
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuperior upconversion fluorescence dopants for highly efficient deep-blue electroluminescent devices†
Yi-Hsiang
Chen
a,
Chih-Chun
Lin
a,
Min-Jie
Huang
*a,
Kevin
Hung
a,
Yi-Ching
Wu
a,
Wei-Chieh
Lin
b,
Ren-Wu
Chen-Cheng
b,
Hao-Wu
Lin
b and
Chien-Hong
Cheng
*a
aDepartment of Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: chcheng@mx.nthu.edu.tw; mjhuang@mx.nthu.edu.tw
bDepartment of Materials Science and Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan
First published on 18th April 2016
Abstract
In this study, we revealed a new approach for the development of new triplet–triplet annihilation (TTA) materials with highly efficient deep-blue fluorescence via the incorporation of a styrylpyrene core and an electron-donating group. The resulting deep-blue emitters (PCzSP, DFASP, and DPASP) exhibit intramolecular charge transfer emissions with remarkably high emission quantum yields. The electroluminescent devices based on these three fluorophores as dopants using CBP as a host exhibit very high device efficiencies; in particular, the DPASP-doped device reveals an extremely high EQE of 12%, reaching the limit of a TTA-based device. The EL characteristics of DPASP-doped CBP-based devices at various doping concentrations (0–5%) suggest that the dopant DPASP is responsible for the TTA-type delayed fluorescence in the device; no delayed fluorescence was observed for the device using CBP as the host emitter. Moreover, when using DMPPP with ambipolar characteristics as the host, the deep-blue DPASP-doped device also gives outstanding performance with an EQE of nearly 11% with an extremely small efficiency roll-off, which was ascribed to the excellent charge balance in the emitting layer of the EL device. The TTA process of the SP-based dopants accounts significantly for the superior efficiencies of the EL devices.
Introduction
Since the first organic electroluminescence (EL) device was developed by Tang and VanSlyke,1 organic light emitting diodes (OLEDs) have garnered considerable attention as a result of their unique applications in displays and lighting.2 Extensive efforts have been made towards the search for RGB luminescent materials with excellent chemical and electrochemical stabilities, colour purities, and quantum efficiencies for highly efficient full-colour displays. Among the known emitting materials, phosphorescent emitters based on transition metal complexes have EL performances four times higher than those of conventional fluorescent materials owing to the harvesting of both singlet and triplet excitons.3 Thus, phosphors are the most promising candidates for high-performance OLEDs. While highly efficient red and green phosphors meet the prerequisites for the commercialization of OLED devices, deep-blue phosphorescent materials suffer from instability, degradation, and serious roll-off in devices, leading to short operational lifetimes.4 As such, the application of deep-blue phosphorescent emitters in commercial OLED products is limited. The development of stable and efficient deep-blue fluorescent materials instead of metal-based blue triplet emitters is considered to be an effective strategy for realizing high-quality blue EL emission.5 Recently, a new generation of fluorescent materials capable of converting non-emissive triplet state excitons into emissive singlet excitons was developed in an attempt to increase the efficiency of fluorescent OLEDs.6 Three mechanisms have been proposed to explain how the triplet excitons are harvested in fluorescent OLEDs, including thermally activated delayed fluorescence (TADF),7 hybridized local and charge-transfer (HLCT),8 and triplet–triplet annihilation (TTA).9 Among the three emission mechanisms, TTA-based deep-blue emitting OLEDs are potential candidates for practical use because they can offer long operational lifetime and low efficiency roll-off at high device luminance. Thus, TTA is considered to be a promising triplet upconversion technique for the realization of efficient and stable deep-blue devices by generating a singlet exciton from the fusion of two triplet excitons, thereby providing a theoretical maximum internal quantum efficiency of 62.5%.In previous studies, most TTA-based materials were used as host or non-doped emitting materials in blue fluorescent OLEDs.10 Unfortunately, the EL performance of a non-doped device is limited by the low quantum yield of the emitting layer because of the self-quenching of the high-concentration emitter. For a TTA material-based host EL device, the device efficiency should be greatly affected by the efficiency of energy transfer including singlet and triplet energy transfer from host to guest. If the triplet energy in the TTA host is efficiently transferred to a non-TTA dopant, the device should not have efficient TTA process and the external quantum efficiency could be distantly below the theoretical value of 12.5%. Recently, a paramount breakthrough was achieved by using a D–A type anthracene-based emitter with TTA characteristics. Kido's group reported an impressive external quantum efficiency (EQE) of 12% with a deep-blue emission in a CBP-based device doped with a D–A type anthracene-based emitter.11 Furthermore, our group also realized a deep-blue TTA-based OLED with an EQE of ca. 10% using a triphenylene-containing molecule as a dopant.12 The use of TTA-based materials as dopant emitters offers another approach to develop highly efficient deep-blue OLEDs. To achieve an efficient TTA-type upconversion emission, the deep-blue emitter used should meet the following criteria: (1) a high fluorescence quantum yield; (2) an appropriate triplet state energy, which is higher than half of its single excited state energy to enable the TTA process; (3) a sufficiently long triplet state lifetime to guarantee occurrence of the TTA mechanism. To date, compounds containing polyaromatic hydrocarbons (PAH) are known to upconvert triplet excitons to singlet excitons through the TTA process.10a–d,13 Nevertheless, in the search for highly efficient blue fluorescent devices, TTA-based dopant emitters that can be used to fabricate deep-blue devices with EQE >10% and low efficiency roll-off at high brightness are still rare.
In this work, we present a novel design strategy to construct three TTA-based deep-blue materials, PCzSP, DFASP, and DPASP (Scheme 1), for highly efficient fluorescent devices, and demonstrate for the first time the significant influence of TTA-type emitters on device EQEs. The use of a styrylpyrene core is expected to result in pronounced quantum yields and TTA behaviour. Moreover, the twisted structure between the pyrene moiety and styryl group is beneficial for greatly reducing the excimer generation of pyrene and thus increasing upconversion quantum efficiency.13b,14 The PhCz and TPA units are introduced to tune the emission toward the deep-blue region. By incorporating a styrylpyrene core with different electron-donating amino moieties, the developed emitters exhibit deep-blue emissions with nearly 100% photoluminescence quantum yields. Of these three blue-emitting materials, the device with DPASP doped in CBP as the emitting layer (EML) shows the best EL performance with a maximum EQE of 12%, close to the theoretical absolute maximum of 12.5% via the TTA process. By using DMPPP as the host, the DPASP-doped device achieves the highest EQE of nearly 11% and shows a very low efficiency roll-off due to the excellent balanced carrier transport in the device. In both devices, the TTA process from SP-based emitters plays the most critical role in the high EQE, as evidenced by the results of transient EL measurements. The device efficiencies are among the best reported for TTA-based deep-blue fluorescent OLEDs.
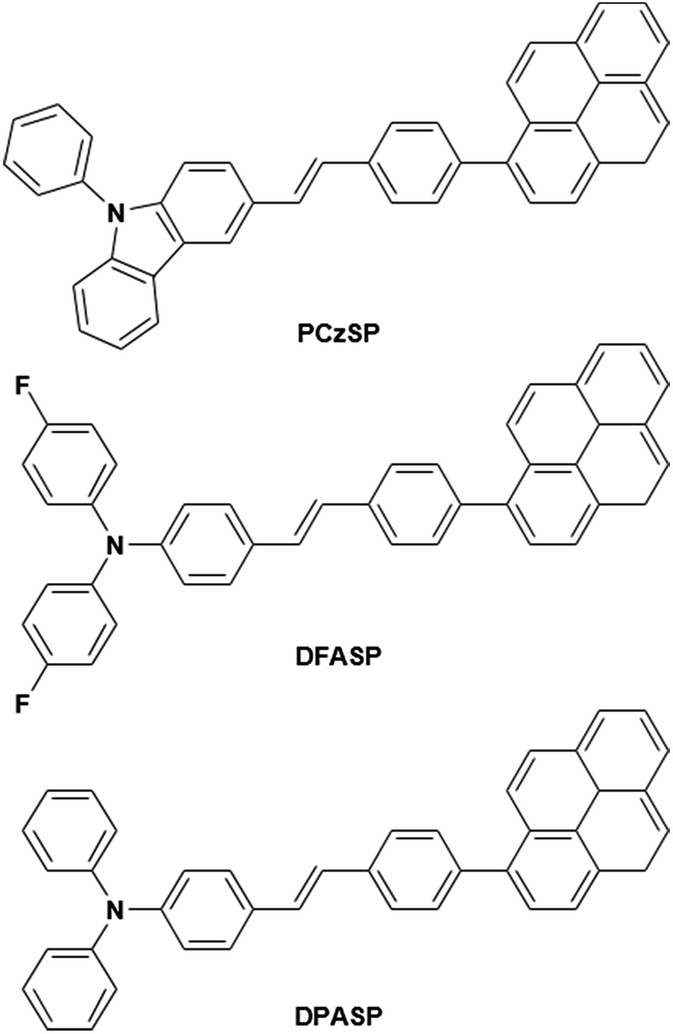 | ||
| Scheme 1 Molecular structures of styrylpyrene-based materials PCzSP, DFASP and DPASP. | ||
Results and discussion
Three styrylpyrene (SP) derivatives, PCzSP, DFASP and DPASP, were easily prepared by Horner–Wadsworth–Emmons (HWE) and Suzuki coupling reactions. The reaction sequences are depicted in Scheme 2. 4-Bromobenzylphosphonate was first reacted with aldehyde derivatives in the presence of potassium t-butoxide to give the corresponding stilbene intermediate. Pd(0)-catalyzed cross-coupling reaction of the stilbene intermediates with 1-pyreneboronic acids gave the desired compounds in excellent isolated yields. The details for the preparation of these compounds are given in the ESI.† The chemical structures of PCzSP, DFASP and DPASP were confirmed by 1H NMR, 13C NMR, high-resolution mass spectrometry and elemental analysis. Their thermal stabilities were evaluated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (see Fig. S1 and S2†); the data are presented in Table 1. All SP-based materials exhibit high thermal decomposition temperatures of 410–451 °C (Td, corresponding to 5% weight loss). The replacement of the diphenylamino unit with a 9H-carbazole moiety leads to an increase in Td due to the planarity and rigidity of the carbazole group. The DSC traces of these materials show that the melting points (Tm) are in the range of 204–270 °C. During repeated heating scans, PCzSP and DPASP revealed distinct glass transition temperatures (Tg) of 99 °C and 96 °C, respectively. The good thermal properties of SP-based materials are conducive to the formation of homogeneous and amorphous films with high morphological stabilities, and thus enhance OLED operational performance.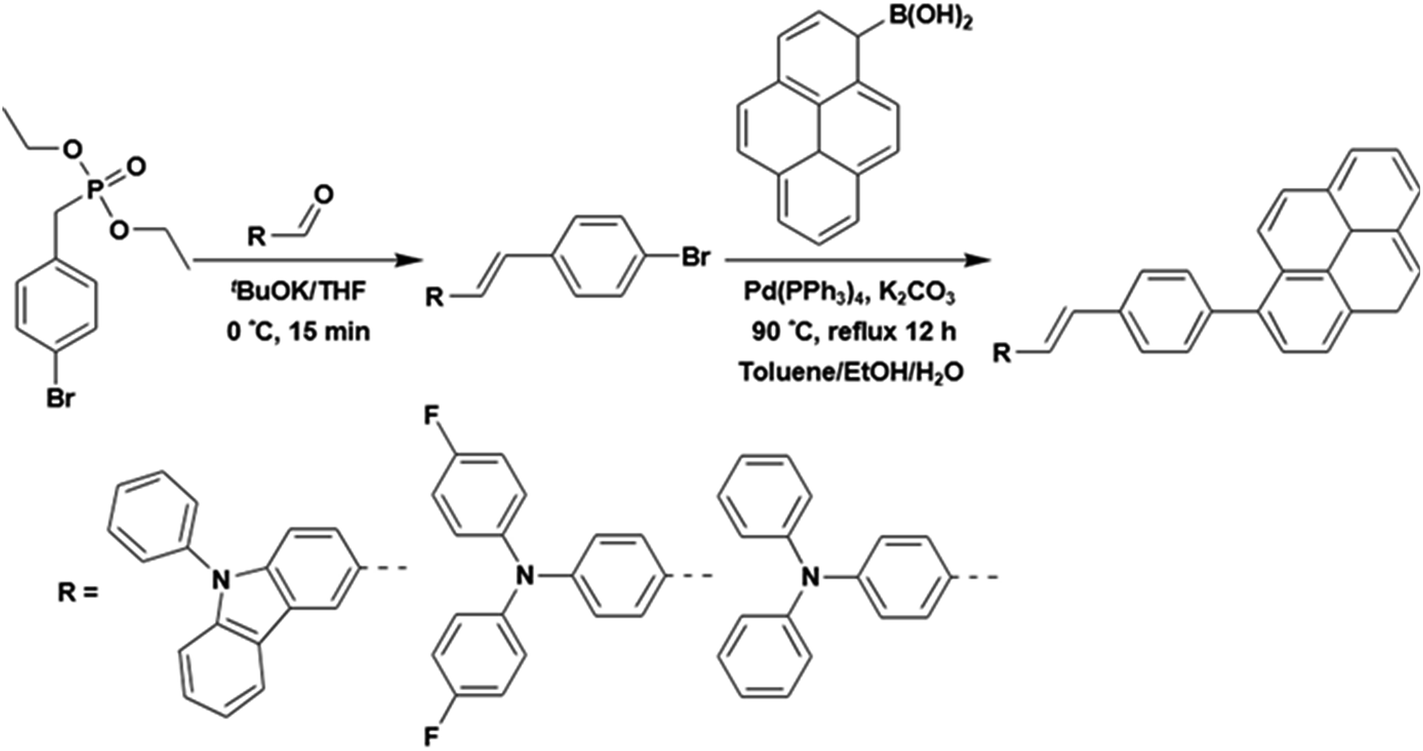 | ||
| Scheme 2 Synthetic routes for PCzSP, DFASP and DPASP. | ||
| Compound | Abssola [nm] | PLsola [nm] | PLfilmb [nm] | HOMO/LUMOc [eV] | E g [eV] | T g/Tm/Tde [°C] | Φ PL |
|---|---|---|---|---|---|---|---|
| a Absorption and fluorescence spectra were measured in toluene solution at 10−5 M. b Measured in the thin film (25 nm). c HOMO values were determined using the oxidative potentials of the materials with Cp2Fe/Cp2Fe+ as the reference. LUMO levels were estimated according to the equation of LUMO = HOMO − Eg. d The energy gap (Eg) was estimated from the absorption threshold. e Thermal behaviour were obtained via TGA and DSC measurements. f The PL quantum yields of the dopants in the DMPPP thin film were carried out by an integral sphere with an excitation wavelength of 320 nm. g N.D.: not detected. | |||||||
| PCzSP | 361 | 434 | 458 | 5.48/2.54 | 2.94 | 99/204/451 | 0.64 |
| DFASP | 381 | 444 | 480 | 5.37/2.49 | 2.88 | N.D.g/246/410 | 0.94 |
| DPASP | 385 | 451 | 472 | 5.30/2.45 | 2.85 | 96/270/439 | 0.99 |
Fig. 1 displays the absorption and fluorescence spectra of SP-based compounds in toluene; the corresponding photophysical properties are summarized in Table 1. All materials show strong absorption bands in the range of 361–385 nm, assigned to π–π* transitions of the molecular backbone. In dilute toluene solutions, their emissions appear in the deep-blue regions. The photoluminescence (PL) peaks of DFASP (444 nm) and DPASP (451 nm) are more red-shifted than that of PCzSP (434 nm), attributed to the stronger electron-donating ability of triphenylamine relative to carbazole. Moreover, the emission spectra of the materials broaden and shift to a longer wavelength in polar solvents, and the PL intensities appear to decrease (Fig. S3†). The obvious positive solvatochromic effect suggests an ICT-based emission for these materials. This phenomenon was also previously demonstrated in PAH-containing materials.15
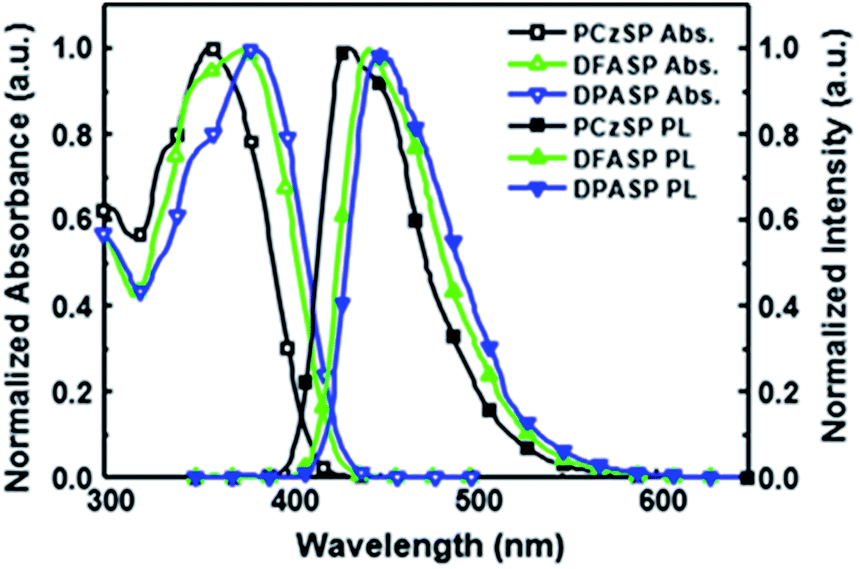 | ||
| Fig. 1 UV-Vis absorption and fluorescence emission spectra of PCzSP, DFASP and DPASP in toluene. | ||
To understand the ICT character in these molecules, the spectroscopic properties of PCzSP, DFASP and DPASP (Table S1†) were quantitatively analyzed utilizing the Lippert–Mataga model,16 which describes the Stokes shift as a function of the solvent polarity and can be expressed as follows:
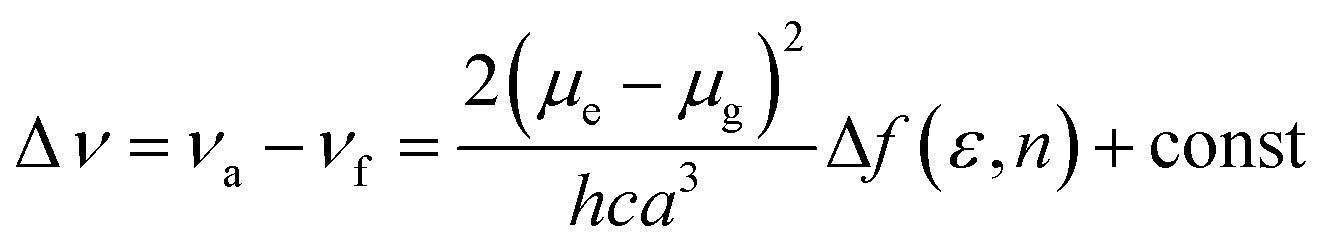
where Δν = νa − νf represents the Stokes shift (Δν) between the absorption (νa) and emission (νf) energies, Δf(ε, n) denotes solvent polarizability, ε and n are the dielectric constant and refractive index of the solvent, respectively, μe and μg stand for the dipole moment of the electronic excited and ground states, respectively, and a represents Onsager radius, which is estimated to be ca. 5.9 Å for the three blue emitters. For these three SP-based materials, the Lippert–Mataga plots displayed in Fig. S3† show a good linear correlation between Stokes shifts and solvent polarizability, suggesting an ICT mechanism. The slopes of the linear fitting obtained for PCzSP, DFASP, and DPASP are 9356, 13
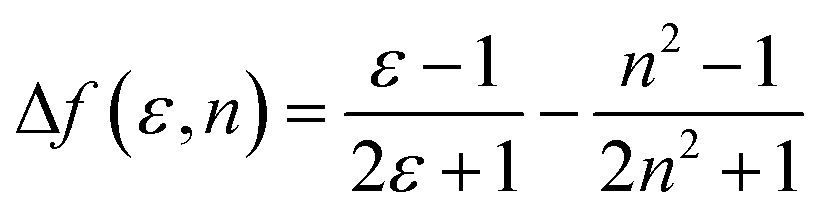
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 061, and 13579 cm−1, corresponding to changes in the dipole moment between the ground and excited state (Δμ = μe − μg) of 20, 23, and 24 D, respectively. Large dipole moment changes are observed for all molecules, confirming that significant CT occurs in the excited states. In addition, the Δμ values of these materials increase in the following order: PCzSP < DFASP < DPASP, which correlates well with electron-donating strength, manifesting that a strong electron-donating group in the molecule results in an excited state with significant ICT character.
061, and 13579 cm−1, corresponding to changes in the dipole moment between the ground and excited state (Δμ = μe − μg) of 20, 23, and 24 D, respectively. Large dipole moment changes are observed for all molecules, confirming that significant CT occurs in the excited states. In addition, the Δμ values of these materials increase in the following order: PCzSP < DFASP < DPASP, which correlates well with electron-donating strength, manifesting that a strong electron-donating group in the molecule results in an excited state with significant ICT character.
The ICT emissions of developed materials can further be demonstrated via quantum mechanical calculations. Density functional theory (DFT) simulations of PCzSP, DFASP, and DPASP were carried out using the B3LYP method with the 6-31G* basis set to model the spatial distributions of the frontier orbitals (Fig. S4†). For the three materials, the LUMO wave functions were primarily located on the pyrene core and styryl π bridge, while the HOMO orbitals are primarily delocalized along the electron-rich unit (i.e., the triphenylamine or carbazole group) and styryl π bridge, except that the HOMO level of PCzSP has a lesser extent on the pyrene core. DFT results suggest that the photoexcitation of these blue emitters redistributes electron density from the triphenylamine or carbazole units to pyrene and thus gives rise to a pronounced ICT to the S1 state, consistent with the solvatochromic PL spectra. In the thin film, the emissions of all synthesized materials shift bathochromically by 21–36 nm with respect to those in solution (Fig. S5†). This red-shift could be ascribed to intermolecular π–π interactions of neighbouring pyrene groups and the extension of π-delocalization arising from the more coplanar configuration of the styrylpyrenes in the neat film.17
The HOMO levels of all materials were estimated according to the equation HOMO = Eox + 4.8 eV, where Eox represents the oxidation potential with regard to the ferrocene/ferrocenium (Fc/Fc+) couple acquired from cyclic voltammetry (Fig. S6†). The corresponding LUMO energy levels were calculated from the HOMO values and the optical band gap determined by the absorption onset. The HOMO/LUMO values of these SP-based materials are reported in Table 1. A significant increase in the HOMO levels from the carbazole- to triphenylamine-containing units was observed, indicating the destabilization of HOMOs when increasing the electron donating ability. To quantitatively evaluate the suitability of styrylpyrene derivatives as blue dopants, their absolute fluorescent quantum yields (ΦPL) were measured by using an integrating sphere system. In the DMPPP host with a singlet energy gap of 3.2 eV, DFASP (ΦPL = 0.94) and DPASP (ΦPL = 0.99) exhibit higher ΦPL as compared to PCzSP (ΦPL = 0.64), due to the better overlap of the absorption with the emission of DMPPP (Fig. S7†), resulting in efficient Förster energy transfer from DMPPP to the emitters. Moreover, the near 100% ΦPL of DFASP and DPASP doped in the DMPPP thin films is attributed to the hindered intramolecular rotation of the dopant imposed by the DMPPP thin film. Suppression of molecular rotation can efficiently reduce the non-radiative pathways in the excited states of emitters and thus increase the fluorescence efficiency.18 The ΦPL of DFASP and DPASP are greater than those of well-known blue emitters DPAVB and BCzVBi,19 suggesting that they are promising blue emissive materials for optoelectronic devices, which prompts us to further explore the characteristics of OLEDs based on these new blue-emitting materials.
To assess the potential of the materials in electroluminescent devices, blue devices A–C were initially constructed by doping PCzSP, DFASP, and DPASP as the emitting layers in a CBP (4,4′-bis(N-carbazolyl)-1,1′-biphenyl) host, respectively. A conventional multilayered architecture composed of ITO/NPB (30 nm)/TCTA (20 nm)/CBP: 5% dopant (30 nm)/TmPyPB (30 nm)/LiF (1 nm)/Al (100 nm) was utilized for the fabrication of devices A–C, where NPB (4,4′-bis[N-(1-naphthyl)-N-phenylamino]biphenyl) and TmPyPB (1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene) are hole and electron transporting materials, respectively. In this device structure, TCTA (tris(4-carbazoyl-9-ylphenyl)amine) was incorporated into the NPB/CBP interface as an exciton-blocking layer and buffer layer to confine the excitons within the EML and improve the hole injection, as depicted in Fig. S11.† The representative performance parameters of these devices and the corresponding device behaviour are presented in Fig. 2 and Table 2. The lack of additional peaks from CBP and neighbouring materials in the EL spectra of fabricated devices suggests a full energy transfer from CBP to the blue emitters and effective exciton blocking by TCTA and TmPyPB. Devices A–C emit deep-blue light with maximum emission peaks at 444, 457, and 462 nm (Fig. S12†) and corresponding CIE coordinates of (0.15, 0.09), (0.14, 0.14), and (0.14, 0.17), respectively. It is worth noting that the emission of device A based on PCzSP is similar to the NTSC (National Television Standards Committee) blue standard (0.14, 0.08). All devices are turned on at a low voltage of ca. 3.3 V and exhibit excellent EL efficiencies. Of the three devices, DFASP- and DPASP-doped devices (B and C) display an amazing performance with maximum external quantum efficiencies (EQEs) of 11.5 and 12.0%, current efficiencies (CEs) of 14.0 and 18.5 cd A−1, and power efficiencies (PEs) of 12.5 and 16.5 lm W−1, respectively, relative to those of PCzSP doped in the CBP as an emitter (EQE = 9.1%, CE = 8.1 cd A−1, and PE = 7.2 l m W−1). The discrepancy in the EL efficiencies probably derives from the higher ΦPL in the thin film doped with DFASP and DPASP. The slightly more violet emission of the PCzSP-doped device also may lead to a lower EL efficiency. More importantly, the EQEs of all fabricated deep-blue devices are much greater than the 5% theoretical limit for conventional fluorescence OLEDs.
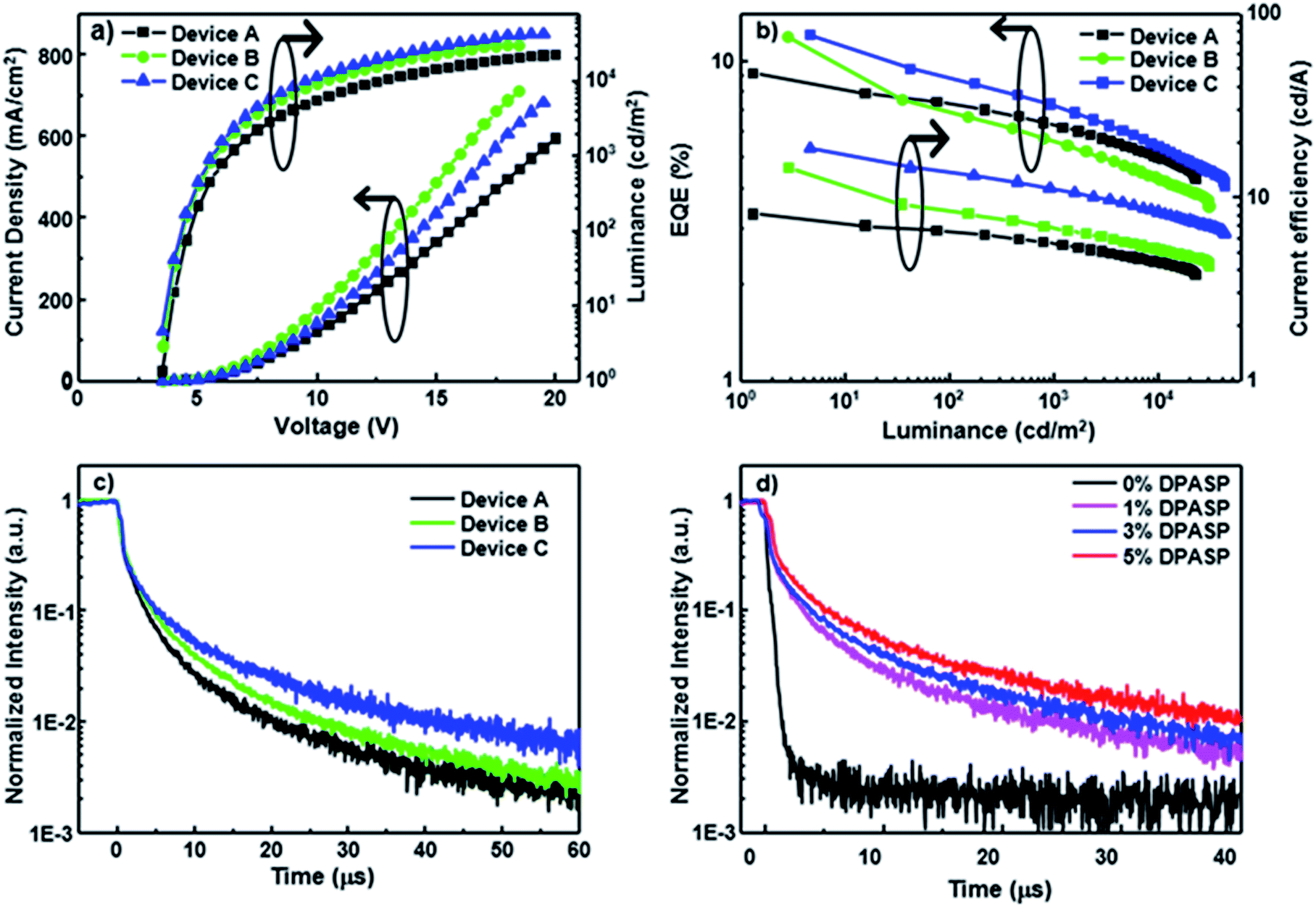 | ||
| Fig. 2 EL characteristics of CBP-based blue devices A, B and C containing PCzSP, DFASP, and DPASP dopants, respectively, in the emitting layers. (a) Current density–luminance–voltage curves, (b) EQE and current efficiency as a function of luminance, and (c) the time-resolved EL decay curves for devices A–C. (d) The transient EL of the devices doped with different concentrations of DPASP. | ||
| Device | Dopant/host | V on (V) | EQEmax (%) | CEmax (cd A−1) | PEmax (lm W−1) | EQE3000b (%) | λ max (nm) | CIEc (x, y) |
|---|---|---|---|---|---|---|---|---|
| a Turn-on voltage at 1 cd m−2. b EQE at 3000 cd m−2. c Recorded at 8 V. d ITO/NPB (30 nm)/TCTA (20 nm)/CBP: dopant (5 wt%, 30 nm)/TmPyPB (30 nm)/LiF (1 nm)/Al (100 nm). e ITO/NPB (60 nm)/NPB: dopant (3 wt%, 10 nm)/DMPPP: dopant (5 wt%, 15 nm)/BAlq (20 nm)/LiF (1 nm)/Al (100 nm). | ||||||||
| Ad | PCzSP/CBP | 3.3 | 9.1 | 8.1 | 7.2 | 5.6 | 444 | (0.15, 0.09) |
| Bd | DFASP/CBP | 3.1 | 11.5 | 14.0 | 12.5 | 4.8 | 457 | (0.14, 0.14) |
| Cd | DPASP/CBP | 3.0 | 12.0 | 18.5 | 16.5 | 6.3 | 462 | (0.14, 0.17) |
| De | PCzSP/DMPPP | 3.0 | 7.7 | 7.0 | 3.5 | 7.7 | 445 | (0.15, 0.10) |
| Ee | DFASP/DMPPP | 2.9 | 10.2 | 11.9 | 6.4 | 10.2 | 456 | (0.14, 0.12) |
| Fe | DPASP/DMPPP | 2.6 | 10.7 | 13.0 | 8.9 | 10.2 | 458 | (0.14, 0.14) |
Generally, EQE of the EL device is governed by four apparent parameters including the light out-coupling efficiency (ηout), electron–hole recombination efficiency (ηrec), fraction of radiative excitons (ηr), and ΦPL based on the equation EQE = ηrecηrΦPLηout. Considering the ηout of 20%, ΦPL of SP-based materials, and full electron–hole recombination (ηrec = 100%), these three devices exhibit ηr values that exceed the 25% limit of spin statistical ratio for fluorescent emitters, indicative of harvesting triplet excitons for emission in the PCzSP-, DFASP-, and DPASP-doped devices. To further understand the origin of the high ηr in these doped blue devices, we analyze the results of optical experiments for PCzSP, DFASP, and DPASP. As shown in Fig. S3,† a linear relationship between Stokes shift and solvent polarity is observed in the Lippert–Mataga plots which indicates the lack of an intercrossed excited state of LE and CT with a HLCT character for SP-based fluorophores. In an oxygen-free environment, the transient PL decay characteristics of CBP films doped with SP-based emitters exhibit a prompt emission lifetime on the nanosecond scale without a delayed component in the time range of microseconds (Fig. S9†). We were unable to measure the phosphorescence spectra of these SPs at 77 K, probably due to the very high fluorescence quantum efficiency of these materials. However when they were doped in the PMMA host using FIrpic as a triplet sensitizer, weak phosphorescence emissions can be obtained as shown in Fig. S10.† The highest energy vibronic sub-bands of the phosphorescence spectra of PCzSP, DFASP, and DPASP appear at 629, 641, and 645 nm (Fig. S10†), corresponding to triplet energy levels (T1) of 1.97, 1.93, and 1.92, respectively. The single energy levels (S1) of PCzSP, DFASP, and DPASP are respectively estimated to be 2.86, 2.79, and 2.75 eV, calculated from the PL peaks in toluene. As a result, we can conclude that these emitters meet the criterion of 2T1 > S1 for TTA-based materials, indicating that they can produce additional singlet excitons through a triplet fusion process. Moreover, the calculated ΔEST values are 0.89, 0.86, and 0.83 eV for PCzSP, DFASP, and DPASP, respectively. Such large ΔEST values dramatically increase the difficulty of the T1 → S1 RISC process and exclude the possibility of TADF behavior. These findings suggest that the observed unusually high efficiency of the doped blue devices are not likely due to TADF and HLCT processes, and therefore are attributed to the efficient upconversion of triplet excitons through TTA.
To gain insight into the influence of delayed fluorescence resulting from TTA on EQE, the time-resolved EL responses of these blue devices were examined under an electrical excitation pulse. The transient EL of devices A–C display a clear microsecond-scale delayed fluorescence after the rapid prompt decay from the short fluorescence lifetime of the SP-based emitters (Fig. 2c). A plot of the inverse of relative transient EL intensity against time exhibits a linear dependence on the microsecond scale for each transient EL curve (Fig. S13†), indicating that the delayed fluorescence intensity decay is second order with respect to exciton concentration. The findings suggest that the delayed fluorescence of these devices is due to TTA-induced emission. Interestingly, the DF components in devices B and C are much greater than that of device A. A greater DF component renders a higher measured EQE for the corresponding device. Therefore, the superior EQEs of DFASP- and DPASP-doped devices must be ascribed to an efficient TTA process.
We also prepared CBP-based devices doped with different concentrations of DPASP in the emitting layer to explore the origin of the TTA mechanism. The fabricated structure is ITO/NPB (30 nm)/TCTA (20 nm)/CBP: x% DPASP (30 nm)/TmPyPB (30 nm)/LiF (1 nm)/Al (100 nm) with doping concentrations of 0%, 1%, 3%, and 5% for devices S1–4, respectively. The detailed performance and characteristics are shown in Table S2 and Fig. S15.† Apparently, no DF component is found in the transient EL (Fig. 2d) of device S1 using CBP only in the EML, indicative of the inability of CBP molecules to harvest triplet excitons via TTA. The device performances increase with increasing doping concentrations for the DPASP-doped devices, and the larger the relative DF intensity, the higher the EQE of the device. These results suggest that the TTA process likely occurs at the dopant DPASP in these devices, as the host CBP does not show TTA-type delayed fluorescence. It is noteworthy that most TTA-based materials were used as hosts for blue fluorescent OLEDs. The results presented herein are probably the only example of TTA from the dopant only. In addition, the findings further demonstrate that the SP-based materials are TTA-based materials capable of harvesting triplet excitons.
Notably, all CBP-based devices doped with SP-based blue emitters exhibit serious efficiency roll-off, probably due to unbalanced carrier injection and transport within the emissive layer. To avoid this issue, SP-doped devices D–F using DMPPP as the host were fabricated. The device structure is ITO/NPB (60 nm)/NPB: dopant (3 wt%, 10 nm)/DMPPP: dopant (5 wt%, 15 nm)/BAlq (20 nm)/LiF (1 nm)/Al (100 nm). A double-emitting layer structure is used to expand the emission zone and decrease the charge accumulation.20 Furthermore, DMPPP exhibits more ambipolar transport properties (see Fig. S16† for details) relative to CBP and was thus chosen as the main host in these devices to balance carrier transport in the EMLs, resulting in a wide recombination zone. The architecture reduces the exciton quenching at high current density and thus leads to low efficiency roll-off. The performances of devices D–F are summarized in Table 2, and the corresponding performance curves are displayed in Fig. 3. All devices show deep-blue EL spectra close to the corresponding PL spectra of their doped films, confirming that the EL spectra predominantly originate from the emission of SP-based materials (Fig. S8†). These saturated deep blue EL emissions with a CIE y coordinate value ≦ 0.14 are quite stable over a wide range of driving voltage. Devices D–F hosted by DMPPP show a relatively low turn-on voltage of ca. 2.8 V and achieve outstanding performance with EQEs of 7.7, 10.2, and 10.7%, corresponding to CE values of 7.0, 11.9, and 13.0 cd A−1 and PE values of 3.5, 6.4, and 8.9 l m W−1 for PCzSP-, DFASP-, and DPASP-based devices, respectively. Importantly, all devices show a TTA-type delayed EL and the DF contributions of these devices correlate very well with their EQE values, manifesting that triplet excitons generated via the TTA process account for the high EQE of DMPPP-based devices. To further explore the TTA phenomena of these DMPPP-based devices, we examined the transient EL of a non-doped device with a device configuration the same as that of devices D–F, except with only DMPPP in the EML. The transient behaviour of this non-doped device (Fig. 3d) clearly shows a microsecond-scale DF, indicating that a DMPPP molecule can also harvest triplet excitons through the TTA mechanism (see the ESI and Fig. S17† for details). As a result, both the dopant and host in devices D–F appear to have TTA behaviour and it is interesting to see which out of the dopant and host dominates the TTA phenomena of these devices. In general, if TTA occurs in the host and then transfers to the dopant, the delayed fluorescence decay curves of the devices with the presence or absence of the dopant should be approximately the same for all three devices, because the transient decay lifetime of DF in the doped device is dominated by that of the TTA-based host.21 It is worth noting that, in the present case, the DF lifetimes for devices D–F are longer than that of the DMPPP only device. Thus, it is unlikely that the TTA process occurs mainly on the DMPPP host molecules in devices D–F. Thus, we can ascribe the DF in devices D–F to the TTA process on the dopant emitters. Moreover, increases in EQE and CE with increasing luminance are observed in the EQE–luminance–CE curves, which plausibly originate from the increased TTA contribution at high current density because the triplet excitons generated by a device are nearly proportional to the current density of the device. As expected, a low efficiency roll-off is obtained at a high brightness of 3000 cd m−2 for these blue devices hosted by DMPPP, owing to the good charge balance and the TTA process in the EML. To verify that the improved roll-off of DMPPP-based devices arises from the bipolar DMPPP host, we fabricated device G, a DPASP-doped device, with the same configuration as device F except using CBP instead of DMPPP as the host. Its device performance is displayed in Fig. S18.† The roll-off of device G is much larger than that of device F. At a luminance of 3000 cd m−2 the roll-off for device F is 4.7%, significantly smaller than that of 39.4% found for the CBP hosted device G (EQEmax = 10.4% and EQE3000 = 6.3%), demonstrating that the use of bipolar DMPPP host effectively reduces the roll-off of the device. Overall, the deep blue devices based on DFASP and DPASP offer excellent efficiencies >10% with extremely small efficiency roll-off at high luminance, and appear to be the best among reported deep-blue fluorescent devices.11,22
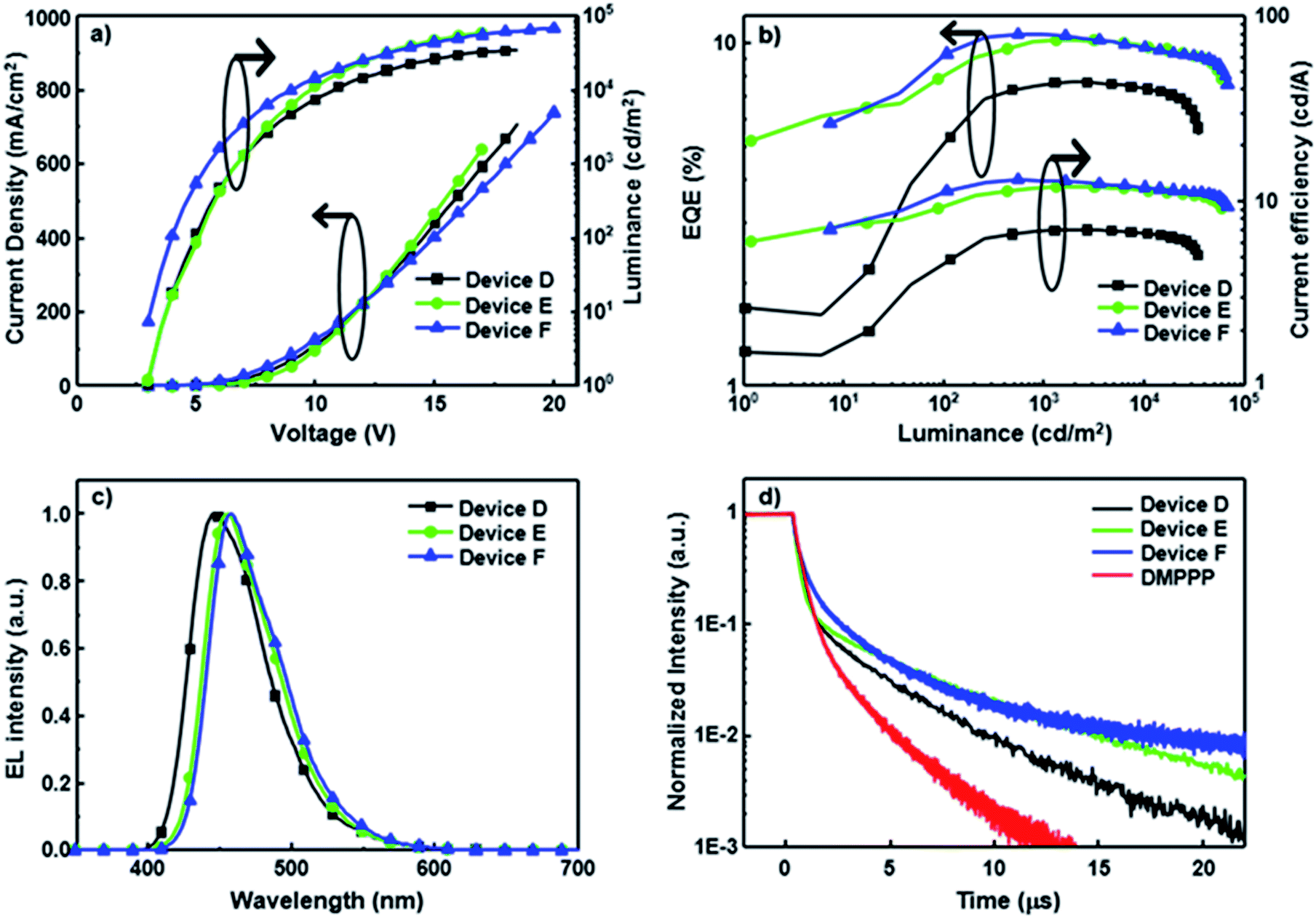 | ||
| Fig. 3 Device performance of blue fluorescent OLEDs hosted by DMPPP. (a) Plots of current density and luminance vs. voltage. (b) EQE–luminance–current efficiency characteristics. (c) EL spectra at 8 V. (d) Transient EL of devices D–F and the dopant-free DMPPP only device. | ||
To compare the EL decay curves with different emission mechanisms, we fabricated a normal fluorescence device using DPVBi: 3% BDAVBi as the host and dopant and a TADF device using mCP: 5% 2CzPN in the emission layer (see Fig. S14† for device structures). The EL decay curves of these two devices and device C are shown in Fig. S14.† For the BDAVBi-doped device, a very sharp EL decay curve is observed, indicative of a very short fluorescence lifetime and no DF contribution from this normal fluorescence device. In contrast, the transient EL of 2CzPN with TADF property shows a relatively flat decay curve with a very high delayed component to prompt ratio, indicating that the TADF process dominates the device emission under electrical excitation. The linearity of the delayed component is best fitted with a first order decay. Different from the normal fluorescence and TADF mechanisms, the DPASP-based TTA device shows an EL decay curve with a clear prompt and delayed component. In addition, the delayed component of this DPASP-based device follows a second order decay pattern (see Fig. S13†).
Conclusions
In conclusion, three deep-blue fluorophores, PCzSP, DFASP, and DPASP, have been successfully designed and synthesized via the integration of a styrylpyrene core with electron-donating moieties PhCz and TPA. These materials possess excellent thermal stability and PL quantum yields. All devices using a SP-based material as the dopant emit deep-blue light with remarkable device efficiency. Notably, an extremely high EQE of up to 12% can be realized for a DPASP-doped device using CBP as the host. Detailed photophysical characterizations and transient EL experiments strongly support that the TTA process results in the superior EL performance of the devices. The EL characteristics of DPASP-doped CBP-based devices at various doping concentrations (0–5%) further indicate that it is the dopant DPASP, not the host CBP, that is responsible for the TTA-type delayed fluorescence in the device. By using DMPPP as the host, the deep-blue device doped with DPASP achieves the EQE as high as ca. 11% with extremely low efficiency roll-off at high luminance. TTA arising from SP-based emitters plays a critical role in increasing the EQE values to more than 10% for these deep-blue devices. These encouraging results demonstrate the feasibility of the styrylpyrene core for highly efficient deep-blue OLEDs with both high quantum yield and TTA characteristics, thus opening a versatile design strategy for the development of highly efficient deep-blue light-emitting materials.Acknowledgements
We acknowledge financial support from the Ministry of Science and Technology of the Republic of China (MOST 104-2633-M-007-001) and are also grateful to the National Center for High-Performance Computing (Account number: u32chc04) for providing the computing time.Notes and references
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- (a) J. Kido, M. Kimura and K. Nagai, Science, 1995, 267, 1332 CrossRef CAS PubMed; (b) M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750 CrossRef CAS PubMed; (c) H. Wu, L. Ying, W. Yang and Y. Cao, Chem. Soc. Rev., 2009, 38, 3391 RSC.
- (a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS; (b) K.-Y. Lu, H.-H. Chou, C.-H. Hsieh, Y.-H. O. Yang, H.-R. Tsai, H.-Y. Tsai, L.-C. Hsu, C.-Y. Chen, I.-C. Chen and C.-H. Cheng, Adv. Mater., 2011, 23, 4933 CrossRef CAS PubMed.
- S. Schmidbauer, A. Hohenleutner and B. Konig, Adv. Mater., 2013, 25, 2114 CrossRef CAS PubMed.
- (a) X. Yang, X. Xu and G. Zhou, J. Mater. Chem. C, 2015, 3, 913 RSC; (b) X. Zhan, N. Sun, Z. Wu, J. Tu, L. Yuan, X. Tang, Y. Xie, Q. Peng, Y. Dong, Q. Li, D. Ma and Z. Li, Chem. Mater., 2015, 27, 1847 CrossRef CAS; (c) W. Qin, Z. Yang, Y. Jiang, J. W. Y. Lam, G. Liang, H. S. Kwok and B. Z. Tang, Chem. Mater., 2015, 27, 3892 CrossRef CAS.
- (a) W.-C. Chen, C.-S. Lee and Q.-X. Tong, J. Mater. Chem. C, 2015, 3, 10957 RSC; (b) Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931 CrossRef CAS PubMed.
- (a) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed; (b) D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050 CrossRef CAS PubMed; (c) K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. V. Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908 CrossRef CAS PubMed; (d) T. Komino, H. Tanaka and C. Adachi, Chem. Mater., 2014, 26, 3665 CrossRef CAS; (e) G. Cheng, G. K.-M. So, W.-P. To, Y. Chen, C.-C. Kwok, C. Ma, X. Guan, X. Chang, W.-M. Kwok and C.-M. Che, Chem. Sci., 2015, 6, 4623 RSC.
- (a) S. Zhang, L. Yao, Q. Peng, W. J. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755 CrossRef CAS; (b) X. Tang, Q. Bai, Q. Peng, Y. Gao, J. Li, Y. Liu, L. Yao, P. Lu, B. Yang and Y. Ma, Chem. Mater., 2015, 27, 7050 CrossRef CAS; (c) M. Liu, X.-L. Li, D. C. Chen, Z. Xie, X. Cai, G. Xie, K. Liu, J. Tang, S.-J. Su and Y. Cao, Adv. Funct. Mater., 2015, 25, 5190 CrossRef CAS; (d) H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li, P. Lu, H. Wang, B. Yang and Y. Ma, Chem. Sci., 2015, 6, 3797 RSC.
- (a) C.-J. Chiang, A. Kimyonok, M. K. Etherington, G. C. Griffiths, V. Jankus, F. Turksoy and A. P. Monkman, Adv. Funct. Mater., 2013, 23, 739 CrossRef CAS; (b) V. Jankus, C.-J. Chiang, F. Dias and A. P. Monkman, Adv. Mater., 2013, 25, 1455 CrossRef CAS PubMed; (c) P. Chen, Z. Xiong, Q. Peng, J. Bai, S. Zhang and F. Li, Adv. Opt. Mater., 2014, 2, 142 CrossRef; (d) Y. Luo and H. Aziz, Adv. Funct. Mater., 2010, 20, 1285 CrossRef CAS; (e) Y. Tamai, H. Ohkita, H. Benten and S. Ito, Chem. Mater., 2014, 26, 2733 CrossRef CAS.
- (a) Y.-J. Pu, G. Nakata, F. Satoh, H. Sasabe, D. Yokoyama and J. Kido, Adv. Mater., 2012, 24, 1765 CrossRef CAS PubMed; (b) B. Kim, Y. Park, J. Lee, D. Yokoyama, J.-H. Lee, J. Kido and J. Park, J. Mater. Chem. C, 2013, 1, 432 RSC; (c) H. Fukagawa, T. Shimizu, N. Ohbe, S. Tokito, K. Tokumaru and H. Fujikake, Org. Electron., 2012, 13, 1197 CrossRef CAS; (d) D. Yokoyama, Y. Park, B. Kim, S. Kim, Y.-J. Pu, J. Kido and J. Park, Appl. Phys. Lett., 2011, 99, 123303 CrossRef; (e) K.-Y. Kay, J.-H. Kim, S.-H. Kim, H.-C. Park and J.-W. Park, Mol. Cryst. Liq. Cryst., 2006, 444, 121 CrossRef CAS; (f) S. Tang, W. Li, F. Shen, D. Liu, B. Yang and Y. Ma, J. Mater. Chem., 2012, 22, 4401 RSC.
- J.-Y. Hu, Y.-J. Pu, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, Adv. Funct. Mater., 2014, 24, 2064 CrossRef CAS.
- P.-Y. Chou, H.-H. Chou, Y.-H. Chen, T.-H. Su, C.-Y. Liao, H.-W. Lin, W.-C. Lin, H.-Y. Yen, I.-C. Chen and C.-H. Cheng, Chem. Commun., 2014, 50, 6869 RSC.
- (a) C. Bohne, E. B. Abuin and J. C. Scaiano, J. Am. Chem. Soc., 1990, 112, 4226 CrossRef CAS; (b) W. Zhao and F. N. Castellano, J. Phys. Chem. A, 2006, 110, 11440 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395 CrossRef CAS PubMed.
- (a) Y. Yuan, J.-X. Chen, F. Lu, Q.-X. Tong, Q.-D. Yang, H.-W. Mo, T.-W. Ng, F.-L. Wong, Z.-Q. Guo, J. Ye, Z. Chen, X.-H. Zhang and C.-S. Lee, Chem. Mater., 2013, 25, 4957 CrossRef CAS; (b) K. R. J. Thomas, N. Kapoor, M. N. K. P. Bolisetty, J.-H. Jou, Y.-L. Chen and Y.-C. Jou, J. Org. Chem., 2012, 77, 3921 CrossRef CAS PubMed; (c) S. Zhuang, R. Shangguan, H. Huang, G. Tu, L. Wang and X. Zhu, Dyes Pigm., 2014, 101, 93 CrossRef CAS.
- (a) C.-C. Lai, M.-J. Huang, H.-H. Chou, C.-Y. Liao, P. Rajamalli and C.-H. Cheng, Adv. Funct. Mater., 2015, 25, 5548 CrossRef CAS; (b) Y. Li, Z. Wang, X. Li, G. Xie, D. Chen, Y.-F. Wang, C.-C. Lo, A. Lien, J. Peng, Y. Cao and S.-J. Su, Chem. Mater., 2015, 27, 1100 CrossRef CAS.
- K.-C. Wu, P.-J. Ku, C.-S. Lin, H.-T. Shih, F.-I. Wu, M.-J. Huang, J.-J. Lin, I.-C. Chen and C.-H. Cheng, Adv. Funct. Mater., 2008, 18, 67 CrossRef CAS.
- (a) G.-F. Zhang, H. Wang, M. P. Aldred, T. Chen, Z.-Q. Chen, X. Meng and M.-Q. Zhu, Chem. Mater., 2014, 26, 4433 CrossRef CAS; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- C. Hosokawa, H. Higashi, H. Nakamura and T. Kusumoto, Appl. Phys. Lett., 1995, 67, 3853 CrossRef CAS.
- (a) T. Zheng and W. C. H. Choy, J. Phys. D: Appl. Phys., 2008, 41, 055103 CrossRef; (b) L. Duan, D. Zhang, K. Wu, X. Huang, L. Wang and Y. Qiu, Adv. Funct. Mater., 2011, 21, 3540 CrossRef CAS.
- (a) T. Suzuki, Y. Nonaka, T. Watabe, H. Nakashima, S. Seo, S. Shitagaki and S. Yamazaki, Jpn. J. Appl. Phys., 2014, 53, 052102 CrossRef; (b) T. Higuchi, H. Nakanotani and C. Adachi, Adv. Mater., 2015, 27, 2019 CrossRef CAS PubMed.
- (a) Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706 CrossRef CAS PubMed; (b) S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402 CrossRef CAS PubMed; (c) S. Wu, M. Aonuma, Q. Zhang, S. Huang, T. Nakagawa, K. Kuwabara and C. Adachi, J. Mater. Chem. C, 2014, 2, 421 RSC; (d) M. Chen, Y. Yuan, J. Zheng, W.-C. Chen, L.-J. Shi, Z.-L. Zhu, F. Lu, Q.-X. Tong, Q.-D. Yang, J. Ye, M.-Y. Chan and C.-S. Lee, Adv. Opt. Mater., 2015, 3, 1215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, 1H and 13C NMR spectra, solvatochromic data and Lippert–Mataga analysis, molecular orbital calculations, cyclic voltammograms, detailed photophysical and thermal characteristics, and EL performance. See DOI: 10.1039/c6sc00100a |
| This journal is © The Royal Society of Chemistry 2016 |