
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Steric control of redox events in organo-uranium chemistry: synthesis and characterisation of U(V) oxo and nitrido complexes†
Nikolaos
Tsoureas
a,
Alexander F. R.
Kilpatrick
 b,
Christopher J.
Inman
a and
F. Geoffrey N.
Cloke
*a
b,
Christopher J.
Inman
a and
F. Geoffrey N.
Cloke
*a
aSchool of Life Sciences, Division of Chemistry, University of Sussex, Falmer, Brighton, BN1 9QJ, UK. E-mail: f.g.cloke@sussex.ac.uk
bChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, OX1 3TA, Oxford, UK
First published on 11th April 2016
Abstract
The synthesis and molecular structures of a U(V) neutral terminal oxo complex and a U(V) sodium uranium nitride contact ion pair are described. The synthesis of the former is achieved by the use of tBuNCO as a mild oxygen transfer reagent, whilst that of the latter is via the reduction of NaN3. Both mono-uranium complexes are stabilised by the presence of bulky silyl substituents on the ligand framework that facilitate a 2e− oxidation of a single U(III) centre. In contrast, when steric hindrance around the metal centre is reduced by the use of less bulky silyl groups, the products are di-uranium, U(IV) bridging oxo and (anionic) nitride complexes, resulting from 1e− oxidations of two U(III) centres. SQUID magnetometry supports the formal oxidation states of the reported complexes. Electrochemical studies show that the U(V) terminal oxo complex can be reduced and the [U(IV)O]− anion was accessed via reduction with K/Hg, and structurally characterised. Both the nitride complexes display complex electrochemical behaviour but each exhibits a quasi-reversible oxidation at ca. −1.6 V vs. Fc+/0.
Introduction
The study of well-defined molecular complexes of uranium is a thriving field of research,1 with significant current interest in the activation of small molecules and organic substrates by U(III) compounds,2 the stabilisation of low oxidation states (i.e. U(II)3) and also the study of higher oxidation state complexes featuring U⋯E (E = main group element) multiple bonds.4 Historically, complexes featuring U⋯O terminal bonds have been dominated by the ubiquitous uranyl moiety,5 partly due to its apparent chemical inertness (although recently disproved6) and its technological relevance to the nuclear cycle.7 In contrast, terminal mono-oxo complexes are much less common partly due to the increased nucleophilicity of the oxo ligand,4u,8 which leads to the formation of dimeric species,8a,9 and stabilisation of monomeric U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O complexes requires the use of bulky supporting ligands.4o,10,11,12 The relative rarity of uranium terminal oxo complexes is paralleled by the case of terminal nitride uranium complexes,13,14 and the majority of unsupported U⋯N bonds are stabilised in dimeric/polymeric structures.15 Indeed, until 2012 no stable, well-defined uranium terminal nitride complex was known,13 although UN triply bonded species had been spectroscopically identified in low temperature matrices,16 and in situ generation and involvement in C–H activation had been proposed and studied computationally.17
O complexes requires the use of bulky supporting ligands.4o,10,11,12 The relative rarity of uranium terminal oxo complexes is paralleled by the case of terminal nitride uranium complexes,13,14 and the majority of unsupported U⋯N bonds are stabilised in dimeric/polymeric structures.15 Indeed, until 2012 no stable, well-defined uranium terminal nitride complex was known,13 although UN triply bonded species had been spectroscopically identified in low temperature matrices,16 and in situ generation and involvement in C–H activation had been proposed and studied computationally.17
We have previously demonstrated the significance of the steric environment around the uranium centre in controlling the reductive coupling of CO2(ref. 18) and CO,19 promoted by U(III) mixed sandwich complexes of the general type [U{η8-C8H6-(1,4-SiR3)2}(η5-CpR′′)THF] (R = iPr (1), Me (2)). In particular, the reductive transformations (i.e. coupling, disproportionation, or reduction) of CO2 using the complexes [U{η8-C8H6-(1,4-SiMe3)2}(η5-CpMe4R′)THF] (A) can be largely controlled by varying the size of R′ (R′ = Me, Et, iPr, tBu).18a Unlike complexes of type A that exhibit a clear trend between the effect of steric environment and the outcome of the possible reductive transformations, when the analogous complexes in which the SiMe3 group had been replaced by the bulkier SiiPr3 group were reacted with CO2, either intractable reaction mixtures were obtained or the reductive disproportionation of CO2 was promoted exclusively.18b In order to better understand this observation, we envisaged that a study of the reactivity towards other heteroallenes (e.g. RNCO) as model substrates for CO2 might be informative.20
Results and discussion
Reaction of a brown-olive green C6D6 solution of [U{η8-C8H6-(1,4-SiiPr3)2}(η5-Cp*)THF] (1) with a slight excess (1.05–1.1 eq.) of tBuNCO under an Ar atmosphere resulted in an immediate colour change to brown red. 1H-NMR spectroscopy showed complete consumption of (1) and the formation of a new uranium species and free tBuNC (further confirmed by GC-MS of the trapped volatiles of the reaction mixture). The 29Si{1H}-NMR spectrum of the product displayed a single resonance at −73 ppm, shifted downfield from −129 ppm in (1) suggesting that a change in the formal oxidation state of the uranium centre of (III) to (V) had taken place, in accordance with the general trend observed by Evans et al.21 The mass spectrum was consistent with the formation of the U(V) terminal oxo complex {U[η8-C8H6(1,4-SiiPr3)2](η5-Cp*)O} (3), and was confirmed by X-ray crystallography (Fig. 1).
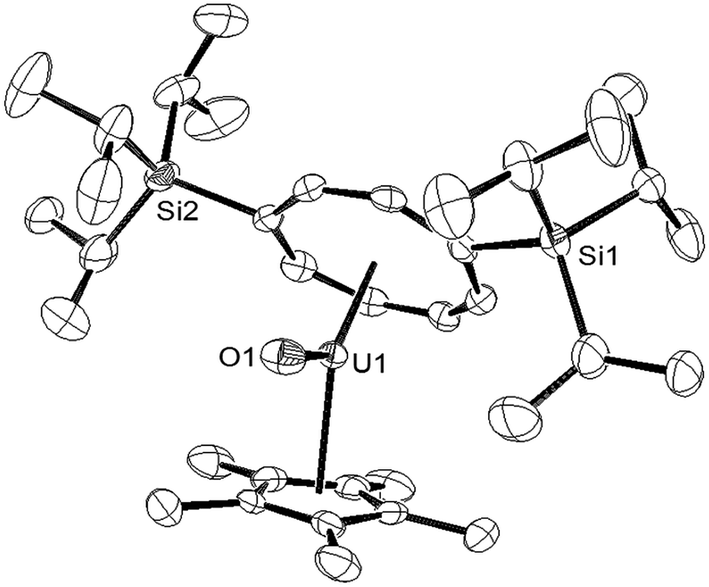 | ||
| Fig. 1 ORTEP-3 diagram of the molecular structure of (3) displaying 50% probability ellipsoids. H atoms have been omitted for clarity. Selected bond-lengths (Å) and angles (°): U1–O1 1.826(3), Ct(COT)–U 1.949(5), Ct(Cp*)–U1 2.492(1); Ct(COT)–U1–Ct(Cp*) 135.27(2), Ct(COT)–U1–O1 162.24(7), Ct(Cp*)–U1–O1 108.08(2). | ||
The U–O bond length in (3) (1.826(3) Å) is shorter than that found in the U(V) terminal oxo complex [Ph3PMe][U(O)(CH2SiMe2NR′)(NR′2)2] (1.847(2) Å),22 but similar within esd's to those in the U(V) complexes [U(O)(NR′2)3] (1.817(1) Å),10 [UTRENTIPS(O)] (1.856(6) Å, TRENTIPS = [N(CH2CH2NSiiPr3)3]3),11a [((RArO)tacn)U(O)] (1.848(8) Å; R = tBu, Ad; tacn = triazacyclononane),4o Cp*2U(O)(ODipp) (1.859(6) Å, Dipp = 2,6-iPr2-C6H3),23 [OU{OSi(OtBu)3}4K] (1.825(2) Å)12 and [NEt4][trans-U(NR2′)3(O)CN].24 The Ct(COT)–U–Ct(Cp*) angle of 135.27(2)° is significantly more acute than those found in U(IV) (137–140°) and U(III) (150–155°) mixed sandwich complexes supported by these ligands;4v,18a,19c,25 the reason for this is not clear but one possible explanation could be to minimise electrostatic repulsion between the anionic ligand and the polarised U–O bond. Compound (3) was further characterised by spectroscopic26 and analytical techniques (see ESI†), and Evans method (C7D8) gave an effective magnetic moment (μeff) of 2.49 μB, very close to the theoretical value of 2.54 μB for an f1 system (see below for further details and SQUID magnetometry).
When the synthesis of (3) was repeated on a larger scale, a second species co-crystallised with (3), and fractional crystallisation produced a small crop of crystals suitable for single crystal X-ray diffraction. The latter revealed the product to be the tBuNC adduct of (3) [U(η8-C8H6{1,4-SiiPr3}2)(η5-Cp*)O(η1-CNtBu)] (4), and the molecular structure is shown in Fig. 2.
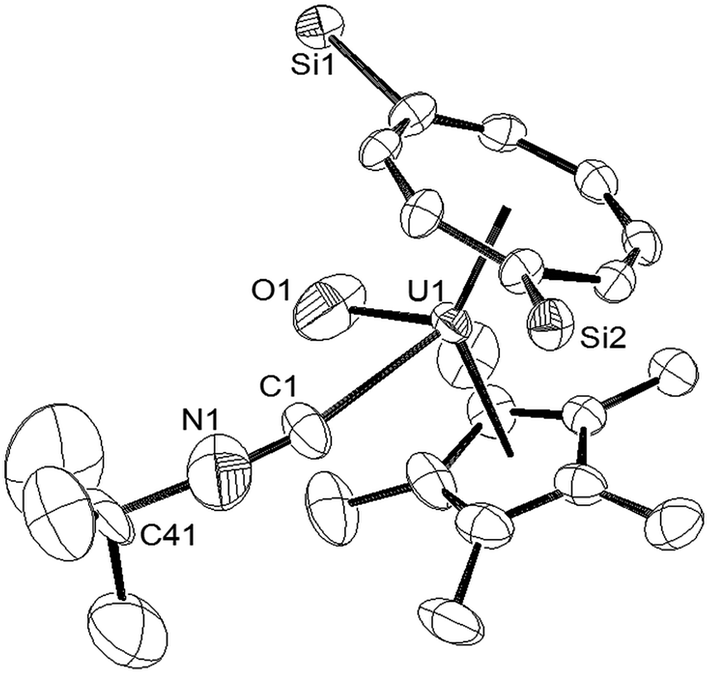 | ||
| Fig. 2 ORTEP-3 diagram of the molecular structure of (4) displaying 50% probability ellipsoids. H atoms and iPr groups have been omitted for clarity. Selected bond-lengths (Å) and angles (°): U1–C1 2.545(8), C1–N1 1.221(12), U1–O1 1.916(8) C41–N1 1.492(13) Ct(COT)–U1 2.005(2), Ct(Cp*)–U1 2.503(1); N1–C1–U1 162.8(9), C1–N1–C41 169.9(12), O1–U1–C1 71.0(4), Ct(COT)–U1–Ct(Cp*) 137.19(2), Ct(COT)–U1–O1 118.85(2), Ct(Cp*)–U1–O1 95.30(2), Ct(COT)–U1–C1 119.21(8), Ct(Cp*)–U1–C1 94.83(3). | ||
The most salient feature of (4) is the elongation of the U–O bond by almost 0.1 Å as compared with that in (3), and also with the U(V)O bonds compared above, with the exception of that in [U(O)(NR′2)3] (R′ = SiMe3).10 The reason for this structural feature is unclear, but a possible explanation could be that the isocyanide ligand acts predominantly as a σ-donor with the extra electron density transferred to π symmetry orbitals of the uranium centre involved in antibonding contributions to the U–O bond. IR spectroscopy (vide infra) revealed νNC at 2179 cm−1 for the isocyanide ligand in (4), a value very close to those observed in [UCp*2(NMe2)(tBuNC)2]BPh4 (ref. 27) and the [UCp3(CNC6H11)(NCMe)]+ cation;28 the short (1.221(12) Å) CN bond in (4) is also comparable (within esd's) to those in the latter complexes, while the small deviation of the C–N–C(tBu) from linearity presumably alleviates steric congestion around the metal centre. The Ct(COT)–U1–Ct(Cp*) angle is slightly more obtuse (ca. 2°) than that in (3), while the Ct(Cp*)–U1 and Ct(COT)–U1 distances are slightly elongated but within the range observed for previously reported complexes supported by these ligands.
Attempts to isolate (4) in better yields from the reaction of (1) with tBuNCO were unsuccessful, leading to mixtures of (3) and (4), and indeed the tBuNC ligand in (4) is very labile and any attempted isolation or manipulation of (4) via operations in vacuo invariably again led to mixtures of (3) and (4). In order to isolate (3) free from (4), the best route involved the reaction of (1) with tBuNCO followed by repeated dissolution in pentane and subsequent evaporation, a method used by Andersen and Evans et al. to obtain base-free Cp* lanthanide complexes,29 which afforded (3) in 55% yield. Reaction of a C6D6 solution of the resultant microanalytically pure (3) with one equivalent of tBuNC resulted in small but discernible shifts of the resonances due to (3) in the 1H-NMR spectrum and which we ascribe to the formation of (4). Similarly, in situ IR spectroscopy showed that, upon reaction of (3) with 1 equivalent of tBuNC in methyl–cyclohexane, two new peaks appeared, one at 2134 cm−1 (νNC in free tBuNC) and one at 2179 cm−1 assigned to νNC in (4).
The above data suggest that the synthesis of the novel U(V) terminal oxo complex (3) proceeds via the isocyanide adduct (4): the use of tBuNCO as an efficient oxygen transfer reagent30 results in the two electron oxidation of (1) and the formation of tBuNC and hence (4) (probably via a concerted reaction), and ultimately (3) after work-up (Scheme 1).
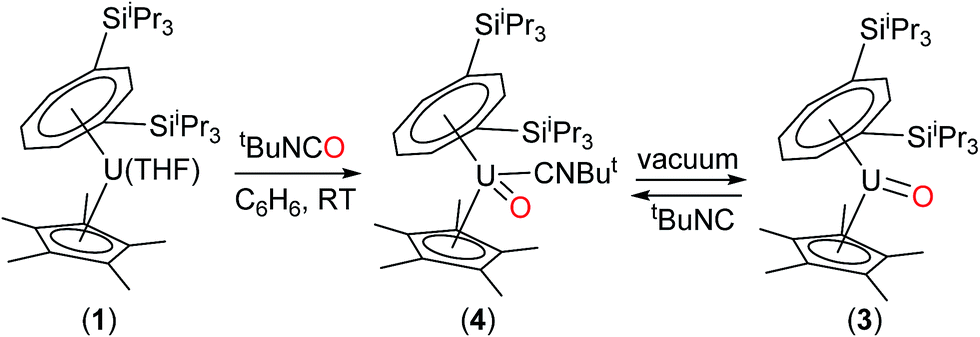 | ||
| Scheme 1 Synthesis of terminal oxo U(V) complexes (3) and (4). | ||
We have previously reported the synthesis of the dimeric, μ-oxo U(IV) complex {U[η8-C8H6(1,4-SiiPr3)2](η5-Cp*)}2(μ-O) (5) from the reaction of (1) with a mixture of NO/CO.31 Given the existence of (5), the isolation of the mononuclear terminal oxo U(V) complex (3) would appear surprising. We therefore decided to investigate whether (3) could be prepared using alternative oxygen transfer reagents. Reaction of (1) with exactly 0.5 equivalents of N2O (administered accurately via a Töepler line) in C7D8 at −78 °C resulted in an immediate colour change to bright red, leading to the clean formation of (5) as evidenced by 1H and 29Si{1H}-NMR spectroscopy, and the μ-oxo complex was isolated as the sole product in very good yields (see Scheme 2 and ESI†).
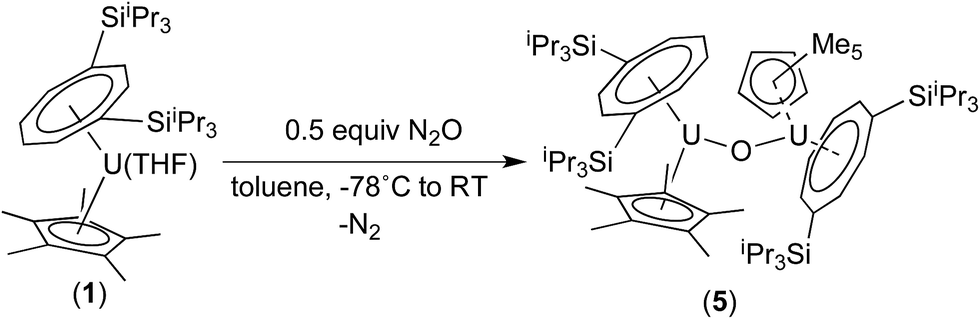 | ||
| Scheme 2 Synthesis of (5). | ||
On the other hand, when equimolar amounts of U(V) terminal oxo complex (3) and the U(III) precursor (1) were mixed in C7D8, no reaction was observed at RT and conversion to the μ-oxo complex (5) (ca. 25% spectroscopic yield relative to (1)) was observed only after heating at 45 °C over three days.32 These experiments in conjunction with the isolation of (4) indicate that these two reactions most likely proceed via different mechanisms. The case of N2O would be consistent with a concerted mechanism involving a dinuclear intermediate in which N2O bridges, and then eliminates N2 leading to a dinuclear μ-oxo product. However for tBuNCO, the formation of mononuclear (4) after the oxo transfer step, stops any further reaction with (1) that could lead to (5), due to the steric congestion imposed by both the TIPS groups and the tBuNC ligand. To further test this hypothesis, the less sterically hindered homologue of (1), [U{η8-C8H6-(1,4-SiMe3)2}(η5-Cp*)THF] (2) was reacted with tBuNCO. In this case the reaction furnished cleanly the dinuclear μ-oxo U(IV) complex {U[η8-C8H6(1,4-SiMe3)2](η5-Cp*)}2(μ-O) (6) as evidenced by its NMR spectroscopic data that were in excellent agreement with those previously reported.18a Compounds (1) and (2) have very similar [U(III)] ↔ [U(IV)] redox potentials (−2.13 V and −2.10 V vs. Fc+/0 respectively, see ESI†), so the clean formation of (6) highlights the importance of the steric hindrance imposed by the silyl substituents on the 8-membered ring in dictating the outcome of the reactions of (1) and (2) with tBuNCO. In the case of (1), reaction with tBuNCO results in a single 2e− oxidation of the metal centre leading to the U(V) complex (4), and hence (3), whereas in the case of (2) this reaction results in two 1e− oxidations leading to the dinuclear U(IV)–U(IV) complex (6) (Scheme 3).
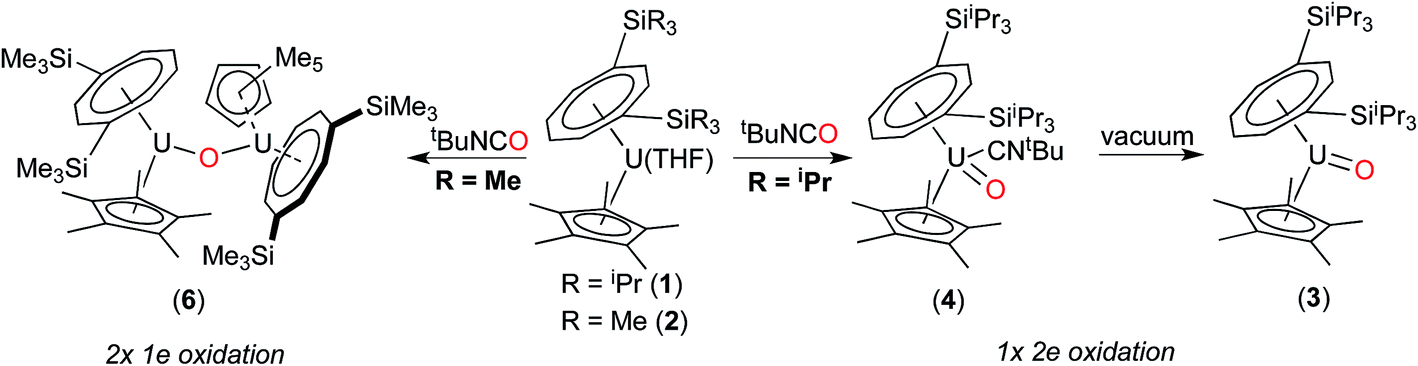 | ||
| Scheme 3 Steric control of the oxidation state of the U centre. | ||
Attempts to generate (3) using other isocyanates (PhNCO, iPrNCO) or oxo transfer reagents (Me3NO, pyridine N-oxide) were uniformly unsuccessful leading to intractable reaction mixtures. Interestingly when Me3SiNCO was reacted with (1), the U(IV) complex {U[η8-C8H6(1,4-Si(iPr)3)2](η5-Cp*)(OSiMe3)} (7) was isolated as the sole product of the reaction.33 The isolation of (7) can reasonably be explained by the formation of a short-lived [U(V)O] complex which, due to the oxophilicity of the SiMe3 group, undergoes a formal reduction to produce the observed U(IV) complex (7) and presumably cyanogen (CN)2 (although formation of the latter was not confirmed). Similar reactivity of UO bonds towards silicon electrophiles has been observed by Andersen et al.8a
Given the similarities between nitride and oxo ligands,14 the successful isolation of the terminal oxo complex (3) suggested that the steric protection afforded by the U[η8-C8H6(1,4-SiiPr3)2](η5-Cp*) mixed sandwich framework might be exploited to access the analogous uranium nitride. The highly reducing nature of (1) (UIII/UIV −2.13 V vs. Fc+/0), suggested reduction of N3− as a possible method for installing the nitride ligand.13a
Reaction of (1) with NaN3 (Scheme 4) in a mixture of C7H8/C4H8O resulted in a slow colour change to brown-red and after work-up and re-crystallisation from Et2O, brown crystals of (9) were isolated in moderate yield (ca. 30%), together with other product(s) which could not be unambiguously characterised despite repeated attempts. X-ray diffraction studies showed (9) to be the nitride complex [U{η8-C8H6-(1,4-SiiPr3)2}(η5-Cp*)(μ-N)(μ-Na{OEt2}2)], best described as a sodium uranium nitride contact ion pair (Fig. 3).
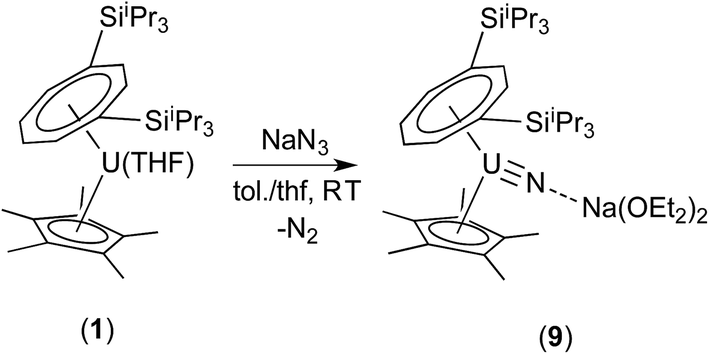 | ||
| Scheme 4 Synthesis of (9). | ||
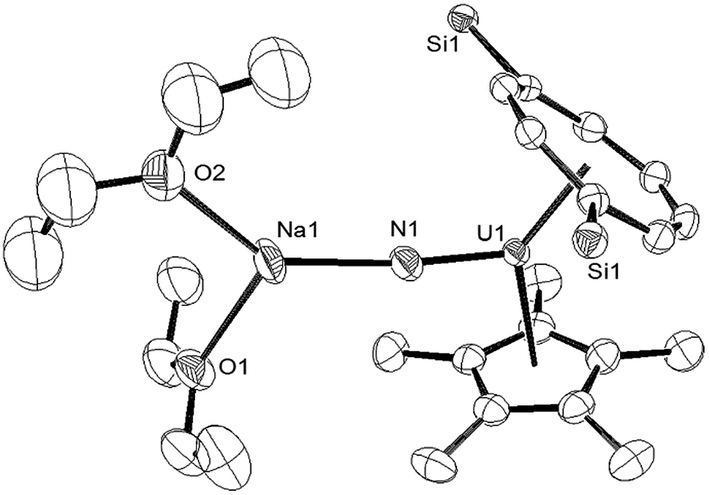 | ||
| Fig. 3 Ortep-3 diagram of the molecular structure of (9) displaying 50% probability ellipsoids. H atoms and iPr groups have been removed for clarity. Selected bond-lengths (Å) and angles (°): U1–N1 1.835(5), N1–Na1 2.244(6), Ct(COT)–U1 2.026(1), Ct(Cp*)–U1 2.548(8); U1–N1–Na1 172.4(3), Ct(COT)–U1-Ct(Cp*) 137.25(7), Ct(COT)–U1–N1 124.73(5), Ct(Cp*)–U1–N1 101.99(1). | ||
Liddle et al. recently described a U(V) terminal nitride anion supported by the TRENTIPS ligand, as well as its U(VI) neutral analogue.13a,b The U–N bond length of 1.835(5) Å in (9) is comparable to that in the U(V) nitride complex [U(TRENTIPS)N]−[Na(12-c-4)2]+ (1.825(15) Å) where the two ions are separated, but is shorter than the one found in [U(TRENTIPS)(μ-N)(μ-Na)]2 (1.883(4) Å) where a N–Na interaction is also present.13a It is also shorter than those in the borane capped nitrido complexes [(C6F5)3BNU(V)(NMestBu)3][NnBu] (1.916(4) Å) and [(C6F5)3BNU(VI)(NMestBu)3] (1.880(4) Å)34 (although the latter two can viewed as borane–imido complexes and the bond distances are more typical of U imido complexes). Compared to the neutral U(VI) complex [U(TRENTIPS)N] the U–N bond in (9) is similar within esd's.13b The Na–N bond length of 2.244(6) Å in (9) is shorter than the ones found in [U(TRENTIPS)(μ-N)(μ-Na)]2 (2.308(5) Å)13a and [U(TRENTIPS)(μ-N)(μ-Na{15-c-5})] (2.291(5) Å),13b and the U–N–Na linkage is close to linear as in the latter. The Ct(Cp*)–U distance in (9) is elongated compared to (3) and (4) while the Ct(COT)–U1–N1 and Ct(Cp*)–U1–N1 angles are significantly more acute than the ones found for the corresponding angles Ct–U1–O1 angles in (3). The reason for these differences is unclear.
Complex (9) was further characterised by spectroscopic35 and analytical techniques (see ESI†), and the μeff (Evans method) was determined to be 2.21 μB (further details below, including SQUID magnetometry), which is in reasonable agreement with the value of 1.99 μB for [U(TRENTIPS)N]−,13a and is within the range of values reported for other U(V) complexes.36
The 23Na NMR spectrum of (9) in THF revealed a single, very broad (Δν1/2 = 8300 Hz) resonance centred at ca. δ 200 ppm suggesting that the interaction of the sodium cation with the paramagnetic uranium centre is maintained in solution (cf. (10), vide infra).
Since the less sterically hindered U(III) complex [U{η8-C8H6-(1,4-SiMe3)2}(η5-Cp*)THF] (2) affords the bridging μ-oxo complex (6), the reaction of (2) with a slight excess of NaN3 (1.5 mol eq.) in a C7H8/THF solvent mixture (ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was explored. Indeed, after work-up and re-crystallisation from THF/Et2O, brown-red crystals of the bridging nitride complex [{U[η8-C8H6(1,4-SiMe3)2](η5-Cp*)}2(μ-N)]−[Na(THF)6]+ (10) suitable for X-ray diffraction studies were isolated in 81% yield (Scheme 5 and Fig. 4).
:1) was explored. Indeed, after work-up and re-crystallisation from THF/Et2O, brown-red crystals of the bridging nitride complex [{U[η8-C8H6(1,4-SiMe3)2](η5-Cp*)}2(μ-N)]−[Na(THF)6]+ (10) suitable for X-ray diffraction studies were isolated in 81% yield (Scheme 5 and Fig. 4).
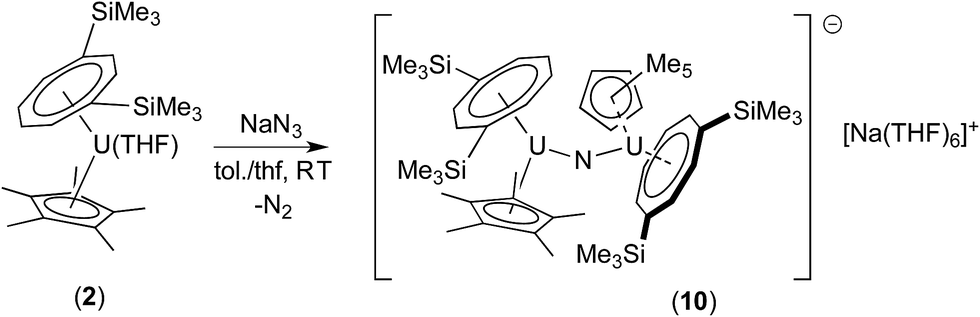 | ||
| Scheme 5 Synthesis of (10). | ||
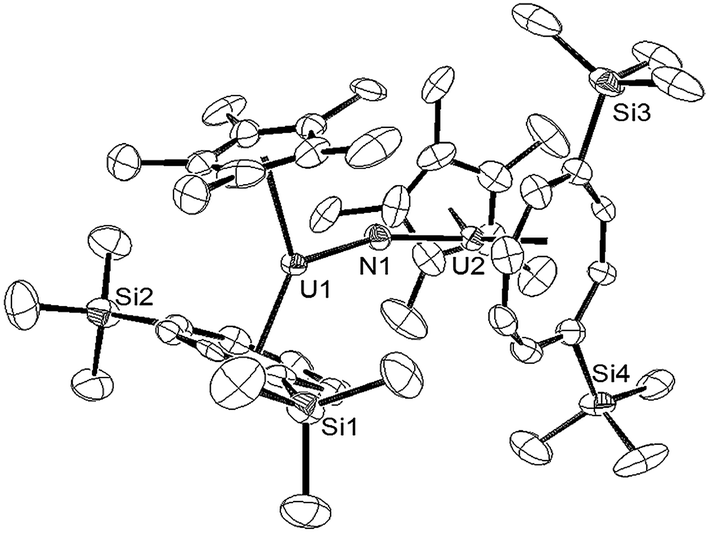 | ||
| Fig. 4 ORTEP-3 diagram of the molecular structure of the anion in (10) displaying 50% probability ellipsoids. H atoms have been removed for clarity. Selected bond-lengths (Å) and angles (°): N1–U1 2.063(5), U2–N1 2.066(5), Ct(COT)–U1 2.033(4), Ct(Cp*)–U1 2.516(2), Ct(COT)–U2 2.038(4), Ct(Cp*)–U2 2.536(2); U1–N1–U2 159.4(3), Ct(COT)–U1–Ct(Cp*) 137.03(2), Ct(COT)–U2–Ct(Cp*) 136.70(2), Ct(COT)–U1–N1 122.83(8), Ct(Cp*)–U1–N1 100.09(1), Ct(COT)–U2–N1 122.75(3), Ct(Cp*)–U2–N1 100.54(2). | ||
The two U–N bond lengths (N1–U1 2.063(5) Å, U2–N1 2.066(5) Å) in the anionic dimer are essentially the same, suggesting a delocalised [U ≃ N ≃ U] bonding interaction as in [Cp*2U(μ-N)(μ-N3)UCp*2]4,15e and also the same within esd's to the ones previously reported for other bridging nitride complexes with the exception of the triply bridging nitride in [{UCp*2(μ-I)2}3(μ3-N)] (2.152(3)–2.138(3) Å).15d Furthermore the length of the bond is in the middle of the range found for complexes with localised U–NU bonding interactions (1.95–2.12 Å).15b,c Compared to the U–N bond length in (9), that in (10) is significantly elongated as expected. The U–N–U bond in (10) has significantly deviated from linearity, which is a common structural motif for many bridging U nitride complexes15f,37 but is less obtuse than those in the [U(IV)–N–U(IV)]−, [U(IV)–N–U(V)] and [U(IV)–N–U(VI)(O)]− complexes supported by bulky silyl amide ligands,15c that in [KU(μ-N)(OSi(OtBu)3)]2 (106.1(2)°),15b as well as in complexes where the nitride ligand bridges more than two U centre.15a,d,f Unsurprisingly, the U–N–U bond angle in (10) is identical to the U–O–U bond angle found in the μ-oxo complex {U[η8-C8H6(1,4-SiMe3)2](η5-Cp*)}2(μ-O) (6)18a – a fact that reflects the effect of the sterically imposed geometry of the complex. As expected the U–N bonds are shorter than the corresponding U–O ones in (6) and that shortening might account for the slightly more acute Ct(COT)–U–Ct(Cp*) angles in (10) compared to the ones found in (6) (139.7(16)° and 140.0(16)°).18a
Complex (10) readily loses its crystallinity due to loss of coordinated THF to yield [{U[η8-C8H6(1,4-SiMe3)2](η5-Cp*)}2(μ-N)]−[Na(THF)2]+ (10′) as a well-defined product, as evidenced by microanalysis. As in the case of its μ-oxo analogue [{U[η8-C8H6(1,4-SiMe3)2](η5-Cp*)}2(μ-O)] (6), the 1H-NMR (C4D8O2) spectrum of (10′) is consistent with a C2-symmetric structure that is retained in solution. In marked contrast to (9), the 23Na NMR spectrum of (10) in THF exhibited a sharp resonance (Δν1/2 = 78 Hz) at δ −7.94 ppm, parameters suggesting no interaction of the [Na(THF)6]+ counterion with the paramagnetic uranium anion.38
Similarly to the reaction of (2) with tBuNCO that yields the μ-oxo complex (6), the bridging nitride complex (10) can be seen as the product of two 1e oxidations of the U(III) precursor (vs. the one 2e oxidation that produces (9) in the case of the bulkier COT substituents), since the formal oxidation state of the uranium centres in (10) is +4. The μeff for 10′ (C4D8O2, Evans method) was determined to be 3.64 μB for the dimer or 2.57 μB per uranium centre, a value consistent with a U(IV) ion (further details including SQUID magnetometry below).
Magnetic studies on (3), (9) and (10′)
Table 1 compares the μeff for complexes (3), (9) and (10′) at 300 K as determined in solution (Evans method), and in the solid state (SQUID under an applied field of 0.1 tesla); the values determined by these two methods are in fair agreement.| Complex | μ eff Evans (μB) | μ eff SQUID (μB) |
|---|---|---|
| (3) | 2.49 | 2.16 |
| (9) | 2.2 | 2.00 |
| (10′) | 3.64 (2.57 per U) | 3.58 (2.53 per U) |
The effective magnetic moment of (3) exhibits a steady decline from the value of 2.16 μB at 300 K to 1.54 μB at 5 K (Fig. 5). This behaviour is typical for a 2F5/2 ion, and is comparable to values reported for molecular U(V) terminal oxo complexes (see ESI† for plots of χm/T, χmT/T and χm−1/T).4o,22,23
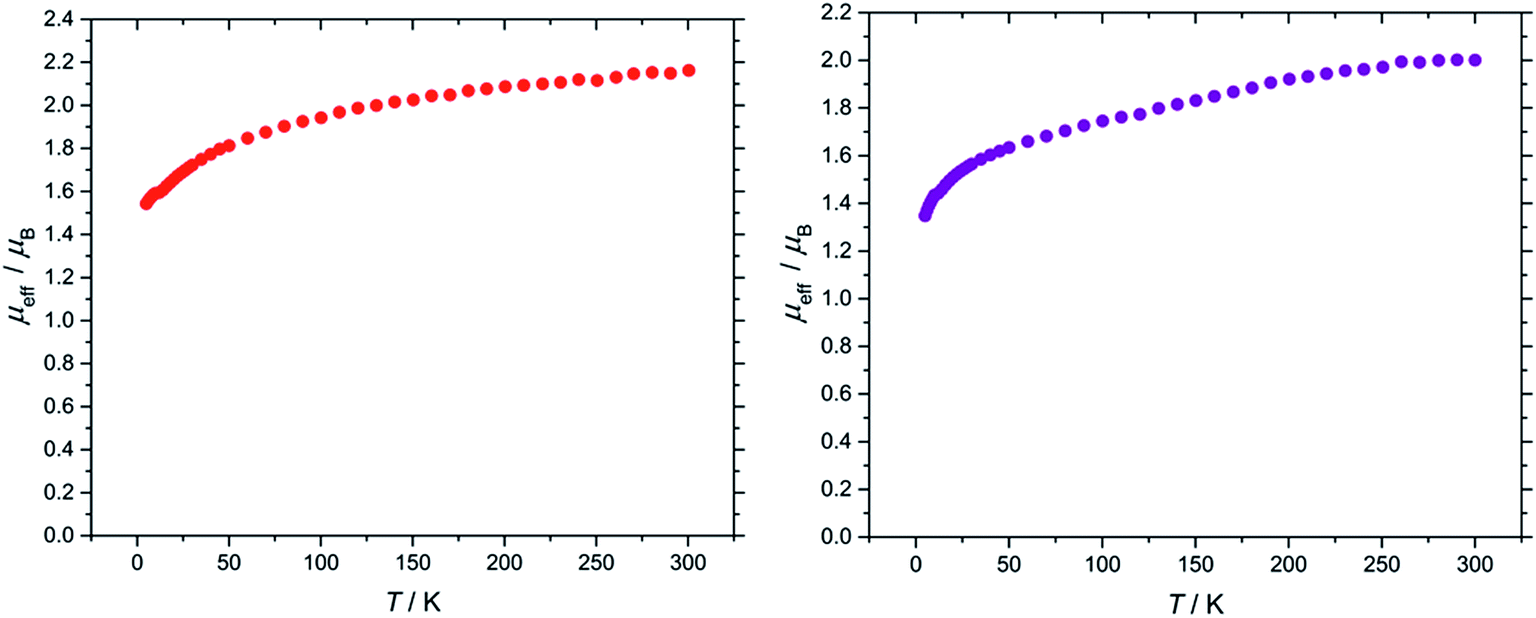 | ||
| Fig. 5 Temperature dependence of the solid state μeff of (3) (left) and (9) (right) at 0.1 tesla. | ||
In the case of the nitride complex (9), its effective magnetic moment was found to be 2.00 μB at 300 K and 1.35 μB at 5 K (Fig. 5). These values are comparable to the effective magnetic moment found for nitride complex [UN(TRENTIPS)][Na(12-crown-4)2] (1.99 μB at 298 K, 1.31 μB at 1.8 K),13a and are in agreement with literature values for molecular U(V) complexes more generally39 (see ESI† for plots of χm/T, χmT/T and χm−1/T).
Magnetic susceptibility data sets for (10′) measured for zero-field cooled and field cooled samples coincided exactly, indicating the absence of long–range interactions between spins on the two U(IV) centres. At 300 K the effective magnetic moment per U is 2.53 μB, and decreases to 0.69 μB at 2 K (Fig. 6a), consistent with two UIV f2 ions. For comparison, the solid state magnetic studies on the di-uranium(IV) dianion [{((nP,MeArO)3tacn)U}2(μ-O)2]2– by Meyer et al. showed a μeff per U of 2.73 μB at 300 K.40 The majority of paramagnetic substances have a molar susceptibility (χm) that obeys the Curie–Weiss law, χm = C/(T − Θ), where C is the Curie constant and Θ is the Weiss constant. The plot of χm−1vs. T (Fig. 6b) follows Curie–Weiss behavior in the range 50–300 K, with C = 0.0289 K−1 mol−1 and Θ = −0.015 K, suggesting that at these temperatures the [{U[η8-C8H6(1,4-SiMe3)2](η5-Cp*)}2(μ-N)]− anion behaves as two non-interacting U(IV) centres. Furthermore, there is no maximum observed in the χmvs. T plot (Fig. 6c), often cited as a definitive indication of antiferromagnetic coupling. The U(IV) ion (3H4 ground term) typically has minimal covalency, hence the two metal centres in 10′ do not participate in exchange coupling.
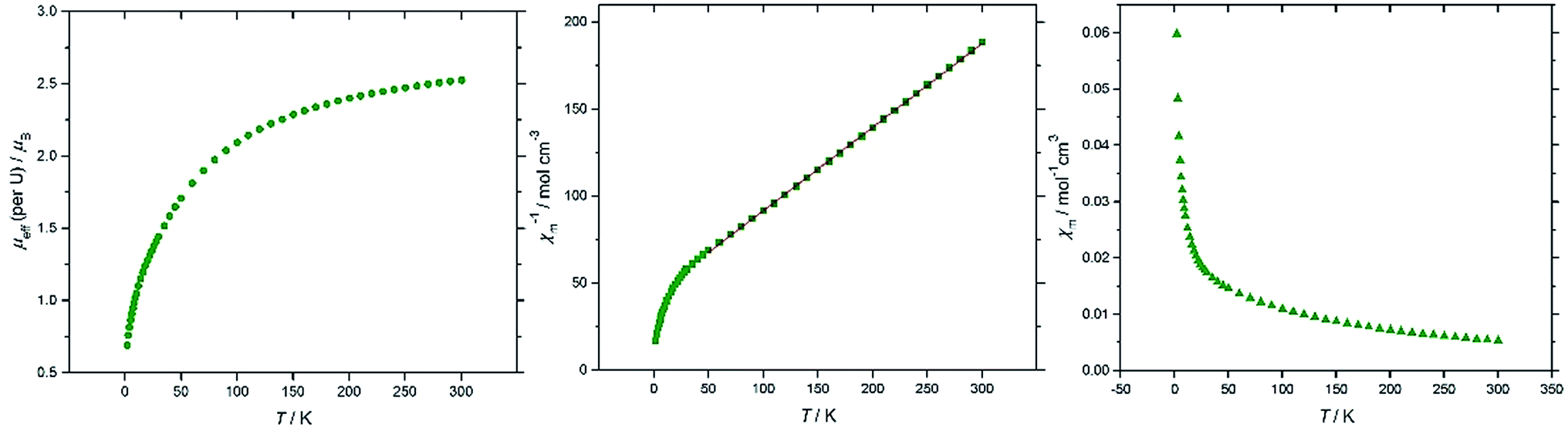 | ||
| Fig. 6 Magnetic data for (10′). From left to right: μeff (per U)/T; χm−1/T (red line is a linear fit to the data in the range 50–300 K); χm/T (see ESI† for the plot of χmT/T). | ||
Finally, magnetic data for all three compounds (3), (9) and (10′) are presented in Fig. 7 for comparison.
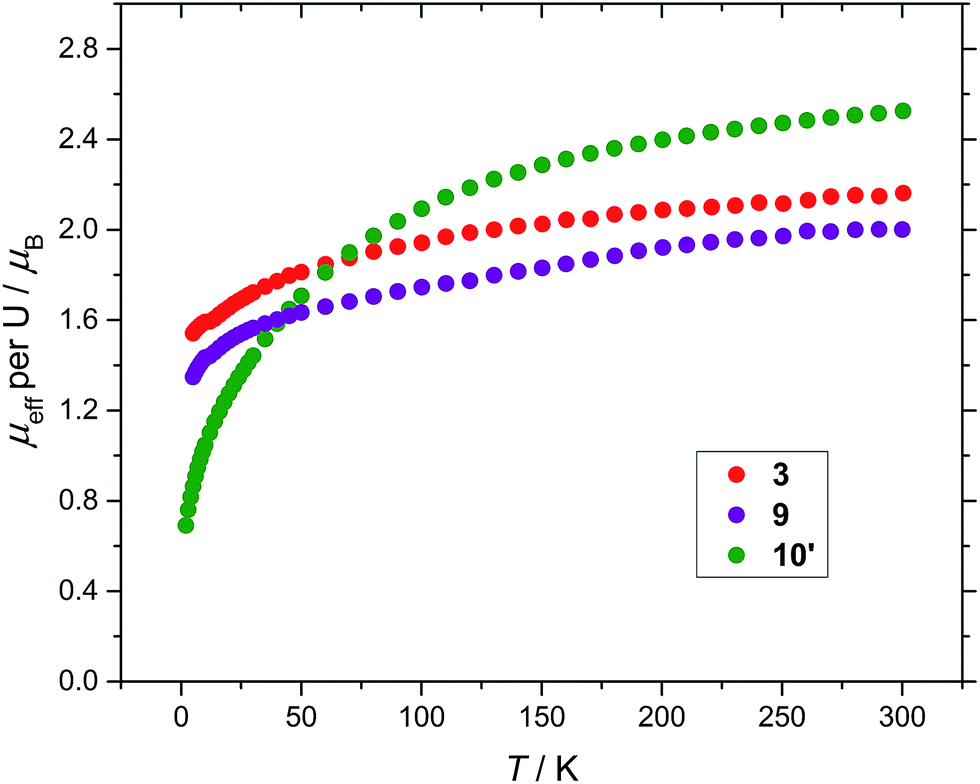 | ||
| Fig. 7 Temperature dependence of the solid state μeff of (3), (9) and (10′) (per U) at 0.1 tesla. | ||
Redox behaviour of (3), (9) and (10)
In order to gauge the potential for accessing terminal oxo and nitrido uranium(VI) complexes, the redox properties of (3), (9) and (10) were studied by cyclic voltammetry (C.V.).In contrast to the terminal oxo [((tBuArO)tacn)U(O)] complex reported by Meyer et al. that features a reversible oxidation,4o the C.V. of the terminal oxo complex (3) revealed only a quasi-reversible reduction process at −1.77 V vs. Fc+/0 (Fig. 8).
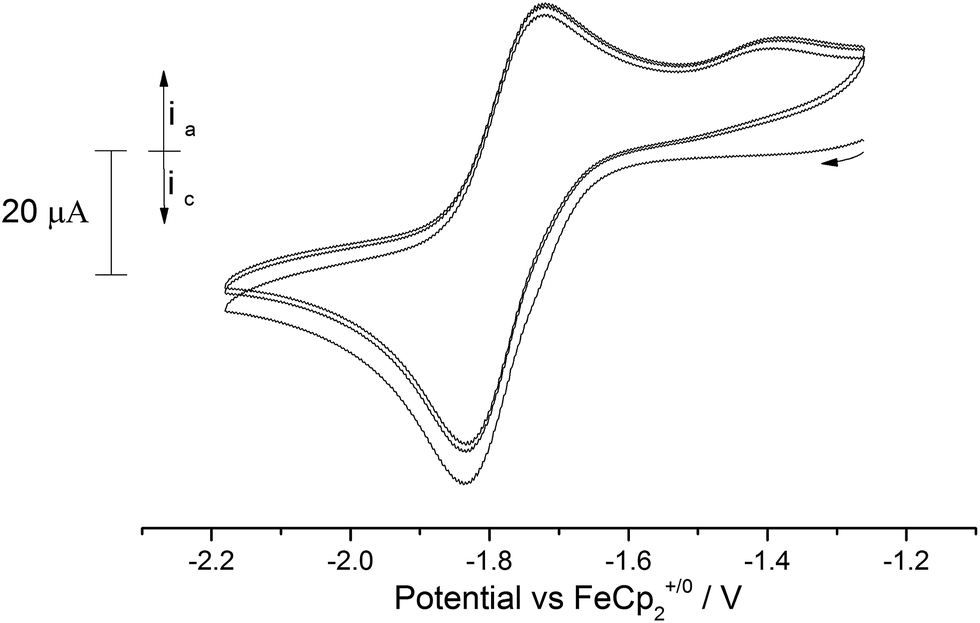 | ||
| Fig. 8 Overlaid CV scans (3 cycles) of (3) in 0.05 M [N(nBu)4][B(C6F5)4]/THF. Scan rate 250 mV s−1. | ||
Changing the scan rate (50–300 mV s−1) did not alter the shape of the observed wave and no other processes were found to occur over the solvent window. This process is assigned to the [U(V)] ↔ [U(IV)] couple, and based on this voltammogram, the reduction of (3) should be a chemically accessible process. Indeed, (3) can be chemically reduced with a slight excess of K/Hg (0.5% w/w) in the presence of 18-crown-6 in n-pentane/Et2O. The almost instantaneous reaction produced a red-pink solid that, after work-up and re-crystallisation from toluene, gave dark red rods suitable for X-ray diffraction studies which showed the product to be the U(IV) complex [U{η8-C8H6-(1,4-SiiPr3)2}(η5-Cp*)(μ-O)K(18-c-6)] (11) (Fig. 9).
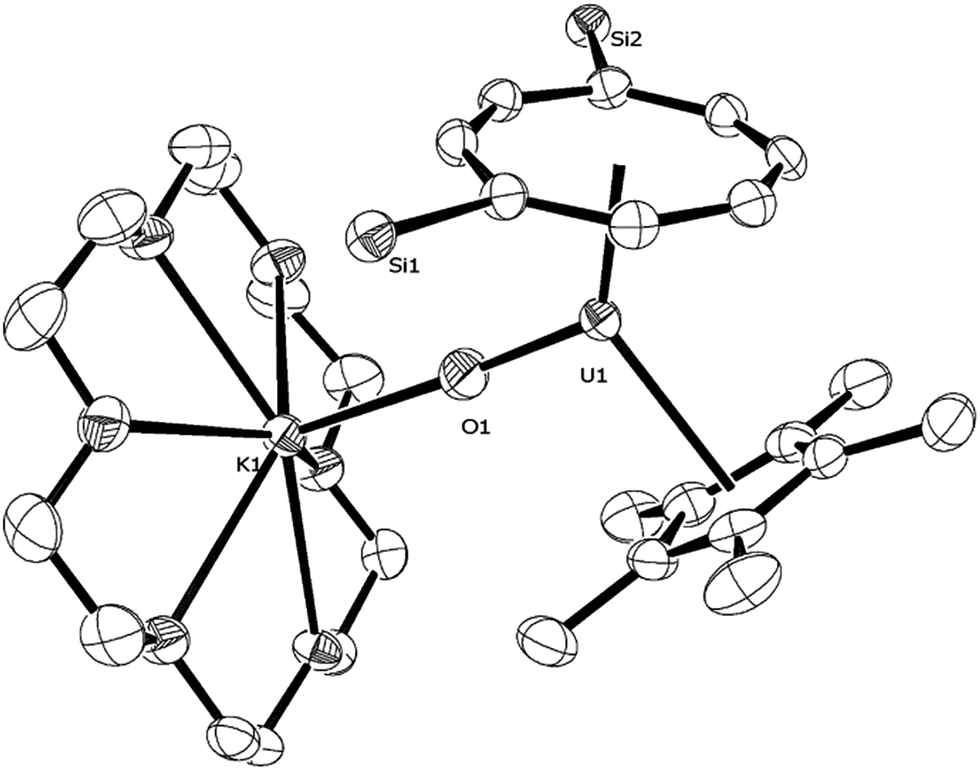 | ||
| Fig. 9 ORTEP-3 diagram of the molecular structure of (11) displaying 50% probability ellipsoids. H atoms, iPr groups and a molecule of toluene have been removed for clarity. Selected bond-lengths (Å) and angles (°): U1–O1 1.891(4), Ct(COT)–U 2.041(1), Ct(Cp*)–U1 2.583(4), O1–K1 2.582(4); U1–O1–K1 177.3(2), Ct(COT)–U1–Ct(Cp*) 133.81(2), Ct(COT)–U1–O1 125.73(6), Ct(Cp*)–U1–O1 100.45(2). | ||
The U–O bond length (1.891(4) Å) in (11) is longer than that in the U(V) complexes (3) (1.826(3) Å) and [U(NR2′)3O]10 (1.826(3) Å), but similar within esd's to the one found in the U(IV) complex (4) (1.916(8) Å). The K1–O1 bond length is as expected shorter than the OUO⋯K bonds (2.60–2.9 Å)41 and is typical of an ionic K–O bond;12 the U–O–K bond is very close to linear.
Complex (11) was fully characterised by spectroscopic and analytical methods (see ESI†); the 29Si{1H}-NMR was of particular diagnostic value as it was shifted upfield to −172.22 ppm (−72.7 ppm for parent (3)), a value that is even more upfield than that for the U(III) complex (1) (−129 ppm), probably due to the anionic nature of (11).
C.V. scans of the nitride complex (9) in the anodic direction over several cycles revealed the existence of several processes in the accessible solvent window (see ESI Fig. SI8† for a full voltammogram). Of these processes, there is a noteworthy quasi-reversible oxidation at −1.63 V vs. Fc+/0 (Fig. 10) which we tentatively assign to the [U(VI)] ↔ [U(V)] couple. As can be seen from Fig. 10, a second process at slightly more cathodic potential (ca. −1.8 V vs. Fc+/0) is also present, which features an asymmetric current response that leads us to conclude that this is probably related to a short lived electrochemically generated species. The shape of the wave at −1.63 V did not change by variation of the scan rate (50–350 mV s−1).
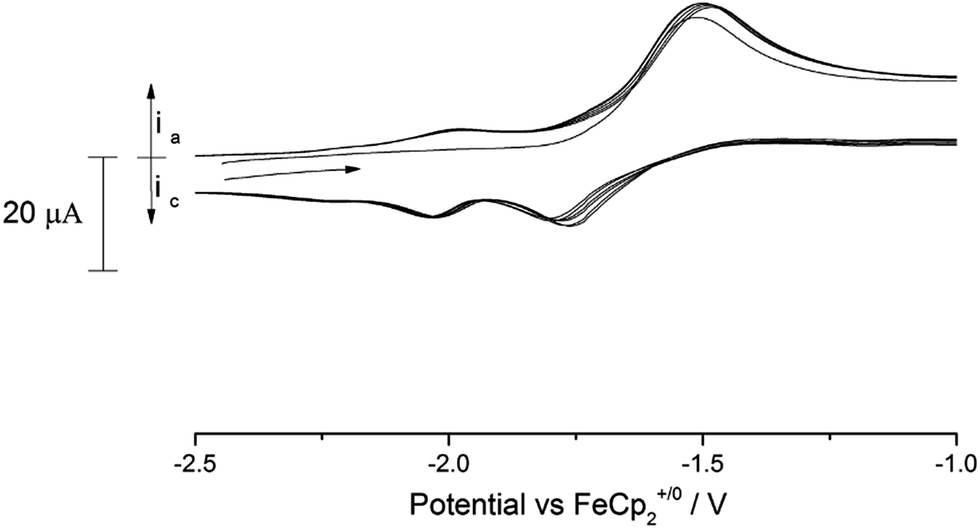 | ||
| Fig. 10 Overlaid scan (4 cycles) of (9) in 0.05 M [N(nBu)4][B(C6F5)4]/THF. Scan rate 100 mV s−1. | ||
In addition to this process, the (full) voltammogram of (9) exhibits also another two irreversible processes: one anodic at 0.7 V and a cathodic one at −2.8 V (both vs. Fc+/0). The nature of these two irreversible processes cannot be unambiguously assigned, but they could be due to ligand activation involving the nitride moiety. Attempts to chemically oxidise (9) by reaction with mild oxidants such as I2 and AgBPh4 have thus far resulted in intractable mixtures from which only ligand decomposition could be observed spectroscopically (1H-NMR).
Similarly, anodic scans of the bridging nitride (10) revealed a quasi-reversible process (peak separation 87 mV) centred at −1.46 V vs. Fc+/0 (Fig. 11). This value is very close to the one observed for complex (9) as well as for the {[U(IV)]N[U(IV)]}− ↔ {[U(V)]N[U(IV)]} couple ([U] = U(NMestBu)3) reported by Cummins et al.15f Based on this, we tentatively assign this process to the {[U(IV)]–N–[U(V)]} ↔ {[U(IV)]–N–[U(IV)]} redox pair.
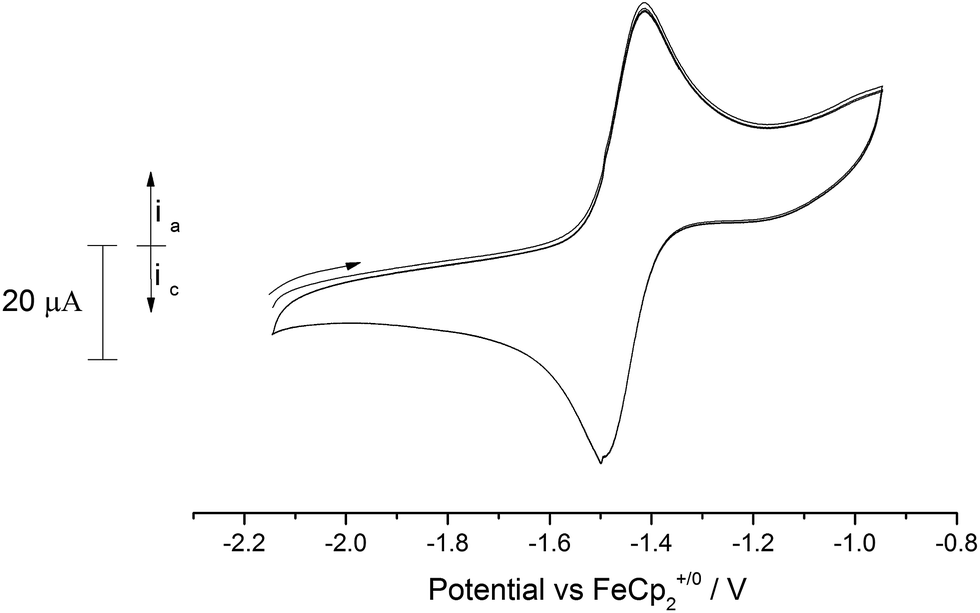 | ||
| Fig. 11 Overlaid CV scans (2 cycles) of (10′) in 0.1 M [N(nBu)4][PF6]/acetonitrile. Scan rate 150 mV s−1. | ||
Apart from this process, the voltammogram also displayed additional irreversible processes centred at anodic voltages (−0.5 V, −0.25 V, 0.35 V; see ESI Fig. SI10†) that are probably due to the formation of higher oxidation state (mixed valence) species (i.e. {[U(V)]–N–[U(V)]}, {[U(VI)]–N–[U(V)]} etc.), although other reasons (e.g. ligand activation) cannot be excluded. As in the case of (9) an irreversible reduction is also observed at ca. −2.5 V vs. Fc+/0 that as above could correspond to a mixed valence species (i.e. {[U(III)]–N–[U(V)]}) or arise from a ligand activation process. Given that similar processes appear in the case of (9), we envisage that they are more likely due to the latter rather than the former.
Conclusion
In summary we have described how the steric environment around the metal centre can manipulate redox events at a uranium centre. This has been demonstrated by the isolation of either mononuclear U(V) or dinuclear U(IV) nitrido/oxo complexes depending on the size of the silyl substituents on the supporting ligands. This has led to the preparation of an anionic uranium(V) nitride complex (9) featuring a U–N triple bond, as well as a neutral U(V) terminal oxo complex (3). Magnetic studies corroborate the formal oxidation states of these complexes further confirming that the 2e− oxidation leads to products featuring either one U(V) or two U(IV) metal centres depending on steric hindrance at the uranium centre. Cyclic voltammetry studies of complex (3) show that it can be readily reduced to the [U(IV)O]− anion (11), which has also been achieved chemically. Unlike (3), cyclic voltammetry studies have shown that the nitride complex (9) might be amenable to oxidation to the U(VI) species although initial attempts to do so have been unsuccessful thus far.
Acknowledgements
We thank the ERC (Project 247390), the EPSRC (EP/M023885/1) and the University of Sussex for financial support. The UK National Crystallography Service (NCS) Southampton are thanked for their assistance with single crystal X-ray data collection.References
- (a) P. L. Arnold, Chem. Commun., 2011, 47, 9005 RSC; (b) T. Andrea and M. S. Eisen, Chem. Soc. Rev., 2008, 37, 550 RSC; (c) M. Ephritikine, Organometallics, 2013, 32, 2464 CrossRef; (d) H. S. La Pierre and K. Meyer, Prog. Inorg. Chem., 2014, 58, 303–416 CrossRef CAS; (e) S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- (a) Recent Advances in Actinide Science, ed. R Alvarez, N. D. Bryan and I. May, RSC Publishing, 2006 Search PubMed; (b) B. M. Gardner and S. T. Liddle, Eur. J. Inorg. Chem., 2013, 22–23, 3753 CrossRef; (c) P. L. Arnold, S. M. Mansell, L. Maron and D. M. McKay, Nat. Chem., 2012, 4, 668–674 CrossRef CAS PubMed; (d) B. Kosog, C. Kefalidis, F. W. Heinemann, L. Maron and K. Meyer, J. Am. Chem. Soc., 2012, 134, 12792–12794 CrossRef CAS PubMed; (e) I. Castro-Rodríguez and K. Meyer, Chem. Commun., 2006, 1353–1368 RSC; (f) P. L. Arnold, M. W. McMullon, J. Rieb and F. E. Kuhn, Angew. Chem., Int. Ed., 2015, 54, 82–100 CrossRef CAS PubMed.
- (a) M. R. MacDonald, M. E. Fieser, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 13310 CrossRef CAS PubMed; (b) H. S. La Pierre, A. Scheurer, F. W. Heinemann, W. Hieringer and K. Meyer, Angew. Chem., Int. Ed., 2014, 53, 7158 CrossRef CAS PubMed.
- (a) S. G. Minasian, J. L. Krinsky, V. A. Williams and J. Arnold, J. Am. Chem. Soc., 2008, 130, 10086–10087 CrossRef CAS PubMed; (b) S. T. Liddle, J. McMaster, D. P. Mills, A. J. Blake, C. Jones and W. D. Woodwul, Angew. Chem., Int. Ed., 2009, 48, 1077–1080 CrossRef CAS PubMed; (c) B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2015, 7, 582–590 CrossRef CAS PubMed; (d) B. M. Gardner, G. Balázs, M. Scheer, A. J. Wooles, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 15250–15254 CrossRef CAS PubMed; (e) B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 4484–4488 CrossRef CAS PubMed; (f) Q.-Y. Wu, J.-H. Lan, C.-Z. Wang, Y.-L. Zhao, Z.-F. Chai and W.-Q. Shi, J. Phys. Chem. A, 2015, 119, 922–930 CrossRef CAS PubMed; (g) S. M. Franke, F. W. Heinemann and K. Meyer, Chem. Sci., 2014, 5, 942–950 RSC; (h) T. Cantat, T. Arliguie, A. Noël, P. Thuéry, M. Ephritikhine, P. Le Floch and N. Mézailles, J. Am. Chem. Soc., 2009, 131, 963–972 CrossRef CAS PubMed; (i) T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Science, 2005, 310, 1941–1943 CrossRef CAS PubMed; (j) M. Porchia, F. Ossola, G. Rossetto, P. Zanella and N. Brianese, J. Chem. Soc., Chem. Commun., 1987, 550 RSC; (k) G. Paolucci, G. Rossetto, P. Zanella and R. D. Fischer, J. Organomet. Chem., 1985, 284, 213 CrossRef CAS; (l) M. Porchia, U. Casellato, F. Ossola, G. Rossetto, P. Zanella and N. Brianese, J. Chem. Soc., Chem. Commun., 1986, 1034 RSC; (m) P. L. Diaconescu, A. L. Odom, T. Agapie and C. C. Cummins, Organometallics, 2001, 20, 4993–4995 CrossRef CAS; (n) M. Porchia, N. Brianese, U. Casellato, F. Ossola, G. Rossetto and P. Zanella, J. Chem. Soc., Dalton Trans., 1989, 677–681 RSC; (o) S. C. Bart, C. Anthon, F. W. Heinmann, E. Bill, N. M. Edelstein and K. Meyer, J. Am. Chem. Soc., 2008, 130, 12536–12546 CrossRef CAS PubMed; (p) I. C. Rodríguez, H. Nakai and K. Meyer, Angew. Chem., Int. Ed., 2006, 25, 2389–2392 CrossRef PubMed; (q) C. J. Windorff and W. J. Evans, Organometallics, 2014, 33, 3786–3791 CrossRef CAS; (r) A.-C. Schmidt, F. W. Heinmann, L. Maron and K. Meyer, Inorg. Chem., 2014, 53, 13142–13153 CrossRef CAS PubMed; (s) N. H. Anderson, S. O. Odoh, Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919–926 CrossRef CAS PubMed; (t) P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 10412–10415 CrossRef CAS PubMed; (u) O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538–1547 RSC; (v) J. A. Higgins-Frey, F. G. N. Cloke and S. Mark Roe, Organometallics, 2015, 34, 2102–2105 CrossRef CAS; (w) D. M. King, J. McMaster, F. Tuna, E. J. L. McInes, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2014, 136, 5619–5622 CrossRef CAS PubMed; (x) J. L. Brown, S. Fortier, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 15468–15475 CrossRef CAS PubMed; (y) N. H. Anderson, H. Yin, J. J. Kiernicki, P. E. Fanwick, E. J. Schelter and S. C. Bart, Angew. Chem., Int. Ed., 2015, 54, 9386–9389 CrossRef CAS PubMed; (z) J. L. Brown, S. Fortier, G. Wu, N. Katsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352–5355 CrossRef CAS PubMed.
- (a) A. E. Commyns, Chem. Rev., 1960, 50, 115 CrossRef; (b) F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef; (c) P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef CAS; (d) Z. Szabo, T. Toraishi, V. Vallet and I. Grenthe, Coord. Chem. Rev., 2006, 250, 816 CrossRef.
- (a) P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221–227 CrossRef CAS PubMed; (b) P. L. Arnold, D. Patel, A. J. Blake, C. Wilson and J. B. Love, J. Am. Chem. Soc., 2006, 130, 9610 CrossRef PubMed; (c) G. M. Jones, P. L. Arnold and J. B. Love, Angew. Chem., Int. Ed., 2012, 51, 12584–12587 CrossRef CAS PubMed.
- The Chemistry of the Actinide Elements, J. J. Katz, G. T. Seaborg and L. R. Morss, Chapman and Hall, New York, 1986 Search PubMed.
- (a) G. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriesden and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS; (b) O. P. Lam, S. C. Bart, F. W. Heinemann and K. Meyer, Chem. Commun., 2010, 46, 3137 RSC.
- (a) L. R. Avens, D. M. Barnhart, C. J. Burns, S. D. McKee and W. H. Smith, Inorg. Chem., 1994, 33, 4245–4254 CrossRef CAS; (b) S. J. Kraft, J. Walensky, P. E. Fanwick, M. B. Hall and S. C. Bart, Inorg. Chem., 2010, 49, 7620–7622 CrossRef CAS PubMed; (c) W. J. Evans, S. A. Kozimor and J. W. Ziller, Polyhedron, 2004, 23, 2689–2694 CrossRef CAS; (d) B. M. Gardner, W. Lewis, A. J. Blake and S. T. Liddle, Inorg. Chem., 2011, 50, 9631–9641 CrossRef CAS PubMed.
- S. Fortier, J. L. Brown, N. Kaltsoyannis, G. Wu and T. W. Hayton, Inorg. Chem., 2012, 51, 1625–1633 CrossRef CAS PubMed.
- (a) D. M. King, F. Tuna, J. McMaster, W. Lewis, A. J. Blake, E. J. L. McInnes and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 4921–4924 CrossRef CAS PubMed; (b) P. Roussel, R. Boaretto, A. J. Kingsley, N. W. Alcock and P. Scott, J. Chem. Soc., Dalton Trans., 2002, 1423 RSC.
- O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed.
- (a) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717 CrossRef CAS PubMed; (b) M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482 CrossRef PubMed.
- The Organometallic Chemistry of the Transition Metals, R. H. Crabtree, John Wiley & Sons, 5th edn, 2009 Search PubMed.
- (a) G. Nocton, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2008, 47, 3040–3042 CrossRef CAS PubMed; (b) C. Camp, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed; (c) S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed; (d) W. J. Evans, K. A. Miller, J. W. Ziller and J. Greaves, Inorg. Chem., 2007, 46, 8008 CrossRef CAS PubMed; (e) W. J. Evans, S. A. Kozimor and J. W. Ziller, Science, 2005, 309, 1835 CrossRef CAS PubMed; (f) A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed; (g) L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed.
- D. W. Green and G. T. Reedy, J. Chem. Phys., 1976, 65, 2921 CrossRef CAS; R. D. Hunt, J. T. Yustein and L. Andrews, J. Chem. Phys., 1993, 98, 6070 CrossRef; L. Andrews, X. Wang, R. Lindh, B. O. Roos and C. J. Marsden, Angew. Chem., Int. Ed., 2008, 47, 5366 CrossRef PubMed.
- R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723–729 CrossRef CAS PubMed.
- (a) N. Tsoureas, L. Castro, A. F. R. Kilpatrick, F. G. N. Cloke and L. Maron, Chem. Sci., 2014, 5, 3777 RSC; (b) O. T. Summerscales, A. S. Frey, F. G. N. Cloke and P. B. Hitchcock, Chem. Commun., 2009, 198 CAS.
- (a) O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829 CrossRef CAS PubMed; (b) O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, J. Am. Chem. Soc., 2006, 128, 9502 CrossRef PubMed; (c) N. Tsoureas, O. T. Summerscales, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 1353 CrossRef CAS.
- (a) H. Werner, Coord. Chem. Rev., 1982, 43, 165–185 CrossRef CAS; (b) K. K. Pandey, Coord. Chem. Rev., 1997, 140, 37–114 CrossRef.
- C. J. Windorff and W. J. Evans, Organometallics, 2014, 33, 3786–3791 CrossRef CAS.
- S. Fortier, N. Kaltsoyannis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 14224–14227 CrossRef CAS PubMed.
- D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1993, 115, 9840–9841 CrossRef CAS.
- A. J. Lewis, K. C. Mullane, E. Nakamaru-Ogiso, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2014, 53, 6944–6953 CrossRef CAS PubMed.
- J. A. Higgins, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 5244–5252 CrossRef CAS.
-
ν
(UO) could not be unequivocally assigned as it coincides with stretches attributed to the TIPS groups.
- C. Boisson, J. C. Berthet, M. Lance, M. Nierlich and M. Ephritikine, J. Organomet. Chem., 1997, 548, 9–16 CrossRef CAS.
- H. Aslan, J. Förster, K. Yünlü and R. D. Fischer, J. Organomet. Chem., 1988, 355, 79 CrossRef CAS.
- (a) M. Schultz, C. J. Burns, D. J. Schwartz and R. A. Andersen, Organometallics, 2000, 19, 781–789 CrossRef CAS; (b) W. J. Evans, L. A. Hughes and T. P. Hanusa, Organometallics, 1986, 5, 1285 CrossRef CAS.
- A. E. Guiducci, C. L. Boyd and P. Mounford, Organometallics, 2006, 25, 1167–1187 CrossRef CAS.
- A. S. P. Frey, F. G. N. Cloke, M. P. Coles and P. B. Hitchcock, Chem.–Eur. J., 2010, 16, 9446 CrossRef CAS PubMed.
- Heating to higher temperature resulted in the formation of a tuck-in complex, see ref. 29.
- The identity of (7) was confirmed by independent synthesis from {U[η8-C8H6(1,4-Si(iPr)3)2](η5-CpMe5)Cl} (8) and NaOSiMe3 in THF. Its formulation was unambiguously confirmed by single crystal XRD. For more details and a brief discussion of metric parameters see the ESI.†.
- A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716–5717 CrossRef CAS PubMed.
- The ν(U
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) could not be unequivocally assigned as it coincides with stretches attributed to the TIPS groups.
N) could not be unequivocally assigned as it coincides with stretches attributed to the TIPS groups. - D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865–8882 CrossRef CAS PubMed.
- L. Maria, I. C. Santos, V. R. Souza and J. Marçalo, Inorg. Chem., 2015, 54, 9115–9126 CrossRef CAS PubMed.
- C. Eaborn, M. S. Hill, P. B. Hitchcock and J. D. Smith, Organometallics, 2000, 19, 5780–5783 CrossRef CAS.
- C. R. Graves and J. L. Kiplinger, Chem. Commun., 2009, 26, 3831 RSC.
- A.-C. Schimdt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef PubMed.
- (a) B. Masci and P. Thuéry, CrystEngComm, 2007, 9, 582–590 RSC; (b) P. Thuéry, B. Masci, M. Takimoto and T. Yamato, Inorg. Chem. Commun., 2007, 10, 795–799 CrossRef; (c) C. Nocton, P. Horegald, V. Vetere, J. Pécaut, L. Dubois, P. Maldivi, N. M. Edelstein and M. Mazzanti, J. Am. Chem. Soc., 2010, 132, 495–508 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and synthetic procedures, additional X-ray crystallographic, cyclic voltammetry and magnetic data. CCDC numbers 1449997–1450002 for compounds (3), (4), (7), (9), (10), and (11). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00632a |
| This journal is © The Royal Society of Chemistry 2016 |