
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen light-induced apoptosis in cancer cells by a tetrapyridyl ruthenium prodrug offering two trans coordination sites†
V. H. S.
van Rixel
a,
B.
Siewert
a,
S. L.
Hopkins
a,
S. H. C.
Askes
a,
A.
Busemann
a,
M. A.
Siegler
b and
Sylvestre
Bonnet
 *a
*a
aLeiden Institute of Chemistry, Universiteit Leiden, Einsteinweg 55 2333 CC, Leiden, Netherlands. E-mail: bonnet@chem.leidenuniv.nl
bSmall Molecule X-ray Crystallography Facility, Johns Hopkins University, 3400N. Charles St, Baltimore, MD 21218, USA
First published on 25th April 2016
Abstract
In this work, two new photopharmacological ruthenium prodrugs are described that can be activated by green light. They are based on the tetrapyridyl biqbpy ligand (6,6′-bis[N-(isoquinolyl)-1-amino]-2,2′-bipyridine), which coordinates to the basal plane of the metal centre and leaves two trans coordination sites for the binding of monodentate sulphur ligands. Due to the distortion of the coordination sphere these trans ligands are photosubstituted by water upon green light irradiation. In vitro cytotoxicity data on A431 and A549 cancer cell lines shows an up to 22-fold increase in cytotoxicity after green light irradiation (520 nm, 75 J cm−2), compared to the dark control. Optical microscopy cell imaging and flow cytometry indicate that the cancer cells die via apoptosis. Meanwhile, very low singlet oxygen quantum yields (∼1–2%) and cell-free DNA binding studies conclude that light-induced cell death is not caused by a photodynamic effect, but instead by the changes induced in the coordination sphere of the metal by light, which modifies how the metal complexes bind to biomolecules.
Introduction
Classical chemotherapy side effects are a burden for patients, limit treatment doses, and lower prognosis. Light-activated anti-cancer prodrugs have appeared as an alternate strategy to increase the selectivity of chemotherapeutic agents.1 Ideally, their inactive form should minimally interact with biological molecules to limit the toxicity of the prodrug to non-irradiated tissues. Upon in vivo light irradiation these prodrugs are locally activated to selectively kill tumour cells. Among light-activated compounds those based on ruthenium(II) have been extensively studied due to their superior light absorption properties and rich photoreactivity. The majority of light-activated ruthenium-based anticancer compounds described to date belong to the class of photodynamic therapeutic agents (PDT agents) that generate singlet oxygen (1O2) as a means to locally kill cancer cells.2 For example, clinical trials recently started with ruthenium-oligothiophene dyads TLD1411 and TLD1433, which are red-light activated, water-soluble, and resistant to photobleaching.2c A less common family of ruthenium compounds consisting of photoactivated chemotherapy agents (PACT agents), where visible light excitation (350–800 nm) leads to the cleavage of a protecting group. This irreversible photoreaction releases a toxic ligand,3 modifies part of it,4 or generates open coordination sites on the metal centre, which enables biological ligands to bind.5 In PACT, a light-induced modification of the interaction between the metal compound and biological molecules triggers cell death.3b,4a,5a,6 The major advantage of this mode of activation, compared to PDT, is that it does not depend on the presence of molecular oxygen, and hence may be applied to treat hypoxic tumour tissues, a type of tumour tissue characterised by low response to standard chemotherapy and faster cancer progression.7Many ruthenium PACT agents known to date contain two bidentate ligands based on the 2,2′-bipyridine scaffold.5a,8 After irradiation, bis-aqua photoproducts are formed with a cis configuration that mimic the binding pattern of cisplatin to DNA.9 Transplatin, on the other hand, is not active in vivo and less cytotoxic than cisplatin in vitro, so that anticancer metallodrugs with a trans geometry, usually based on platinum(II), have not been considered until recently.10 New trans platinum(IV) compounds have also been prepared as PACT agents that can be activated with UVA (320–400 nm) or high-energy visible light (400–450 nm).5b,11 This type of light is, however, harmful to cells12 and penetrates biological tissues sub-optimally.13 We embarked on developing ruthenium-based PACT agents with a trans geometry that can be activated at higher wavelengths, i.e., closer to the phototherapeutic window.14
Here we report two trans-ruthenium-based PACT compounds that can be activated using green light. The two ruthenium complexes, [Ru(biqbpy)(dmso)Cl]Cl ([1]Cl, biqbpy = 6,6′-bis[N-(isoquinolyl)1-amino]-2,2′-bipyridine) and [Ru(biqbpy)(Amet)(HAmet)]PF6 ([2]PF6, HAmet = N-acetyl-L-methionine, Amet− = deprotonated N-acetyl-L-methionine, see Scheme 1), are based on a tetrapyridyl ligand (biqbpy) specifically developed to coordinate in the basal plane of octahedral metal complexes and to leave two trans positions for the coordination of monodentate ligands.15 In order to minimize interactions of the metal centre with biomolecules in the dark, sulphur-based monodentate ligands were selected, i.e., one dmso in [1]Cl, and two Amet− ligands in [2]PF6, which can be removed by visible light irradiation.16 The synthesis, photochemistry, and biological properties of these compounds are reported, which demonstrates that they can trigger apoptosis in human cancer cell lines upon green light irradiation.
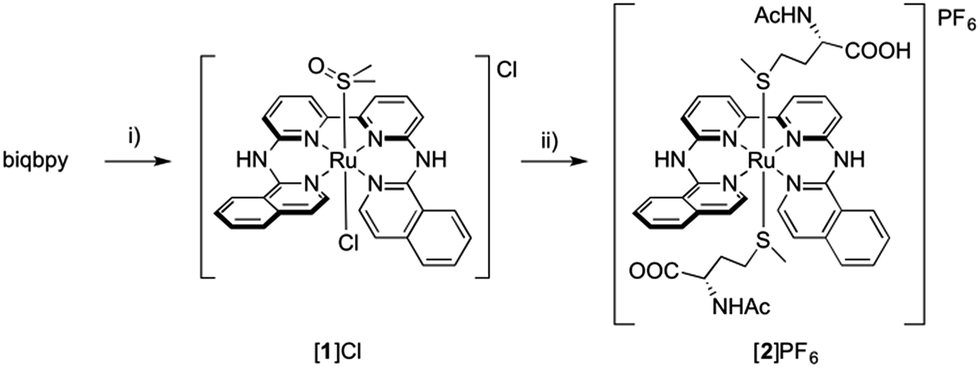 | ||
| Scheme 1 Synthesis of [1]Cl and [2]PF6. Conditions: (i) 1.1 eq. [Ru(DMSO)4Cl2], 80 °C, in EtOH under argon, 16 h, yield 43%; (ii) 20 eq. HAmet, 80 °C, in water under argon, 16 h, yield, 43%. | ||
Results and discussion
Synthesis and characterization
Complex [1]Cl was synthesized by reacting biqbpy with 1.1 equivalents of [Ru(dmso)4Cl2] in ethanol overnight at 80 °C (Scheme 1). After filtration [1]Cl was obtained as a red brown powder. Slow vapour diffusion of a methanol solution containing [1]Cl into ethyl acetate gave ruby-coloured crystals suited for X-ray diffraction (Fig. 1). In the structure of ([1]Cl·MeOH) the ligand biqbpy is coordinated to ruthenium(II) in a highly distorted fashion with an N1–N3–N4–N6 torsion angle of 12.78°. The difference between the bond angle N1–Ru1–N6 = 97.80° at the open-ended site of the complex and the angle N4–Ru1–N3 = 80.78° at the bpy site highlights the distortion of the coordination octahedron. Strain is caused by the repulsion between the hydrogen atoms borne by C1 and C28, and forces [1]+ to assume a helical, thus chiral configuration. The crystal structure of ([1]Cl·MeOH) is a racemate containing both the right-handed (P) and left-handed (M) helices.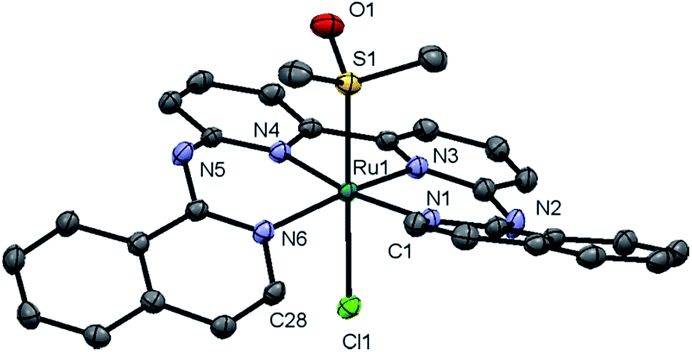 | ||
| Fig. 1 Displacement ellipsoid of cationic M-[1]+ (50% probability level) as observed in the crystal structure of ([1]Cl·MeOH)2. Chloride counter-anions, H atoms, lattice MeOH, and disorder, have been omitted for clarity. | ||
Reacting [1]Cl with 20 equivalents of HAmet in water overnight at 80 °C was required to substitute both trans ligands by the monodentate thioethers (Scheme 1). Anion exchange to the PF6 salt increased the lipophilicity of [2]+ allowing extraction of the compound using ethyl acetate. Purification using size exclusion chromatography resulted in analytically pure [2]PF6. Coordination of two N-acetylmethionine (Amet) ligands was confirmed using high-resolution mass spectrometry (HRMS), NMR, and elemental analysis (see ESI†).
Dark stability
Testing the dark stability of anticancer metallodrugs in conditions relevant for biological testing is critical for interpreting uptake and cytotoxicity data. Stability assays were thus performed in the dark in aqueous and DMSO solutions. Like for cisplatin the dark stability of [1]Cl in aqueous solution depends on chloride concentration. According to 1H NMR (Fig. S3†) and mass spectrometry upon dissolution in deionized water or D2O the chloride ligand of [1]+ was immediately hydrolysed to afford [Ru(biqbpy)(dmso)(H2O)]2+ ([1a]2+, see Scheme 2). Upon adding chlorides the concentration of [1]+ increased, to reach a ratio [1]2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[1a]+ in solution of 1:3 at 0.15 M of NaCl. By contrast, a DMSO solution of compound [1]Cl was stable in the dark at −20 °C for at least six months (Fig. S5†), which allowed storage in stock solutions for all biological studies. Dissolving [1]Cl in DMSO first, and adding in a second step a physiological relevant NaCl aqueous solution (0.11 M), led to a 2:3 mixture of [1]+:[1a]2+ (Fig. S7†). Whether [1]Cl was in aqueous or DMSO solutions, the Ru–S bond with the dmso ligand remained stable in the dark at 298 K. The dark behaviour of [2]PF6 was quite different. Although the protonation of one HAmet ligand in the solid state is corroborated by elemental analysis, in aqueous solution at neutral pH the complex is deprotonated into the neutral species [2a] (Scheme 2). In D2O, this species remained stable in the dark for 6 weeks (Fig. S4†). In pure DMSO, however, [2a] degraded over 16 h, also at −20 °C (Fig. S6†). Thus, DMSO-containing stock solutions of [2]PF6 were freshly prepared, or, the compound being soluble in water, DMSO should simply be avoided. Overall, in aqueous solution [2]PF6 appears as a “protected” version of [1]Cl, since the hydrolysable Ru–Cl bond of [1]Cl was replaced by thermally stable Ru–S bonds.
:[1a]+ in solution of 1:3 at 0.15 M of NaCl. By contrast, a DMSO solution of compound [1]Cl was stable in the dark at −20 °C for at least six months (Fig. S5†), which allowed storage in stock solutions for all biological studies. Dissolving [1]Cl in DMSO first, and adding in a second step a physiological relevant NaCl aqueous solution (0.11 M), led to a 2:3 mixture of [1]+:[1a]2+ (Fig. S7†). Whether [1]Cl was in aqueous or DMSO solutions, the Ru–S bond with the dmso ligand remained stable in the dark at 298 K. The dark behaviour of [2]PF6 was quite different. Although the protonation of one HAmet ligand in the solid state is corroborated by elemental analysis, in aqueous solution at neutral pH the complex is deprotonated into the neutral species [2a] (Scheme 2). In D2O, this species remained stable in the dark for 6 weeks (Fig. S4†). In pure DMSO, however, [2a] degraded over 16 h, also at −20 °C (Fig. S6†). Thus, DMSO-containing stock solutions of [2]PF6 were freshly prepared, or, the compound being soluble in water, DMSO should simply be avoided. Overall, in aqueous solution [2]PF6 appears as a “protected” version of [1]Cl, since the hydrolysable Ru–Cl bond of [1]Cl was replaced by thermally stable Ru–S bonds.
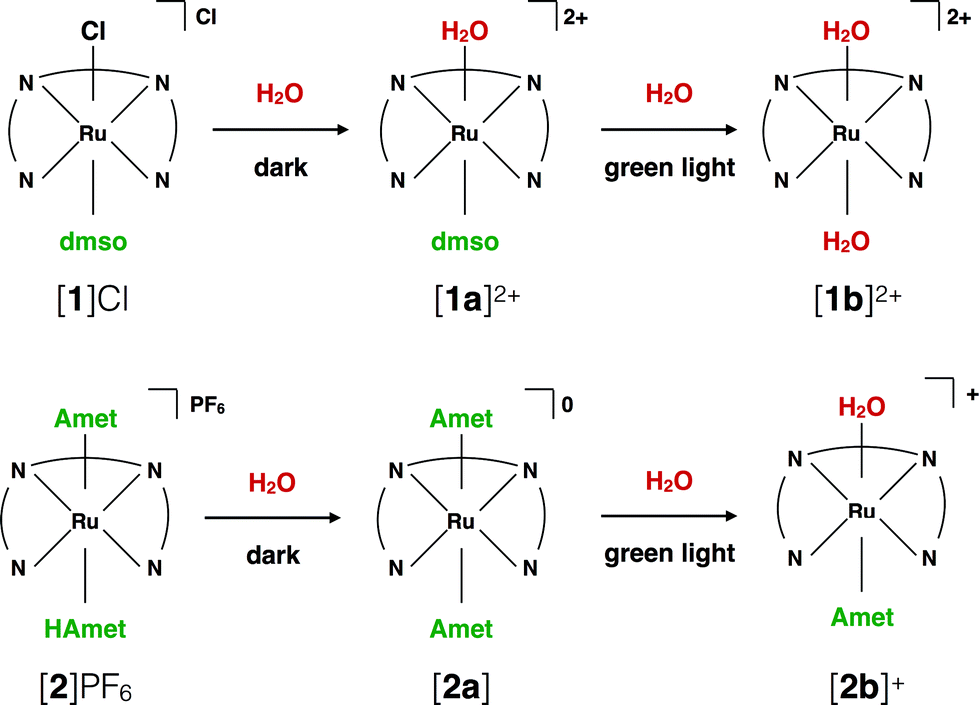 | ||
| Scheme 2 Ligand exchange processes upon dilution of [1]Cl and [2]PF6 in aqueous solutions, and upon green light irradiation. | ||
Photoreactivity of [1]Cl and [2]PF6
Under green light irradiation (λirr = 520 nm) and under argon a solution of [1]Cl in water, which mostly contains [1a]2+, resulted in a shift of the absorption maximum from 305 nm to 320 nm, and a slight increase of the absorbance in the visible region (Fig. 2a). Mass spectrometry after light irradiation showed new peaks at m/z = 288.7 corresponding to [Ru(biqbpy)(H2O)2]2+ ([1b]2+ in Scheme 2, calc. m/z = 288.8). Thus, the dmso ligand was photosubstituted by water (Scheme 2). This reactivity is typical of geometrically distorted ruthenium(II) compounds that possess low-lying triplet metal-centred (3MC) excited states with a strongly dissociative character.171H NMR confirmed this analysis, as a new resonance at 2.72 ppm, characteristic of free dmso, appeared after green light irradiation, but not in the dark (Fig. S16†). Similar evolutions were observed under blue light irradiation (450 nm, Fig. S10 and S11†), which also allowed measuring a photosubstitution quantum yield (ΦPS) of 0.3% (see ESI†). Overall, cleavage of the Ru–S bond of [1]+ is a photochemical process, and compound [1]Cl can be seen as a semi-protected light-activated prodrug. One of the two trans ligands is thermally labile in water, while the other is only labile under visible light irradiation.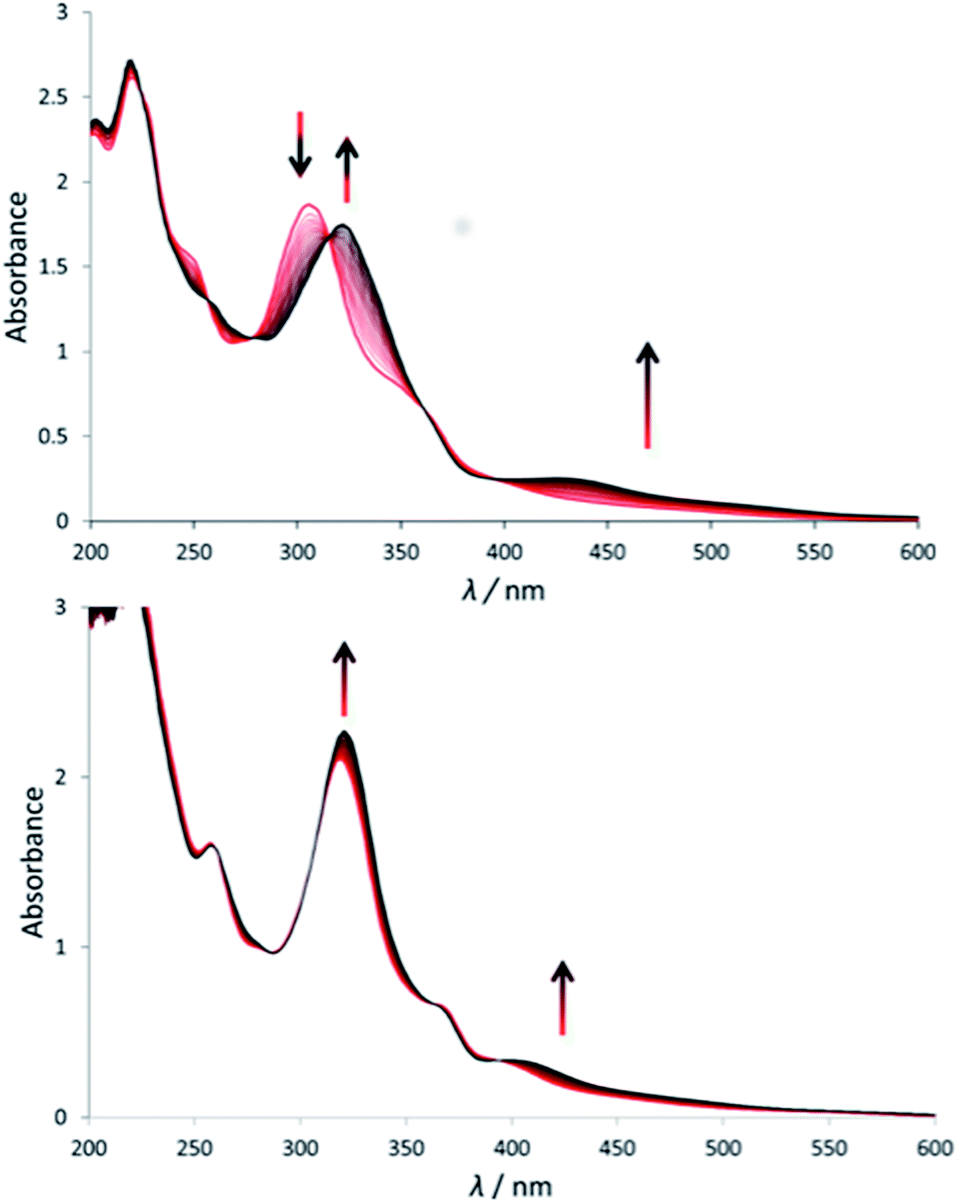 | ||
| Fig. 2 Evolution of the electronic absorption spectra of a solution of [1]Cl (top) and [2](PF6) (bottom) in demi-water upon green light irradiation under argon (λ = 530 nm, Δλ1/2 = 25 nm, 3.02 mW, 2.1 × 10−8 einstein per s). Time: 0 min (red curve) to 120 min (black curve, a) or 160 min (black curve, b). Conditions [Ru]0 = 7.5 × 10−5 (a), 7.8 × 10−5 (b), irradiated volume was 3.0 mL at 298 K. | ||
For [2]PF6, green light irradiation in aqueous solution under argon (Fig. 2b) was accompanied by increased intensity of the metal-to-ligand charge transfer (MLCT) absorption band near 400 nm and of the transition near 325 nm, and several isosbestic points. Mass spectrometry gave a clearer indication about the photoreaction occurring in such conditions. The initial peak at m/z = 923.4 characteristic for [2]+ (calc. m/z = 923.2) was gradually replaced by a signal at m/z = 732.4 characteristic for [Ru(biqbpy)(Amet)]+ (calc. m/z = 732.1), which showed the formation of [Ru(biqbpy)(Amet)(OH2)]+, [2b]+. A signal at m/z = 605.1 for [Ru(biqbpy)(MeOH)(OMe)]+ (calc. 605.1) or m/z = 386.6 for [Ru(biqbpy)(CH3CN)2]2+ (calc. 387.1) could only be obtained under extensive blue light irradiation (450 nm, see Fig. S9, S12, and S13;† MeOH and CH3CN were solvents used for MS and HPLC, respectively). Irradiation with high-energy visible light was hence necessary to form the bis-aqua complex [1b]2+ from [2b]+ (Scheme 2). In our conditions the formation of [1b]2+ under green light irradiation was too slow to be observed. This result was confirmed by 1H-NMR (Fig. S17†), as only one ligand was photosubstituted by water under green light irradiation at a dose of 75 J cm−2. In conclusion, complex [2]PF6 is a water-soluble, fully protected complex: both trans N-acetyl-L-methionine ligands remain coordinated to the metal in the dark, while one of them is cleaved off by green light irradiation, and the second one is removed by high doses of blue light.
(Photo)cytotoxicity studies
The cytotoxicity of compounds [1]Cl and [2]PF6 was investigated against three cell lines, i.e., A549 (human adenocarcinoma human alveolar basal epithelial cells), A431 (human epidermoid carcinoma cells), and MRC-5 (noncancerous human foetal lung fibroblasts). The effective concentrations (EC50), defined as the compound concentration that reduces cell viability by 50% compared to untreated wells, were measured, in the dark and after light activation, following a protocol described in detail in Hopkins et al.12a These studies aimed at establishing whether the photosubstitution reactions observed in a chemical environment may translate into in vitro light activation. Although both blue and green light resulted in photosubstitution, green light (520 nm) was chosen for the photocytotoxicity tests because it is much less toxic to human cell lines than blue light12a and it penetrates further into biological tissues. Preliminary studies in a 96-well plate (Fig. S14 and S15†) demonstrated that in the conditions of our cell-irradiation setup (21 mW cm−2) a 60 min irradiation time, corresponding to a dose of 75 J cm−2, was necessary to activate 0.8–1.6 nmol of the compounds (the maximum amount present in each well for concentrations of 40–80 μM). The EC50 of complexes [1]Cl, [2]PF6, and cisplatin, against A431, A549, and MRC-5 cell lines, measured in the dark and after green light irradiation, are reported in Table 1.| Cell line | t incubation (h) | Light dose (J cm−2) | [1]Cl | [2]PF6 | Cisplatin | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (μM) | ±CI (95%) | PI | EC50 (μM) | ±CI (95%) | PI | EC50 (μM) | ±CI (95%) | PI | |||
| a “Light” = green light irradiation (520 nm, 60 min, 75 J cm−2). b Incubation time is the time the Ru complex is incubated (37 °C, 7% CO2) with cells in the dark before light irradiation. c n.d. stands for not determined. | |||||||||||
| A431 | 6 | 0 | 13.0 | 1.30 | 22 | 38.0 | 8.8 | 4.9 | 4.3 | 1.5 | 0.9 |
| 75 | 0.60 | 0.05 | 7.80 | 1.0 | 4.6 | 1.5 | |||||
| 24 | 0 | 10.0 | 0.59 | 11 | 30.0 | 4.3 | 2.1 | 4.8 | 1.6 | 1.0 | |
| 75 | 0.88 | 0.24 | 14.0 | 1.1 | 4.9 | 1.6 | |||||
| A549 | 6 | 0 | 9.30 | 2.30 | 16 | 20.0 | 6.1 | 5.6 | 3.3 | 0.55 | 1.0 |
| 75 | 0.58 | 0.08 | 3.60 | 1.0 | 3.3 | 0.54 | |||||
| 24 | 0 | 6.20 | 0.86 | 9.5 | 11.0 | 1.0 | 2.2 | 3.1 | 0.55 | 0.8 | |
| 75 | 0.65 | 0.0 | 5.00 | 0.4 | 3.6 | 0.77 | |||||
| MRC-5 | 6 | 0 | 13.0 | 1.3 | 8.1 | 8.50 | 3.5 | >8.5 | 3.8 | 1.5 | n.dc |
| 75 | 1.60 | 2.4 | <1 | n.d.c | |||||||
| 24 | 0 | 8.30 | 1.0 | 4.9 | 18.3 | 1.4 | >18 | 6.9 | 1.2 | n.dc | |
| 75 | 1.70 | 2.3 | <1 | n.dc | |||||||
In the dark, the EC50 values of ∼10 and ∼35 μM were observed for [1]Cl and [2]PF6, respectively in A431 cells (Table 1). For the A549 cell line similar trends were observed with EC50 values of 6–9 μM for [1]Cl and 11–20 μM for [2]PF6. Thus, [1]Cl has similar cytotoxicity in the dark as cisplatin (Table S1†), whereas the two thioether ligands in [2]PF6 decreased the cytotoxicity by a factor of two to four compared to [1]Cl. This result suggested that coordination of the sulphur ligands may slow down or diminish the cellular response to these ruthenium compounds.
Although an identical dose of green light did not induce photocytotoxicity by itself (Fig. S23†), nor modify the cytotoxicity of cisplatin (Fig. S24 and S25,†Table 1), a dramatically decreased cell population was observed when the cells were incubated with compound [1]Cl or [2]PF6 for 6 h or 24 h, and then irradiated with 75 J cm−2 of green light (Table 1, Fig. 4 and S26–28†). For complex [1]Cl, EC50 values close to 1 μM or lower were observed for all cell lines independently of when irradiation was performed. For A549 cells treated with complex [2]PF6, the EC50 decreased from 20 μM to 3.6 μM when irradiation occurred 6 h after treatment, and from 11 μM to 5 μM when it was done 24 h after treatment. Similar trends were observed for A431 cells. After green light irradiation, complex [2]PF6 showed cytotoxicity comparable to the dark toxicity of [1]Cl, although compound [2]PF6 was less toxic in the dark than [1]Cl. For both compounds, the photo index (PI) increased when irradiation occurred 6 h after treatment, compared to 24 h. This effect was mostly a consequence of lower EC50 values in the dark after 24 h incubation, which suggested a higher degree of thermal activation with longer dark incubation times. Overall, these results suggest that the sulphur ligands of [1]Cl (dmso) and of [2]PF6 (Amet−) partially inhibit the cytotoxicity of the ruthenium centre in the dark, and that ligand photosubstitution is accompanied by an increase of the cytotoxicity of the compound. In other words, selectivity was obtained by light irradiation.
Singlet oxygen production
Due to the long-lived triplet excited states of many photostable ruthenium polypyridyl complexes, singlet oxygen (1O2) generation is often a dominant pathway upon light irradiation.1e,18 In fact, promising photodynamic therapeutic agents also include ruthenium-based sensitizers.2c,19 However, photosubstitution reactions observed with distorted ruthenium(II) complexes often lead to quenching of their long-lived 3MLCT states by nearby 3MC excited states, which lowers the quantum yields of phosphorescence and 1O2 generation. These trends represent a unique opportunity for PACT, as the hypoxic conditions in many tumour tissues, requires new oxygen-independent photoactivation strategies. In order to test whether compounds [1]Cl and [2]PF6 would qualify better as PDT or as PACT agents their quantum yields of 1O2 generation (Φ1O2) were measured under 450 nm excitation by direct detection of the 1274 nm infrared emission of 1O2 in CD3OD. The prototypical [Ru(bpy)3]Cl2 complex was used as a reference (Φref = 73%).20Φ1O2 values of 1.3% and 2.3% were found for [1]Cl and [2]PF6, respectively (Table S1 and Fig. S19†). According to these results, both [1]Cl and [2]PF6 are extremely poor 1O2 generators, and the photoactivation observed in vitro is not a PDT effect.Light-induced apoptosis
To investigate which type of cell death occurred, the morphology of A549 cells was inspected in the dark and after green light irradiation using bright field microscopy (Fig. 3, S29 and 30†). Directly after irradiation (520 nm, 75 J cm−2), cells treated with [1]Cl (1.5 μM) displayed cell shrinkage, loss of cell–cell contact, and membrane blebbing as depicted in Fig. 3b. An enhanced effect was detected when the cells were incubated for an additional 24 h after light irradiation (Fig. 3d). The changes in cell morphology are characteristic for apoptotic cell death.21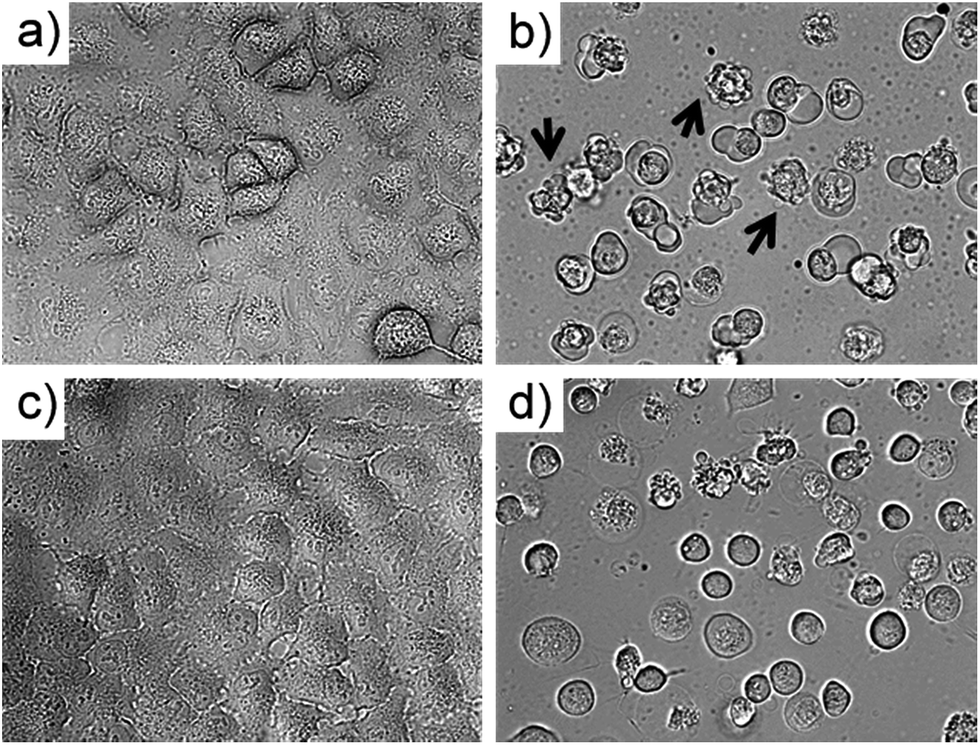 | ||
| Fig. 3 Bright field microscopy images (40× magnification) of A549 cells treated with [1]Cl (1.5 μM) for 6 + 1 h in the dark (a) and 6 h in the dark followed by 1 h green light irradiation ((b) 520 nm, 75 J cm−2). Images (c) and (d) show sample (a) and (b) after an additional 24 h incubation in the dark. Arrows in (b) show examples of membrane-blebbing, which is characteristic for early apoptosis. | ||
To confirm that a majority of the A549 cells treated with [1]Cl or [2]PF6 and irradiated with green light died by apoptosis, their fate was investigated using the Annexin V–propidium iodide assay and analysed using flow cytometry (FC).22Fig. 5 shows representative density plots of non-irradiated A549 cells treated with [1]Cl (1.5 μM, Fig. 4a) or [2]PF6 (10 μM, Fig. 4c). The majority of the cells are in the lower left quadrant, i.e., alive (see also Fig. S31†). However, upon green light irradiation (1 h, 75 J cm−2) a clear shift of the cell population to the bottom right quadrant indicates, for both [1]Cl and [2]PF6, Annexin V binding, thus apoptotic cells.
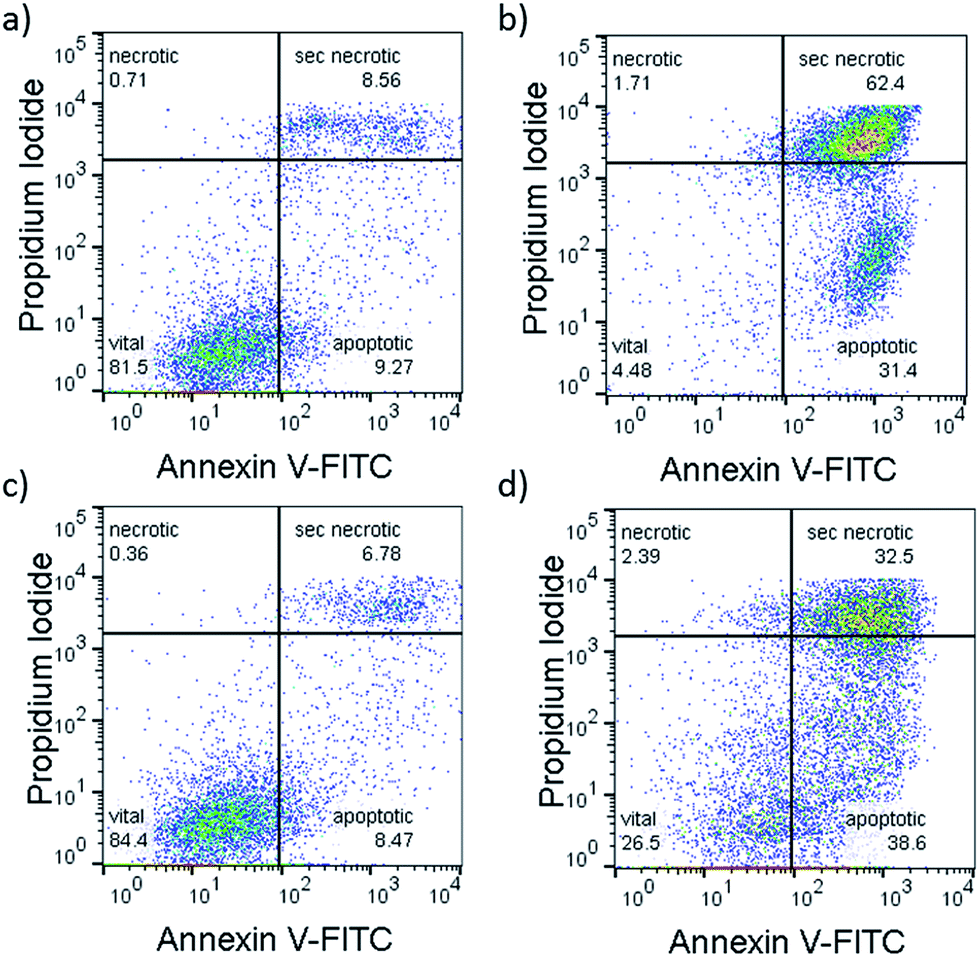 | ||
| Fig. 4 Representative flow cytometry density plots (Annexin V-FITC (525 nm)/propidium iodide (670 nm) of A549 cells incubated with [1]Cl (1.5 μM) in the dark for 6 + 1 + 24 h (a), or in the dark for 6 h, followed by irradiation with green light for 1 h, followed by 24 h incubation (b) or treated with [2]PF6 (10 μM) and left in the dark for 31 h (c) or irradiated 1 h with green light 6 h after treatment and further incubated for 24 h in the dark (d). Irradiation conditions: 520 nm, 60 min, 75 J cm−2. Quantification: see Fig. S20.† | ||
The lack of cells in the top left quadrant indicates the absence of purely necrotic cells. Cells in the top right are commonly referred to as “secondary necrotic”, and are a known artefact in in vitro assays.
According to the flow cytometry data, the photocytoxicity of [1]Cl and [2]PF6 occurs via apoptosis without any sign of necrosis. In addition, confocal microscopy of A549 cells stained with tetramethylrhodamine ethyl ester (mitochondria) and DRAQ5 (nuclear DNA) showed that light irradiation diminished the mitochondrial membrane potential and induced chromosomal condensation, especially for [1]Cl (Fig. S31†). All of the tested cellular responses clearly demonstrate that compounds [1]Cl and [2]PF6 belong to a rare sub-family of metallodrugs that can trigger apoptosis with green light.23
Intracellular distribution and uptake
In order to gather information on the intracellular localisation of [1]Cl and [2]PF6, and to investigate whether the difference in cytotoxicity between [1]Cl and [2]PF6 in the dark was due to differences in cell-uptake and/or of intracellular distribution, cell fractionation was performed. For this experiment, A549 cells were incubated with [1]Cl or [2]PF6 for 6 h in the dark at concentrations corresponding to the EC50 value. The cells were then harvested, the cytosol, membrane, nuclei, and cytoskeleton fractions were separated (see ESI†), and the ruthenium concentration in each fraction was measured by ICP-MS (Fig. S32†). The observed total uptake of [2]PF6 (7.5 ng per 106) was significantly lower compared to that of [1]Cl (16 ng/106 cells). As the effect of both treatments was identical (i.e., reducing the cell population by 50%), [2]PF6 seems to be more potent than [1]Cl, although larger EC50 values were found for [2]PF6. This result suggests that the dark cytotoxicity of [2]PF6 might be limited by a lower uptake. In terms of intracellular distribution both complexes were found in all fractions, with a slight ([2]PF6) to strong ([1]Cl) preference for the membrane fraction, and to a lesser extent in the nuclear fractions. The membrane fraction does not only contain the cell membrane but also mitochondria, endosomes, lysosomes, etc. These results are in agreement with contemporary literature suggesting an endocytosis-dependent uptake mechanism for polypyridyl metal complexes (thus high Ru content in endosomes and lysosomes), and accumulation of lipophilic cationic species in the mitochondrial membranes.24Cell-free DNA binding studies
Thermal and photoinduced DNA binding studies were performed to establish whether the photolabile sulphur-based ligands in [1]Cl and [2]PF6 were protecting the compounds from interaction with biomolecules. The pUC19 plasmid used for this study (2686 bp) exists in three forms: supercoiled (SC, most condensed form, migrates the fastest), single-nicked open circular (OC, relaxed form of the SC, migrates in between the SC and LD) and linear dimer (LD, largest form at 5372 bp, migrates the slowest). For both the thermal and photoinduced DNA binding studies, chloride-free phosphate buffer was used to model a pseudo intracellular environment. For the dark thermal binding experiments, [1]Cl and [2]PF6 were incubated at varied DNA base pair (BP) to metal complex (MC) ratios for 24 h (Fig. S33†). Both [1]Cl and [2]PF6 showed negligible binding (minimal change in migration of the OC or SC forms), even at the largest concentration of metal complex (5:1 BP:MC ratio). Cisplatin was included as a positive control (5:1 BP:MC ratio) and displayed typical DNA binding results as those observed in literature (Fig. S34†).25 In the dark, [1]Cl and [2]PF6 have a low affinity and negligible association with any of the forms of the plasmid DNA.
In a second experiment, the ruthenium complexes and cisplatin were photolysed (λirr = 520 nm) for different amounts of time (0–60 min) in the presence of pUC19 plasmid (Fig. 5). For these experiments, a 50:1 BP:MC ratio was used, which displayed insignificant dark thermal binding. However, following green light irradiation complex [1]Cl showed significant retardation of the SC form (Fig. 5a, lanes 5–9) compared to [2]PF6 (Fig. 5b, lanes 5–9), which itself showed slight changes in the OC and SC forms compared to the control. Additionally, a change in the intensity of the staining indicates that increased photoinduced binding of the metal complexes interferes with the intercalation of ethidium bromide. These studies clearly show that after light activation, [1]Cl interacts strongly with the pUC19 plasmid, whereas [2]PF6 interacts less but still significantly more than in the dark. Clearly, 1O2-based DNA cleavage was not observed under irradiation in presence of either ruthenium compound. Although these simple results neither allow to specify in detail the binding mode of [1]Cl and [2]PF6 to DNA, nor to say whether this interaction is relevant for cell death, they clearly demonstrate that the photosubstitution reactions occurring under green light irradiation critically changes the way these two ruthenium compounds interact with biomolecules.26
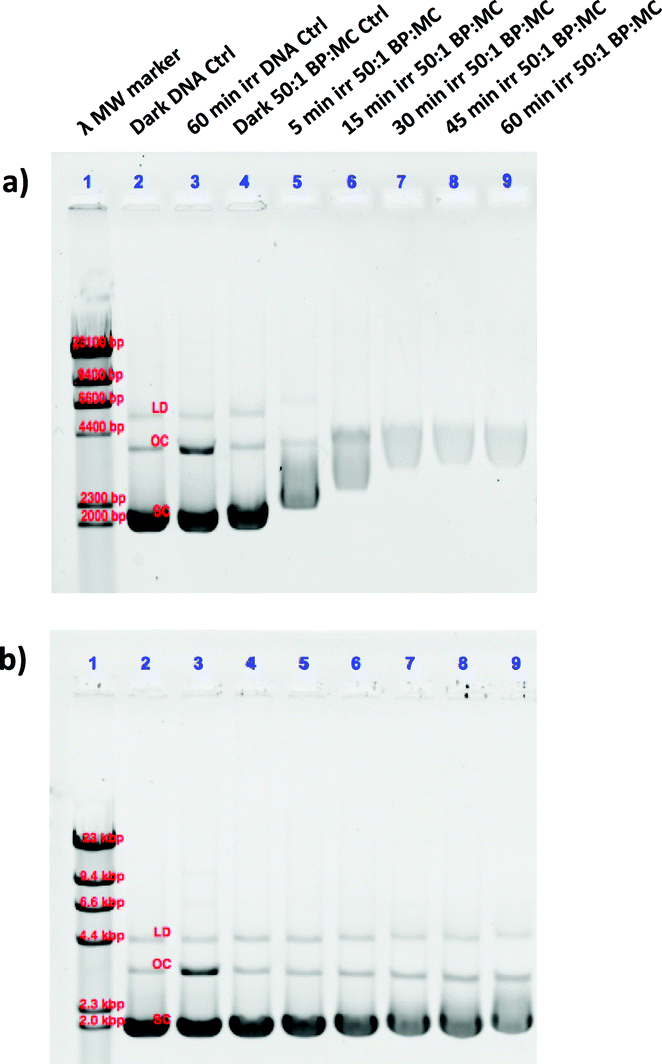 | ||
| Fig. 5 Photoinduced binding of [1]Cl (a) and [2]PF6 (b) to pUC19 plasmid DNA. The lanes correspond to (1) lambda DNA MW marker, (2) dark DNA only control, (3) irradiated DNA only control, (4) dark 50:1 BP:MC control, (5–9) 5, 15, 30, 45, and 60 min irradiated 50:1 BP:MC samples, respectively. The bands of the lambda MW marker correlate to 23, 9.4, 6.6, 4.4, 2.3, and 2.0 kpb. The dark DNA control bands are labelled according to the form, linear dimer (LD), open circular (OC), and supercoiled (SC). | ||
Conclusions
Complexes [1]Cl and [2]PF6 are the first light-activated trans ruthenium-based anticancer prodrugs. In the dark these water-soluble complexes are well taken up and display mild cytotoxicity to A431 and A549 cancer cells. However, upon green light irradiation, [1]Cl and [2]PF6 are activated resulting in highly cytotoxic therapeutics, with EC50 values below 1 μM and photo-indices of up to 22. Clearly the combination of these compounds and green light irradiation induces apoptosis, and the low singlet oxygen generation efficiency and the absence of DNA photocleavage conclude that cell death is not due to a photodynamic effect. The dose of light necessary to activate [1]Cl and [2]PF6in vitro (75 J cm−2) is somewhat higher compared to values published for other photoactivated ruthenium or trans-platinum complexes (typically 10 J cm−2). However, the green light used in this work (520 nm) is much less harmful to cells than the shorter wavelength (UV or blue light) reported previously,4a,5b,12a,c,19a,27 so that high doses do not represent per se a problem. Green light also penetrates deeper into the skin,28 which makes it more relevant for phototherapy.Overall, the data presented in this article suggests that the activation mechanism for this new type of trans-ruthenium polypyridyl complexes relies on ligand photosubstitution reactions. The ruthenium species [2a] bound to two sulphur protecting ligands is the least cytotoxic, followed by the two mono-protected species [1a]2+ and [2b]2+ bound to a single sulphur ligand, while the bis-aqua, fully deprotected species [1b]2+ shows the highest cytotoxicity. Although cell-free DNA studies showed clear photoinduced DNA-binding by [1]Cl and, to a lesser extent, by [2]PF6, DNA only represents one of the possible biological target(s) of these compounds, as they distribute in the whole cell. It will be necessary to follow for example chemical biological methods described by Hartinger et al.,29 to determine which interaction with which biomolecule is actually responsible for the green light-induced apoptosis observed with[1]Cl and [2]PF6.
Acknowledgements
The European Research Council is kindly acknowledged for a Starting Grant to S. B. This work was also supported by the Dutch Organisation for Scientific Research (NWO-CW) via a VIDI grant to S. B.. Prof. E. Bouwman is kindly acknowledged for support and input. The COST action CM1105 “Functional metal complexes that bind to biomolecules" is gratefully acknowledged for stimulating scientific discussion.References
- (a) N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 48, 10690–10701 RSC; (b) J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282–283, 110–126 CrossRef CAS PubMed; (c) S. Yano, S. Hirohara, M. Obata, Y. Hagiya, S.-i. Ogura, A. Ikeda, H. Kataoka, M. Tanaka and T. Joh, J. Photochem. Photobiol., C, 2011, 12, 46–67 CrossRef CAS; (d) D. Crespy, K. Landfester, U. S. Schubert and A. Schiller, Chem. Commun., 2010, 46, 6651–6662 RSC; (e) U. Schatzschneider, Eur. J. Inorg. Chem., 2010, 2010, 1451–1467 CrossRef; (f) W. A. Velema, W. SzymaŃski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- (a) H.-J. Yu, S.-M. Huang, L.-Y. Li, H.-N. Jia, H. Chao, Z.-W. Mao, J.-Z. Liu and L.-N. Ji, J. Inorg. Biochem., 2009, 103, 881–890 CrossRef CAS PubMed; (b) H. Huang, P. Zhang, B. Yu, C. Jin, L. Ji and H. Chao, Dalton Trans., 2015, 44, 17335–17345 RSC; (c) G. Shi, S. Monro, R. Hennigar, J. Colpitts, J. Fong, K. Kasimova, H. Yin, R. DeCoste, C. Spencer, L. Chamberlain, A. Mandel, L. Lilge and S. A. McFarland, Coord. Chem. Rev., 2015, 282–283, 127–138 CrossRef CAS; (d) J. Fong, K. Kasimova, Y. Arenas, P. Kaspler, S. Lazic, A. Mandel and L. Lilge, Photochem. Photobiol. Sci., 2015, 14, 2014–2023 RSC; (e) S. P. Foxon, M. A. H. Alamiry, M. G. Walker, A. J. H. M. Meijer, I. V. Sazanovich, J. A. Weinstein and J. A. Thomas, J. Phys. Chem. A, 2009, 113, 12754–12762 CrossRef CAS PubMed; (f) H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS PubMed; (g) M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC; (h) P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed; (i) C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem.–Eur. J., 2014, 20, 14421–14436 CrossRef CAS PubMed.
- (a) R. Sharma, J. D. Knoll, P. D. Martin, I. Podgorski, C. Turro and J. J. Kodanko, Inorg. Chem., 2014, 53, 3272–3274 CrossRef CAS PubMed; (b) M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2013, 135, 11274–11282 CrossRef CAS PubMed.
- (a) T. Joshi, V. Pierroz, C. Mari, L. Gemperle, S. Ferrari and G. Gasser, Angew. Chem., Int. Ed., 2014, 53, 2960–2963 CrossRef CAS PubMed; (b) A. Presa, R. F. Brissos, A. B. Caballero, I. Borilovic, L. Korrodi-Gregório, R. Pérez-Tomás, O. Roubeau and P. Gamez, Angew. Chem., Int. Ed., 2015, 54, 4561–4565 CrossRef CAS PubMed.
- (a) B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed; (b) N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905–8908 CrossRef CAS PubMed.
- (a) S. Bonnet, B. Limburg, J. D. Meeldijk, R. J. M. K. Gebbink and J. A. Killian, J. Am. Chem. Soc., 2010, 133, 252–261 CrossRef PubMed; (b) A.-C. Laemmel, J.-P. Collin and J.-P. Sauvage, Eur. J. Inorg. Chem., 1999, 1999, 383–386 CrossRef; (c) G. Ragazzon, I. Bratsos, E. Alessio, L. Salassa, A. Habtemariam, R. J. McQuitty, G. J. Clarkson and P. J. Sadler, Inorg. Chim. Acta, 2012, 393, 230–238 CrossRef CAS; (d) F. Barragan, P. Lopez-Senin, L. Salassa, S. Betanzos-Lara, A. Habtemariam, V. Moreno, P. J. Sadler and V. Marchan, J. Am. Chem. Soc., 2011, 133, 14098–14108 CrossRef CAS PubMed.
- J. P. Cosse and C. Michiels, Anti-Cancer Agents Med. Chem., 2008, 8, 790–797 CrossRef CAS PubMed.
- (a) A. N. Hidayatullah, E. Wachter, D. K. Heidary, S. Parkin and E. C. Glazer, Inorg. Chem., 2014, 53, 10030–10032 CrossRef CAS PubMed; (b) R. N. Garner, J. C. Gallucci, K. R. Dunbar and C. Turro, Inorg. Chem., 2011, 50, 9213–9215 CrossRef CAS PubMed; (c) O. Filevich and R. Etchenique, Photochem. Photobiol. Sci., 2013, 12, 1565–1570 RSC; (d) L. Salassa, C. Garino, G. Salassa, R. Gobetto and C. Nervi, J. Am. Chem. Soc., 2008, 130, 9590–9597 CrossRef CAS PubMed.
- (a) F. Arnesano, M. Losacco and G. Natile, Eur. J. Inorg. Chem., 2013, 2013, 2701–2711 CrossRef CAS; (b) P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652 CrossRef CAS PubMed.
- (a) U. Kalinowska-Lis, J. Ochocki and K. Matlawska-Wasowska, Coord. Chem. Rev., 2008, 252, 1328–1345 CrossRef CAS; (b) A. G. Quiroga, J. Inorg. Biochem., 2012, 114, 106–112 CrossRef CAS PubMed; (c) S. M. Aris and N. P. Farrell, Eur. J. Inorg. Chem., 2009, 2009, 1293–1302 CrossRef PubMed; (d) Y. Zhao, J. A. Woods, N. J. Farrer, K. S. Robinson, J. Pracharova, J. Kasparkova, O. Novakova, H. Li, L. Salassa, A. M. Pizarro, G. J. Clarkson, L. Song, V. Brabec and P. J. Sadler, Chem.–Eur. J., 2013, 19, 9578–9591 CrossRef CAS PubMed.
- F. S. Mackay, J. A. Woods, P. Heringová, J. Kašpárková, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed.
- (a) S. L. Hopkins, B. Siewert, S. H. C. Askes, P. Veldhuizen, R. Zwier, M. Heger and S. Bonnet, Photochem. Photobiol. Sci., 2016 10.1039/c1035pp00424a; (b) T. J. McMillan, E. Leatherman, A. Ridley, J. Shorrocks, S. E. Tobi and J. R. Whiteside, J. Pharm. Pharmacol., 2008, 60, 969–976 CrossRef CAS PubMed; (c) S. Wäldchen, J. Lehmann, T. Klein, S. van de Linde and M. Sauer, Sci. Rep., 2015, 5, 15348 CrossRef PubMed.
- S. L. Jacques, Phys. Med. Biol., 2013, 58, 5007–5008 CrossRef.
- S. Bonnet, Comments Inorg. Chem., 2015, 35, 179–213 CrossRef CAS.
- Z. Arcis-Castíllo, S. Zheng, M. A. Siegler, O. Roubeau, S. Bedoui and S. Bonnet, Chem.–Eur. J., 2011, 17, 14826–14836 CrossRef PubMed.
- (a) A. Bahreman, B. Limburg, M. A. Siegler, E. Bouwman and S. Bonnet, Inorg. Chem., 2013, 52, 9456–9469 CrossRef CAS PubMed; (b) R. E. Goldbach, I. Rodriguez-Garcia, J. H. van Lenthe, M. A. Siegler and S. Bonnet, Chem.–Eur. J., 2011, 17, 9924–9929 CrossRef CAS PubMed.
- P. C. Ford, Coord. Chem. Rev., 1982, 44, 61–82 CrossRef CAS.
- (a) O. J. Stacey and S. J. A. Pope, RSC Adv., 2013, 3, 25550–25564 RSC; (b) A. Ruggi, F. W. B. van Leeuwen and A. H. Velders, Coord. Chem. Rev., 2011, 255, 2542–2554 CrossRef CAS.
- (a) C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC; (b) R. Lincoln, L. Kohler, S. Monro, H. Yin, M. Stephenson, R. Zong, A. Chouai, C. Dorsey, R. Hennigar, R. P. Thummel and S. A. McFarland, J. Am. Chem. Soc., 2013, 135, 17161–17175 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- (a) Z. Darzynkiewicz, G. Juan, X. Li, W. Gorczyca, T. Murakami and F. Traganos, Cytometry, 1997, 27, 1–20 CrossRef CAS PubMed; (b) G. Melino and D. Vaux, Cell Death, Wiley, 2010 Search PubMed.
- M. van Engeland, L. J. W. Nieland, F. C. S. Ramaekers, B. Schutte and C. P. M. Reutelingsperger, Cytometry, 1998, 31, 1–9 CrossRef CAS PubMed.
- (a) M. Dickerson, Y. Sun, B. Howerton and E. C. Glazer, Inorg. Chem., 2014, 53, 10370–10377 CrossRef CAS PubMed; (b) L. He, Y. Huang, H. Zhu, G. Pang, W. Zheng, Y.-S. Wong and T. Chen, Adv. Funct. Mater., 2014, 24, 2754–2763 CrossRef CAS.
- (a) M. Groessl, O. Zava and P. J. Dyson, Metallomics, 2011, 3, 591–599 RSC; (b) S. M. Zeman and D. M. Crothers, in Drug-Nucleic Acid Interactions, Elsevier, 2001, vol. 340, pp. 51–68 Search PubMed.
- M. V. Babak, S. M. Meier, K. V. M. Huber, J. Reynisson, A. A. Legin, M. A. Jakupec, A. Roller, A. Stukalov, M. Gridling, K. L. Bennett, J. Colinge, W. Berger, P. J. Dyson, G. Superti-Furga, B. K. Keppler and C. G. Hartinger, Chem. Sci., 2015, 6, 2449–2456 RSC.
- B. Zhang, S. Seki, K. Akiyama, K. Tsutsui, T. Li and K. Nagao, Acta Med. Okayama, 1992, 46, 427–434 CAS.
- (a) G. M. Findlay, Lancet, 1928, 212, 1070–1073 CrossRef; (b) C. Kielbassa, L. Roza and B. Epe, Carcinogenesis, 1997, 18, 811–816 CrossRef CAS PubMed; (c) B. H. Mahmoud, C. L. Hexsel, I. H. Hamzavi and H. W. Lim, Photochem. Photobiol., 2008, 84, 450–462 CrossRef CAS PubMed; (d) C. Opländer, S. Hidding, F. B. Werners, M. Born, N. Pallua and C. V. Suschek, J. Photochem. Photobiol., B, 2011, 103, 118–125 CrossRef PubMed; (e) M. Frasconi, Z. Liu, J. Lei, Y. Wu, E. Strekalova, D. Malin, M. W. Ambrogio, X. Chen, Y. Y. Botros, V. L. Cryns, J.-P. Sauvage and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 11603–11613 CrossRef CAS PubMed.
- L. J. Steven, Phys. Med. Biol., 2013, 58, R37 CrossRef PubMed.
- M. V. Babak, S. M. Meier, K. V. M. Huber, J. Reynisson, A. A. Legin, M. A. Jakupec, A. Roller, A. Stukalov, M. Gridling, K. L. Bennett, J. Colinge, W. Berger, P. J. Dyson, G. Superti-Furga, B. K. Keppler and C. G. Hartinger, Chem. Sci., 2015, 6, 2449–2456 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1420778. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00167j |
| This journal is © The Royal Society of Chemistry 2016 |