley, J. Am. Chem. Soc., 2010, 132, 2309–2320 CrossRef CAS PubMed; (c) W. Jiang, D. Sattler, K. Rissanen and C. A. Schalley, Org. Lett., 2011, 13, 4502–4505 CrossRef CAS PubMed; (d) W. Jiang, K. Nowosinski, N. L. Löw, E. V. Dzyuba, F. Klautzsch, A. Schäfer, J. Huuskonen, K. Rissanen and C. A. Schalley, J. Am. Chem. Soc., 2012, 134, 1860–1868 CrossRef CAS PubMed; (e) B. Zheng, F. Klautzsch, M. Xue, F. H. Huang and C. A. Schalley, Org. Chem. Front., 2014, 1, 532–540 RSC; (f) Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- C. J. Zhang, S. J. Li, J. Q. Zhang, K. L. Zhu, N. Li and F. Huang, Org. Lett., 2007, 9, 5553–5556 CrossRef CAS PubMed.
- A. Wolf, E. Moulin, J. J. Cid, A. Goujon, G. Du, E. Busseron, G. Fuks and N. Giuseppone, Chem. Commun., 2015, 51, 4212–4215 RSC.
- C. J. Bruns, M. Frasconi, J. Iehl, K. J. Hartlieb, S. T. Schneebeli, C. Cheng, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 4714–4723 CrossRef CAS PubMed.
- B. Zheng, M. Zhang, S. Dong, J. Liu and F. Huang, Org. Lett., 2012, 14, 306–309 CrossRef CAS PubMed.
- H. Li, X. Li, Y. Wu, H. Ågren and D. H. Qu, J. Org. Chem., 2014, 79, 6996–7004 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental procedures and characterization data for all compounds. See DOI: 10.1039/c5sc04844c |
| ‡ X. F. and Q. Z. contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |