
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic radical reduction in aqueous solution via oxidation of biologically-relevant alcohols†
Yamin
Htet
a and
Andrew G.
Tennyson
*abc
aDepartment of Chemistry, Clemson University, Clemson, SC 29634, USA. E-mail: atennys@clemson.edu
bDepartment of Materials Science and Engineering, Clemson University, Clemson, SC 29634, USA
cCenter for Optical Materials Science and Engineering Technologies, Anderson, SC 29625, USA
First published on 10th March 2016
Abstract
Metalloenzymes that normally perform catalytic antioxidant or radical-degrading functions, as well as small-molecule complexes that mimic them, can also exert pro-oxidant or radical-forming effects depending on the identity of the terminal reductant. Because nitroxyl radicals function as redox active cocatalysts in the aerobic oxidation of alcohols, we hypothesized that catalytic radical reduction could be achieved via the oxidation of biologically-relevant alcohols. Herein we report an organoruthenium complex (Ru1) that catalyzed reduction of 2,2′-azino-bis(3-ethylbenzo-thiazoline-6-sulfonate) radical monoanion (ABTS˙−) to ABTS2− in phosphate buffered saline (pH 7.4) using MeOH, EtOH, i-PrOH, serine, threonine, glucose, arabinose, methyl lactate or dimethyl malate as the terminal reductant. Replacing either the C–H or O–H groups of a –CHOH– moiety resulted in the loss of ABTS˙− reducing ability. Moreover, in conjunction with an alcohol terminal reductant, Ru1 was able to inhibit the oxidation of ABTS2− by H2O2 and horseradish peroxidase, even after multiple successive challenges with excess H2O2 or ABTS˙−. Collectively, these results demonstrate that Ru1 inhibits the oxidative formation of and catalyzes the reduction of radicals in aqueous solution via oxidation of biologically-relevant alcohols.
Introduction
Reactive oxygen species (ROS) such as superoxide (O2−) and hydrogen peroxide (H2O2) can alter the structures and functions of biomolecules1,2via DNA base lesion formation,3 transition metal ion release from proteins,4 and membrane phospholipid chain degradation.5 Antioxidants can prevent or attenuate this damage by reacting with ROS to yield less oxidizing, more stable radical or non-radical products. Superoxide dismutase (SOD) and catalase, for example, are metalloenzymes that protect cells by catalyzing the disproportionation of O2− and H2O2, respectively.6 Synthetic Mn–salen and Mn–porphyrin complexes have demonstrated both SOD and catalase activity,7 and some have entered phase 1 clinical trials as therapeutics for neurodegenerative and pulmonary diseases.8Recent studies have shown that these classes of synthetic Mn complexes can also exert pro-oxidant effects at low ROS concentrations,9,10 dichotomous behavior that is consistent with the enzymes they mimic. Catalase converts 2H2O2 into O2 + 2H2O (Scheme 1-i),11 wherein the first equivalent of H2O2 is reduced to H2O and generates an Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O˙+ intermediate, which then oxidizes the second equivalent of H2O2 to O2 and releases H2O.12 At low H2O2 concentrations, this Fe(IV)O˙+ intermediate can abstract H˙ from other molecules (i.e., A–H) and thus exhibit peroxidase-like reactivity (Scheme 1-ii).13 Conversely, peroxidase can exhibit catalase-like reactivity at low A–H/high H2O2 concentrations.14 Because the Mn–salen and Mn–porphyrin complexes proceed through high-valent Mn oxo/hydroxo intermediates15–18 with oxidizing power comparable to Fe(IV)O˙+, their reactivity will mimic catalase or peroxidase based on the availability of H2O2. Therefore, the function of catalase and its biomimetic Mn complexes as either antioxidants or pro-oxidants will be determined by the nature of the terminal reductant.
O˙+ intermediate, which then oxidizes the second equivalent of H2O2 to O2 and releases H2O.12 At low H2O2 concentrations, this Fe(IV)O˙+ intermediate can abstract H˙ from other molecules (i.e., A–H) and thus exhibit peroxidase-like reactivity (Scheme 1-ii).13 Conversely, peroxidase can exhibit catalase-like reactivity at low A–H/high H2O2 concentrations.14 Because the Mn–salen and Mn–porphyrin complexes proceed through high-valent Mn oxo/hydroxo intermediates15–18 with oxidizing power comparable to Fe(IV)O˙+, their reactivity will mimic catalase or peroxidase based on the availability of H2O2. Therefore, the function of catalase and its biomimetic Mn complexes as either antioxidants or pro-oxidants will be determined by the nature of the terminal reductant.
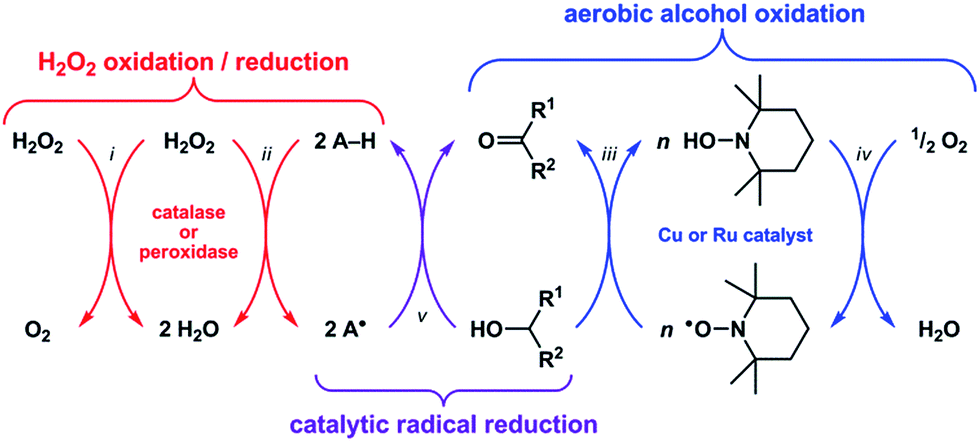 | ||
| Scheme 1 Catalase normally catalyzes the disproportionation of 2H2O2 → O2 + 2H2O (i), but at low H2O2 concentrations it can abstract H˙ from other molecules (A–H) and produce radicals (A˙) in a manner similar to peroxidase (ii). Copper or ruthenium complexes can catalyze the oxidation of R1–CHOH–R2 to R1–C(O)–R2 using a nitroxyl radical (e.g., TEMPO, n = 1 for Cu, n = 2 for Ru) as the H˙ abstracting reagent (iii) and O2 as the terminal oxidant (iv). By a similar mechanism, a complex could catalyze radical reduction using a biologically relevant non-tertiary alcohol as the terminal reductant (v). | ||
A catalytic oxidation reaction important in synthetic chemistry is the aerobic oxidation of R1–CHOH–R2 to R1–C(O)–R2 that does not cause any (1) over-oxidation to R–COOH or (2) undesired reactions at other functional groups.19–23 One strategy employs a copper or ruthenium catalyst in conjunction with a nitroxyl radical, such as 2,2,6,6-tetramethylpiperidinyl-N-oxyl (TEMPO), to oxidize R1–CHOH–R2 to R1–C(O)–R2 with concomitant formation of TEMPO–H (Scheme 1-iii). Subsequent regeneration of the TEMPO radical occurs with the oxidation of TEMPO–H by O2, which functions as the terminal oxidant for this reaction (Scheme 1-iv). Because the TEMPO radical is reduced by R1–CHOH–R2 in the aerobic alcohol oxidation catalytic cycle, we hypothesized that the catalytic reduction of other radicals could also be achieved via the oxidation (i.e., dehydrogenation) of non-tertiary alcohols (Scheme 1-v). Herein we report the validation of this hypothesis with an organoruthenium complex that catalyzes the reduction and inhibits the formation of radicals in buffered aqueous solutions via the oxidation of biologically-relevant non-tertiary alcohols. Given the successes of group 8 transfer hydrogenation catalysts as chemotherapeutics24,25 in particular and the burgeoning scope of medicinal applications for transition metal-based catalytic systems26–28 in general, we anticipate that the reduction of radicals via the Ru-catalyzed oxidation of alcohols will lead to new strategies for protecting against ROS in vivo.
Results and discussion
Approach and rationale
We recently reported a Ru complex supported by a chelating N-heterocyclic carbene–carboxylate ligand (Ru1, Scheme 2A) that catalyzed the hydrogenation of CO, CN and CC bonds, in which the H2 transferred was obtained from the oxidation of i-PrOH to acetone.29 Based on this catalytic chemical reactivity observed with Ru1 and previous reports of other Ru complexes with chelating carboxylate ligands exerting antioxidant effects in cells, which derived from irreversible stoichiometric reactions with nitric oxide,30,31 we hypothesized that Ru1 would function as a catalytic antioxidant, reducing radicals using non-tertiary alcohols as terminal reductants. Using methods previously reported by our group,29,32 the N-heterocyclic carbene (NHC) ligand precursor [1H2][Br] was treated with Ag2O to afford the Ag–NHC complex [Ag(1)]n (2). The NHC ligand was subsequently transferred to Ru via the reaction of 2 with [{RuCl(η6-cymene)}2(μ-Cl)2], which yielded the Ru–NHC complex [RuCl(1)(η6-cymene)] (Ru1).
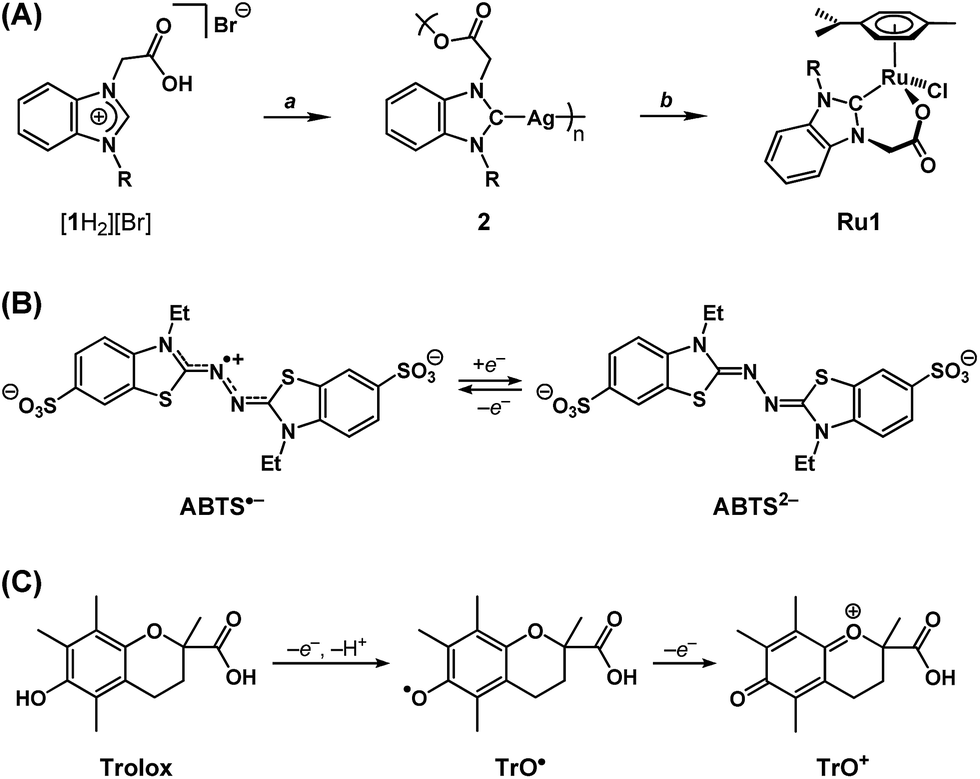 | ||
Scheme 2 (A) Synthesis of [1H2][Br], 2 and Ru1. Reagents and conditions: (a) Ag2O (1.5 equiv.) or (b) [{RuCl(η6-cymene)}2(μ-Cl)2] (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Ag/Ru), CH2Cl2, room temperature, 24 h. R = p-tolyl. (B) One-electron redox interconversion between ABTS˙− and ABTS2− (NH4+ counterions omitted for clarity). (C) First and second one-electron oxidations of Trolox. :1 Ag/Ru), CH2Cl2, room temperature, 24 h. R = p-tolyl. (B) One-electron redox interconversion between ABTS˙− and ABTS2− (NH4+ counterions omitted for clarity). (C) First and second one-electron oxidations of Trolox. | ||
As a radical substrate to probe antioxidant activity, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate) radical monoanion (ABTS˙−, Scheme 2B) was selected on the basis of its large extinction coefficient (ε) at long wavelengths (ε750 = 1.6 × 104 M−1 cm−1 in EtOH and ε734 = 1.5 × 104 M−1 cm−1 in aqueous buffer),33 its reversible one-electron reduction to ABTS2− (E1/2 = 0.68 V vs. NHE) falling within the range of potentials accessible in biological systems (−0.4 to +0.8 V vs. NHE),34 and its stability in aerobic, protic media. The longer absorption wavelength and greater extinction coefficient for ABTS˙− enables spectroscopic analysis of radical-degrading and -forming reactions (1) with less interference from other optically absorbing species present in biological systems and (2) at lower concentrations‡ more relevant to oxidative stress than would be possible with direct measurements of O2˙− (ε250 = 1.9 × 103 M−1 cm−1) or H2O2 (ε240 = 43.6 M−1 cm−1).35,36 Furthermore, the 1e− reduction potential for ABTS˙− is lower than or comparable to the standard reduction potentials for several of the oxidizing species associated with oxidative stress in biological systems (e.g., E° = 1.78 V for H2O2, 1.6 V for RO˙, 1.0 V for ROO˙, 0.92 V for Cys–S˙, 0.70 for O2, etc.),34 therefore ABTS˙− is a reasonable substrate for approximating biologically-relevant oxidants.
As a non-catalytic antioxidant control, Trolox (Scheme 2C) was employed given its use as a benchmark in a variety of radical degradation and antioxidant studies.37 Trolox can serve as a 1e− or 2e− reductant, whereby the first 1e− oxidation is accompanied by rapid H+ loss to form a phenoxyl radical (TrO˙), which can then undergo a second 1e− oxidation to form a phenoxonium cation (TrO+). However, in methanol (MeOH) or ethanol (EtOH) solutions, these processes converge into a single 2e− oxidation.38 Subsequent hydrolysis of TrO+ can then cleave the tertiary carbon–oxonium bond, which renders the 2e− oxidation of Trolox irreversible.
Because ROS and other oxidants can cause damage to critical biomolecules, inhibiting the formation of these oxidizing species in biological systems can potentially prevent the onset of harmful pathologies.2 As a model system, we selected the oxidation of ABTS2− to ABTS˙− by horseradish peroxidase (HRP) and H2O2 (i.e., Scheme 1-ii).39 However, before studying the ability of Ru1 to inhibit ABTS˙− formation, it was first necessary to explore the fundamental reactivity of Ru1 with ABTS˙− under controlled conditions and achieve the catalytic reduction of ABTS˙− to ABTS2− in buffered aqueous solution.
Catalytic radical reduction in EtOH
Addition of 5 μM Ru1 (CH3CN stock) to 50 μM ABTS˙− in EtOH caused a 100% decay in ABTS˙− concentration within 15 min (Fig. 1, red line), accompanied by the formation of 50 μM ABTS2− (Fig. S1†), indicating the 1:1 conversion of ABTS˙− to ABTS2− by Ru1. No ABTS˙− degradation occurred when CH3CN alone was added (Fig. 1, dotted green line), indicating that Ru1 was essential for the reduction of ABTS˙− to ABTS2− and that CH3CN did not impact ABTS˙− stability. No ABTS˙− formation was observed in an aerobic solution containing 5 μM Ru1 and 50 μM ABTS2−, indicating that Ru1 does not oxidize ABTS2− to ABTS˙− under these conditions. In contrast to Ru1, the addition of 5 μM Trolox caused a rapid (within mixing time) 22% decrease in ABTS˙− concentration (Fig. 1, blue line), which corresponded to the reduction of 10 μM ABTS˙− (2 equiv. vs. Trolox) and was consistent with the ability of Trolox to serve as a non-catalytic 2e− reductant in EtOH solution.38
 | ||
| Fig. 1 Plot of relative [ABTS˙−] vs. time in EtOH following the addition of Ru1 (CH3CN stock, red line), Trolox (CH3CN stock, blue line), or CH3CN alone (dotted green line). Conditions: [Ru1]0 or [Trolox] = 5 μM, [ABTS˙−]0 = 50 μM, EtOH, 25 °C; [ABTS˙−] determined using absorbance measured at 750 nm and ε750 = 1.6 × 104 M−1 cm−1 (ref. 33). | ||
To assess its catalytic potential and corresponding regeneration, the reactivity of Ru1 with multiple sequential aliquots of excess ABTS˙− was examined. After the reduction of 50 μM ABTS˙− by 5 μM Ru1 was complete (Fig. 2, red line), 2 additional 50 μM ABTS˙− aliquots were introduced§ and complete ABTS˙− reduction was observed in each instance, indicating that Ru1 remained catalytically competent for the complete reduction of 150 μM ABTS˙− (30 turnovers). Unlike Ru1, the addition of successive 10 μM aliquots§ of ABTS˙− following the initial reduction by 5 μM Trolox (Fig. 2, blue line) only caused the radical absorbance to increase by amounts equivalent to the concentration of ABTS˙− added, indicating that the antioxidant capacity of Trolox had been exhausted after the reduction of only 2 equiv. of ABTS˙−. However, subsequent addition of 5 μM Ru1 to the exhausted Trolox solution containing 70 μM ABTS˙− resulted in 100% radical reduction within 30 min.
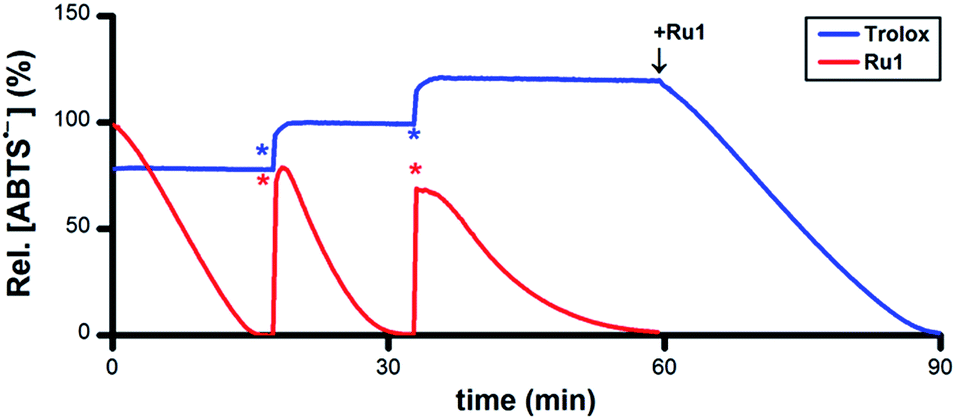 | ||
| Fig. 2 Plot of relative [ABTS˙−] vs. time which shows the reduction of ABTS˙− after adding Ru1 (red line) followed by 2 additional 50 μM ABTS˙− aliquots (*) and Trolox (blue line) followed by 2 additional 10 μM ABTS˙− aliquots (*), then 5 μM Ru1. Conditions: [Ru1]0 or [Trolox]0 = 5 μM, [ABTS˙−]0 = 50 μM, EtOH, 25 °C; [ABTS˙−] determined using absorbance measured at 750 nm and ε750 = 1.6 × 104 M−1 cm−1 (ref. 33). | ||
Addition of 5 μM ascorbate (AscH−) or 5 μM glutathione (GSH) to a solution of 50 μM ABTS˙− in EtOH caused the ABTS˙− concentration to decrease by 5% or 60% (Fig. S2 and S3†), values that were consistent with their ability to function as 1e− or 6e− reductants, respectively, under these experimental conditions.40–42 Similar to the experiments with Trolox, subsequent addition of ABTS˙− only afforded increases in radical absorbance equal to the concentration of ABTS˙− added, which likewise indicated that the antioxidant capacity of AscH− and GSH had been exhausted. Quantitative radical reduction could nonetheless be achieved via addition of 5 μM Ru1 to the exhausted AscH− or GSH solutions that contained excess ABTS˙−. Collectively, these data demonstrate that Ru1 can catalytically reduce ABTS˙− and, because it is regenerated after each reaction cycle, that a catalytic antioxidant can offer significantly greater protection than traditional stoichiometric antioxidants, such as Trolox, AscH− and GSH.
Although ABTS˙− degradation has been previously observed with other Ru complexes,43–45Ru1 is the first to demonstrate catalytic ABTS˙− reduction. It is important to note that the previous studies measured percentage of ABTS˙− degradation with respect to Ru complex concentration, for the purpose of determining the dose dependence of radical degradation. However, the reported absorbance vs. time plots displayed significantly slower ABTS˙− degradation for the Ru complexes compared to Trolox (much like Ru1vs. Trolox, Fig. 1) and 100% ABTS˙− degradation at multiple Ru concentrations (demonstrating dose independence). Thus, it is possible that a previously reported Ru complex may have degraded ABTS˙− catalytically, but its significantly slower reactivity compared to Trolox created the appearance that the percent of ABTS˙− degraded was dependent on Ru complex concentration and led to the conclusion that the observed radical degradation was non-catalytic.
Catalytic radical reduction in aqueous buffer
As a catalyst for the 1e− reduction of ABTS˙− to ABTS2−, Ru1 itself cannot serve as the terminal reductant for this reaction, a role most likely played by the EtOH solvent for the experiments displayed in Fig. 1. To test this hypothesis and identify the chemical features required for terminal reductant function, it was first necessary to determine experimental conditions under which no ABTS˙− degradation occurred in the presence of Ru1 alone. Phosphate-buffered saline (PBS, pH 7.4) was selected as a suitable reaction medium because neither the solvent (H2O) nor the buffer components (Na2HPO4, KH2PO4, NaCl, KCl) would undergo oxidation to supply the electrons necessary for the reduction of ABTS˙− (i.e., Fig. 3-i). No degradation of ABTS˙− in PBS was observed after treatment with 5 μM Ru1 (Fig. 3-ii), indicating this solution lacked a suitable terminal reductant. Subsequent addition of 50 mM EtOH to this PBS solution containing 5 μM Ru1 and 50 μM ABTS˙− caused the ABTS˙− concentration to decrease (Fig. 3-iii), evidence that the electrons needed for the ABTS˙− reduction observed in Fig. 1 were ultimately supplied by the EtOH solvent.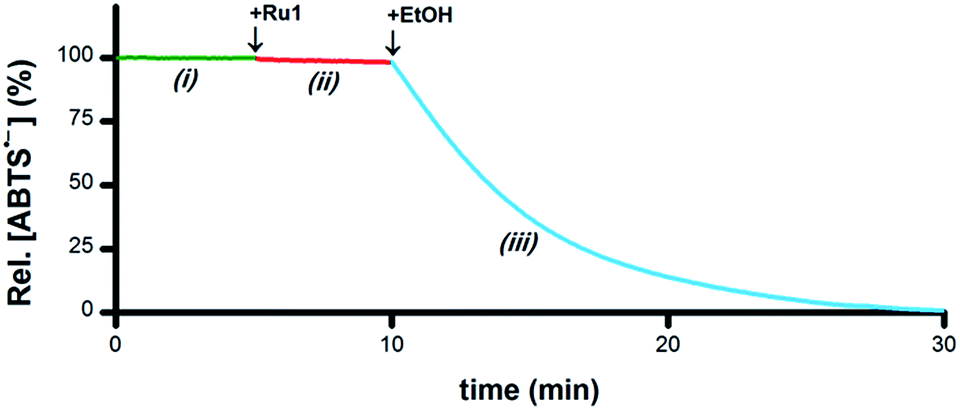 | ||
| Fig. 3 Plot of relative [ABTS˙−] vs. time which shows the catalytic reduction of ABTS˙− by Ru1 in PBS. By itself, ABTS˙− was stable in PBS (i, green line). Addition of Ru1 did not cause ABTS˙− reduction (ii, red line). Subsequent addition of EtOH caused the absorbance to decrease (iii, blue line), which indicated that EtOH functioned as a terminal reductant. Conditions: [Ru1]0 = 5 μM, [ABTS˙−]0 = 50 μM, [EtOH]0 = 50 mM, PBS (pH 7.4), 25 °C; [ABTS˙−] determined using absorbance measured at 734 nm and ε734 = 1.5 × 104 M−1 cm−1 (ref. 33). | ||
If the hypothesis that the oxidation of R1–CHOH–R2 to R1–C(O)–R2 provides the electrons necessary for the reduction of ABTS˙− to ABTS2− is correct (i.e., Scheme 1-v), then both the O–H and C–H groups in a –CHOH– moiety will be essential for terminal reductant function. To test this hypothesis, PBS solutions containing 5 μM Ru1 and 50 μM ABTS˙− were treated with 50 mM MeOH, i-PrOH, t-BuOH, ethylene glycol (EG) or 1,2-dimethoxyethane (DME) and the ABTS˙− concentration was measured. With MeOH, i-PrOH and EG, the ABTS˙− concentration began to decrease immediately following addition of the alcohol, indicating that these alcohols could serve as terminal reductants (Fig. S4–S6†). In contrast, no ABTS˙− reduction was observed with DME or t-BuOH (e.g., Fig. 4-ii). However, when the addition of 50 mM t-BuOH was followed by 50 mM EtOH, ABTS˙− reduction did occur (Fig. 4-iii), revealing that the lack of reactivity with t-BuOH was not due to catalyst deactivation but rather the inability of t-BuOH to serve as a terminal reductant. Collectively, these results show that the C–H and O–H groups of a –CHOH– moiety are both essential for terminal reductant function, which is consistent with the hypothesis that oxidation of R1–CHOH–R2 to R1–C(O)–R2 provides the reducing equivalents necessary to convert ABTS˙− to ABTS2−. Replacing either the C–H group with C–Me or the O–H group with O–Me precludes this oxidation and thus the ability to function as a terminal reductant, which is supported by the lack of ABTS˙− reduction observed with t-BuOH and DME.
 | ||
| Fig. 4 Plot of relative [ABTS˙−] vs. time which shows the catalyst is not deactivated by t-BuOH. In a PBS solution containing only Ru1, ABTS˙− was stable (i, red line). Addition of t-BuOH did not cause any ABTS˙− reduction (ii, orange line). Subsequent addition of EtOH resulted in ABTS˙− degradation (iii, blue line), indicating that the lack of reactivity with t-BuOH was not due to catalyst deactivation. Conditions [Ru1]0 = 5 μM, [ABTS˙−]0 = 50 μM, [t-BuOH]0 = 50 mM, [EtOH]0 = 50 mM, PBS (pH 7.4), 25 °C; [ABTS˙−] determined using absorbance measured at 734 nm and ε734 = 1.5 × 104 M−1 cm−1 (ref. 33). | ||
Biologically-relevant terminal reductants
A wide variety of biomolecules comprise –CHOH– groups, therefore we hypothesized that sugars, amino acids and citric acid cycle metabolites could serve as suitable terminal reductants in Ru1-catalyzed ABTS˙− reduction. To test this hypothesis, PBS solutions containing 5 μM Ru1 and 50 μM ABTS˙− were treated with 50 mM serine, threonine, glucose, arabinose, malic acid or lactic acid. Reduction of ABTS˙− was markedly slower following the addition of the amino acids compared to the sugars, consistent with the greater number of CH–OH groups per molecule in the latter (Fig. 5). No significant differences in reactivity were observed between serine and threonine or between glucose and arabinose.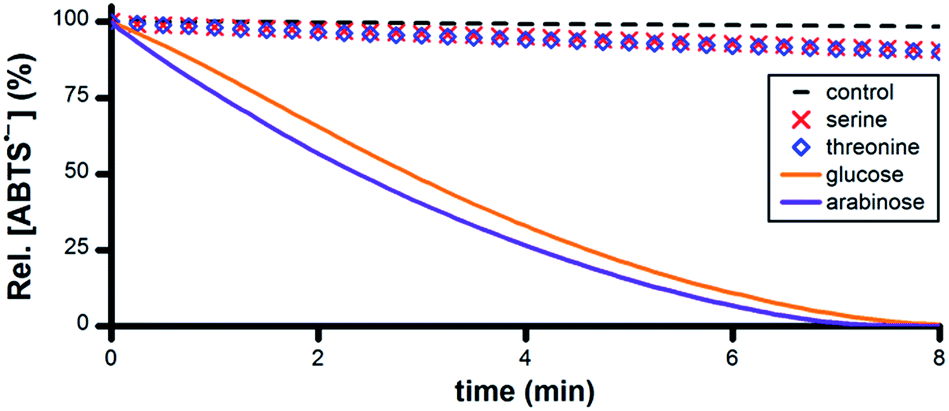 | ||
| Fig. 5 Plot of relative [ABTS˙−] vs. time following the addition of amino acid or sugar (t = 0) to PBS solutions containing ABTS˙− and Ru1. Conditions: [Ru1]0 = 5 μM, [ABTS˙−]0 = 50 μM, [ROH]0 = 50 mM, PBS (pH 7.4), 25 °C; [ABTS˙−] determined using absorbance measured at 734 nm and ε734 = 1.5 × 104 M−1 cm−1 (ref. 33). | ||
Malic acid and lactic acid are metabolites of the citric acid cycle and each comprises a –CHOH– group. However, no ABTS˙− reduction occurred following the addition of 50 mM malic acid or lactic acid to 5 μM Ru1 and 50 μM ABTS˙− in PBS (Fig. 6). Malate and lactate will be the predominant species in pH 7.4 PBS, and these carboxylate anions can function as chelating bidentate ligands, which could potentially disrupt the catalytic activity of Ru1. To prevent catalyst deactivation, the methyl esters of these substrates were used. Addition of 50 mM methyl lactate (Me-lactate) or dimethyl malate (Me2-malate) to PBS solutions containing 5 μM Ru1 and 50 μM ABTS˙− caused the ABTS˙− concentration to decrease rapidly. None of the biologically-relevant alcohols afforded ABTS˙− reduction in the absence of Ru1, and no oxidation of ABTS2− to ABTS˙− by Ru1 was observed under any experimental conditions in PBS.
 | ||
| Fig. 6 Plot of relative [ABTS˙−] vs. time following the addition of lactate, malate or their methyl esters (t = 0) to PBS solutions containing ABTS˙− and Ru1. Conditions: [Ru1]0 = 5 μM, [ABTS˙−]0 = 50 μM, [ROH]0 = 50 mM, PBS (pH 7.4), 25 °C; [ABTS˙−] determined using absorbance measured at 734 nm and ε734 = 1.5 × 104 M−1 cm−1 (ref. 33). | ||
Inhibition of oxidative radical formation
Having obtained unambiguous evidence that Ru1 catalyzes ABTS˙− reduction using biologically-relevant alcohols as terminal reductants, we next sought to demonstrate that Ru1 could also inhibit radical formation. As the model system, we utilized the in situ oxidation of ABTS2− to ABTS˙− by horseradish peroxidase (HRP) and H2O2 (i.e., Scheme 1-ii).39 Addition of 10 μM H2O2 to a PBS solution of 10 nM HRP, 20 μM ABTS2−, 5 μM Ru1, and 50 mM EtOH caused the ABTS˙− concentration to increase immediately. The ABTS˙− concentration peaked at 1.0 μM after 2.3 min, which then gradually declined and complete ABTS˙− reduction was observed 13 min after the peak (Fig. 7A, red line). No ABTS˙− formation occurred when 10 μM H2O2 was added to 5 μM Ru1 and 20 μM ABTS2− in PBS, indicating that Ru1 did not cause any pro-oxidant reactions. Because Ru1 and Trolox were added as 30 μL aliquots from 5.0 mM stock solutions in CH3CN, 10 μM H2O2 was added to a PBS solution containing 10 nM HRP, 20 μM ABTS2− and 0.19 M CH3CN to control for the influence of this solvent. The radical absorbance immediately began to increase and reached a maximum of 17.9 μM after 50 min (Fig. 7A, dotted green line and Fig. S7†). In contrast, the addition of 10 μM H2O2 to a PBS solution of 5 μM Trolox, 10 nM HRP and 20 μM ABTS2− resulted in no change in ABTS˙− concentration for the first 7.6 min, demonstrating that Trolox completely halted ABTS˙− formation (Fig. 7A, blue line). After 7.6 min, however, the ABTS˙− concentration began to increase and reached a maximum of 9.3 μM at 12 min after the onset of ABTS˙− formation. Notably, the maximum [ABTS˙−] observed in the presence of Trolox was 8.6 μM lower than the control, which was consistent with the ability of Trolox to function as a 2e− reductant (cf., the 22% decrease observed with Trolox in Fig. 1A). For the control and Trolox experiments, the decline in ABTS˙− concentration after peaking was consistent with normal ABTS˙− thermal decay.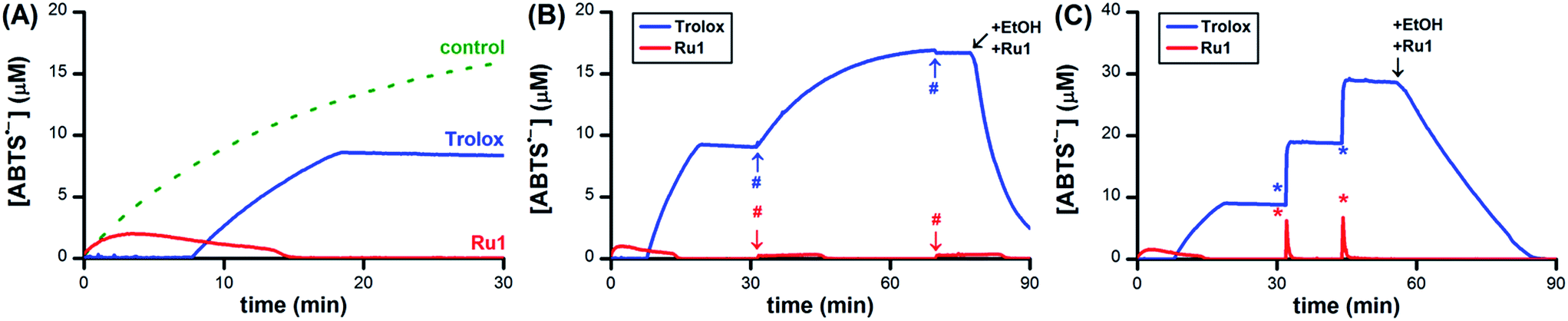 | ||
| Fig. 7 (A) Plot of relative [ABTS˙−] vs. time which shows the in situ oxidation of ABTS2− to ABTS˙−via HRP and H2O2 in the presence of Ru1 and EtOH (red line), Trolox (blue line) or 0.19 M CH3CN as a control (dotted green line). Plot of relative [ABTS˙−] vs. time which shows the (B) ABTS˙− formation by HRP in the presence of Ru1 and EtOH (red line) or Trolox (blue line) followed by two additional aliquots of 10 μM H2O2 (#) or (C) two 10 μM aliquots of chemically synthesized ABTS˙− (*). For the Trolox experiments shown in (B) or (C), 50 mM EtOH and 5 μM Ru1 were added after the final aliquot of H2O2 or ABTS˙−, respectively. Conditions: [HRP]0 = 10 nM [Ru1]0 or [Trolox]0 = 5 μM, [H2O2]0 = 10 μM, [ABTS2−]0 = 20 μM, [EtOH]0 = 50 mM, PBS (pH 7.4) at 25 °C; [ABTS˙−] determined using absorbance measured at 734 nm and ε734 = 1.5 × 104 M−1 cm−1 (ref. 33). | ||
To determine if Ru1 remained catalytically competent after the complete reduction of ABTS˙− formed by 10 nM HRP and 10 μM H2O2, two additional 10 μM aliquots of H2O2 were introduced (Fig. 7B, red line). Impressively, the concentration of ABTS˙− never exceeded 0.4 μM and decreased back to zero in less than 16 min after the introduction of the second and third H2O2 aliquots. In contrast, addition of the second 10 μM H2O2 aliquot to the Trolox experiment caused the ABTS˙− concentration to gradually increase by 7.9 μM over the course of 38 min (Fig. 7B, blue line), effectively resulting in complete oxidation of ABTS2− to ABTS˙− (96% of the 17.9 μM observed for control). It was therefore unsurprising that the third 10 μM H2O2 aliquot produced no change in radical absorbance. Subsequent addition of 50 mM EtOH and 5 μM Ru1 caused the ABTS˙− concentration to decrease rapidly, affording quantitative ABTS˙− reduction in less than 30 min.
To confirm that the results obtained with Ru1 arose from its ability to catalyze ABTS˙− reduction, rather than inhibiting HRP or another chemical reaction, two 10 μM aliquots of chemically synthesized ABTS˙− were introduced after the ABTS˙− concentration had decreased to zero following the initial addition of 10 μM H2O2 (Fig. 7C, red line). The first and second ABTS˙− aliquots produced immediate increases in ABTS˙− concentration corresponding to 6.3 and 6.7 μM, respectively, followed by rapid decreases in radical absorbance that resulted in quantitative radical reduction within 75 s of aliquot addition. With the Trolox experiment, however, the first and second ABTS˙− aliquots caused [ABTS˙−] to increase by 9.9 and 9.7 μM, respectively, with no observable changes in radical absorbance other than thermal decay (Fig. 7C, blue line). Addition of 50 mM EtOH and 5 μM Ru1 after the second ABTS˙− aliquot afforded complete ABTS˙− reduction within 30 min, similar to the experiment with multiple H2O2 aliquots. Both AscH− and GSH inhibited the HRP-induced oxidation of ABTS2−, albeit for shorter periods of time and less effectively than Trolox. The introduction of additional H2O2 (Fig. S8 and S9†) or ABTS˙− aliquots (Fig. S10 and S11†) only afforded increases in ABTS˙− concentration that mirrored the behaviors observed for Trolox (i.e., Fig. 7B and C). Subsequent addition of 50 mM EtOH and 5 μM Ru1 to the AscH− and GSH solutions containing excess ABTS˙− produced quantitative radical reduction, consistent with the Trolox experiments.
Conclusions
An organoruthenium complex (Ru1) that was previously reported to catalyze the hydrogenation of unsaturated organic substrates, using the oxidation of i-PrOH to acetone to supply the needed H2, has now been shown to also catalyze the reduction of ABTS˙− to ABTS2− in buffered aqueous solution. By itself, Ru1 was unreactive towards ABTS˙− and the presence of a non-tertiary alcohol was essential for ABTS˙− reduction to occur. Replacing either the C–H or O–H group in a –CHOH– moiety with C–Me or O–Me, respectively, resulted in the loss of ABTS˙− reducing ability. Given the previously reported transfer hydrogenation activity of Ru1 and the fact that Ru1-catalyzed ABTS˙− reduction required a reactant comprising a –CHOH– moiety, we deduced that the reducing equivalents necessary for the 1e− reduction of ABTS˙− to ABTS2− were supplied by the oxidation of R1–CHOH–R2 to R1–C(O)–R2. Consistent with this deduction, a diverse array of biologically-relevant non-tertiary alcohols, including amino acids, sugars and citric acid cycle metabolites, were found to be suitable terminal reductants for the reduction of ABTS˙− catalyzed by Ru1. Furthermore, in conjunction with an alcohol terminal reductant, Ru1 was able to inhibit the oxidation of ABTS2− to ABTS˙− by H2O2 and horseradish peroxidase. In contrast, non-catalytic antioxidants (Trolox, ascorbate, glutathione), only offered limited protection that, once exhausted, had no impact on ABTS˙− formation or stability. Impressively, no ABTS˙− formation or other pro-oxidant effects by Ru1 were observed under any experimental conditions. The mechanism for the catalytic reduction of ABTS˙− by Ru1 and non-tertiary alcohols, along with the biological applications, will be detailed in subsequent reports.
Acknowledgements
We are grateful to A. Mangalum for preceding work and helpful insight into the synthesis of 2 and Ru1.Notes and references
- M. P. Murphy, Biochem. J., 2009, 417, 1–13 CrossRef CAS PubMed.
- M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS PubMed.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, FASEB J., 2003, 17, 1195–1214 CrossRef CAS PubMed.
- W. Maret, Antioxid. Redox Signaling, 2006, 8, 1419–1441 CrossRef CAS PubMed.
- R. Stocker and J. F. Keaney Jr, Physiol. Rev., 2004, 84, 1381–1478 CrossRef CAS PubMed.
- T. Fukai and M. Ushio-Fukai, Antioxid. Redox Signaling, 2011, 15, 1583–1606 CrossRef CAS PubMed.
- S. Miriyala, I. Spasojevic, A. Tovmasyan, D. Salvemini, Z. Vujaskovic, D. S. Clair and I. Batinic-Haberle, Biochim. Biophys. Acta, 2012, 1822, 794–814 CrossRef CAS PubMed.
- I. Batinic-Haberle, A. Tovmasyan, E. R. H. Roberts, Z. Vujaskovic, K. W. Leong and I. Spasojevic, Antioxid. Redox Signaling, 2014, 20, 2372–2415 CrossRef CAS PubMed.
- S. D. Amaral and B. P. Espósito, Biometals, 2008, 21, 425–432 CrossRef PubMed.
- M. K. Evans, A. Tovmasyan, I. Batinic-Haberle and G. R. Devi, Free Radical Biol. Med., 2014, 68, 302–314 CrossRef CAS PubMed.
- P. Chelikani, I. Fita and P. C. Loewen, Cell. Mol. Life Sci., 2004, 61, 192–208 CrossRef CAS PubMed.
- H.-P. Hersleth, U. Ryde, P. Rydberg, C. H. Görbitz and K. K. Andersson, J. Inorg. Biochem., 2006, 100, 460–476 CrossRef CAS PubMed.
- H. N. Kirkman and G. F. Gaetani, Trends Biochem. Sci., 2007, 32, 44–50 CrossRef CAS PubMed.
- J. Vlasits, C. Jakopitsch, M. Bernroitner, M. Zamocky, P. G. Furtmüller and C. Obinger, Arch. Biochem. Biophys., 2010, 500, 74–81 CrossRef CAS PubMed.
- R. Kubota, S. Imamura, T. Shimizu, S. Asayama and H. Kawakami, ACS Med. Chem. Lett., 2014, 5, 639–643 CrossRef CAS PubMed.
- M. A. Sharpe, R. Olloson, V. C. Stewart and J. B. Clark, Biochem. J., 2002, 366, 97–107 CrossRef CAS PubMed.
- M. Itoh, K.-i. Motoda, K. Shindo, T. Kamiusuki, H. Sakiyama, N. Matsumoto and H. Ōkawa, J. Chem. Soc., Dalton Trans., 1995, 3635–3641 RSC.
- Y. Naruta and K. Maruyama, J. Am. Chem. Soc., 1991, 113, 3595–3596 CrossRef CAS.
- R. A. Sheldon, Catal. Today, 2015, 247, 4–13 CrossRef CAS.
- B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed.
- Y. Seki, K. Oisaki and M. Kanai, Tetrahedron Lett., 2014, 55, 3738–3746 CrossRef CAS.
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC.
- M. J. Schultz and M. S. Sigman, Tetrahedron, 2006, 62, 8227–8241 CrossRef CAS.
- J. J. Soldevila-Barreda, I. Romero-Canelón, A. Habtemariam and P. J. Sadler, Nat. Commun., 2015, 6, 6582 CrossRef CAS PubMed.
- Y. Fu, M. J. Romero, A. Habtemariam, M. E. Snowden, L. Song, G. J. Clarkson, B. Qamar, A. M. Pizarro, P. R. Unwin and P. J. Sadler, Chem. Sci., 2012, 3, 2485–2494 RSC.
- J. J. Soldevila-Barreda and P. J. Sadler, Curr. Opin. Chem. Biol., 2015, 25, 172–183 CrossRef CAS PubMed.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef CAS PubMed.
- A. Mangalum, C. D. McMillen and A. G. Tennyson, Inorg. Chim. Acta, 2015, 426, 29–38 CrossRef CAS.
- S. P. Fricker, E. Slade, N. A. Powell, O. J. Vaughan, G. R. Henderson, B. A. Murrer, I. L. Megson, S. K. Bisland and F. W. Flitney, Br. J. Pharmacol., 1997, 122, 1441–1449 CrossRef CAS PubMed.
- B. R. Cameron, M. C. Darkes, H. Yee, M. Olsen, S. P. Fricker, R. T. Skerlj, G. J. Bridger, N. A. Davies, M. T. Wilson, D. J. Rose and J. Zubieta, Inorg. Chem., 2003, 42, 1868–1876 CrossRef CAS PubMed.
- A. Mangalum, Y. Htet, D. A. Roe, C. D. McMillen and A. G. Tennyson, Inorg. Chim. Acta, 2015, 435, 320–326 CrossRef CAS.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed.
- U. Jungwirth, C. R. Kowol, B. K. Keppler, C. G. Hartinger, W. Berger and P. Heffeter, Antioxid. Redox Signaling, 2011, 15, 1085–1127 CrossRef CAS PubMed.
- B. H. J. Bielski and A. O. Allen, J. Phys. Chem., 1977, 81, 1048–1050 CrossRef CAS.
- K. Yusa and K. Shikama, Biochemistry, 1987, 26, 6684–6688 CrossRef CAS PubMed.
- D. Huang, B. Ou and R. L. Prior, J. Agric. Food Chem., 2005, 53, 1841–1856 CrossRef CAS PubMed.
- M. Karbarz and J. Malyszko, Electroanalysis, 2008, 20, 1884–1890 CrossRef CAS.
- L. Pitulice, I. Pastor, E. Vilaseca, S. Madurga, A. Isvoran, M. Cascante and F. Mas, Biocatal. Biotransform., 2013, 2, 1–5 Search PubMed.
- C. Creutz, Inorg. Chem., 1981, 20, 4449–4452 CrossRef CAS.
- J. M. Pullar, M. C. M. Vissers and C. C. Winterbourn, J. Biol. Chem., 2001, 276, 22120–22125 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Chem. Res. Toxicol., 2009, 22, 35–40 CrossRef CAS PubMed.
- P. Sathyadevi, P. Krishnamoorthy, N. S. P. Bhuvanesh, P. Kalaiselvi, V. V. Padma and N. Dharmaraj, Eur. J. Med. Chem., 2012, 55, 420–431 CrossRef CAS PubMed.
- Y. Liu, X. Zhang, R. Zhang, T. Chen, Y.-S. Wong, J. Liu and W.-J. Zheng, Eur. J. Inorg. Chem., 2011, 1974–1980 CrossRef CAS.
- J. M. Alfaro, A. Prades, M. del Carmen Ramos, E. Peris, J. Ripoll-Gómez, M. Poyatos and J. S. Burgos, Zebrafish, 2010, 7, 13–21 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc00651e |
| ‡ In PBS, an ABTS˙− concentration of 6.7 μM would give rise to an absorbance value of 0.1. Achieving a similar value with O2˙− or H2O2 would require a concentration of 53 μM or 2.29 mM, respectively. |
| § The concentration of the successive ABTS˙− aliquots was selected based on the concentration of the initial 50 μM ABTS˙− that was degraded by the antioxidant. Because Ru1 degraded all of the initial 50 μM ABTS˙− and Trolox only degraded 10 μM, the concentration of the multiple sequential aliquots added was 50 μM with Ru1 and 10 μM with Trolox. |
| This journal is © The Royal Society of Chemistry 2016 |