
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe auxiliary [4Fe–4S] cluster of the Radical SAM heme synthase from Methanosarcina barkeri is involved in electron transfer†
Melanie
Kühner
a,
Peter
Schweyen
b,
Martin
Hoffmann
b,
José Vazquez
Ramos
c,
Edward J.
Reijerse
d,
Wolfgang
Lubitz
d,
Martin
Bröring
b and
Gunhild
Layer
*ac
aInstitute of Microbiology, Technische Universität Braunschweig, Spielmannstr. 7, 38106 Braunschweig, Germany
bInstitute of Inorganic and Analytical Chemistry, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany
cInstitute of Biochemistry, Leipzig University, Brüderstraße 34, 04103 Leipzig, Germany. E-mail: gunhild.layer@uni-leipzig.de
dMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany
First published on 30th March 2016
Abstract
The heme synthase AhbD catalyzes the oxidative decarboxylation of two propionate side chains of iron-coproporphyrin III to the corresponding vinyl groups of heme during the alternative heme biosynthesis pathway occurring in sulfate-reducing bacteria and archaea. AhbD belongs to the family of Radical SAM enzymes and contains two [4Fe–4S] clusters. Whereas one of these clusters is required for substrate radical formation, the role of the second iron–sulfur cluster is not known. In this study, the function of the auxiliary cluster during AhbD catalysis was investigated. Two single cluster variants of AhbD from M. barkeri carrying either one of the two clusters were created. Using these enzyme variants it was shown that the auxiliary cluster is not required for substrate binding and formation of the substrate radical. Instead, the auxiliary cluster is involved in a late step of AhbD catalysis most likely in electron transfer from the reaction intermediate to a final electron acceptor. Moreover, by using alternative substrates such as coproporphyrin III, Cu-coproporphyrin III and Zn-coproporphyrin III for the AhbD activity assay it was observed that the central iron ion of the porphyrin substrate also participates in the electron transfer from the reaction intermediate to the auxiliary [4Fe–4S] cluster. Altogether, new insights concerning the completely uncharacterized late steps of AhbD catalysis were obtained.
Introduction
In almost all living organisms heme serves as an essential cofactor in enzymes, redox proteins involved in electron transport chains or gas sensing regulators. Depending on the organism, heme is either acquired from external sources or synthesized by the organism itself. In eukaryotes and most bacteria, heme is formed via the so-called classical biosynthesis pathway, whereas in sulfate-reducing bacteria and archaea an alternative route is operative.1–7 In both pathways heme is synthesized from the common precursor uroporphyrinogen III (UROGEN III). During the classical route, heme is then formed via the intermediates coproporphyrinogen III, protoporphyrinogen IX, and protoporphyrin IX (PROTO IX).8 In contrast, during the alternative pathway UROGEN III is converted to precorrin-2 which is subsequently oxidized to sirohydrochlorin. The insertion of iron leads to the formation of siroheme which is then decarboxylated to 12,18-didecarboxysiroheme. This intermediate is transformed into iron-coproporphyrin III (Fe-COPRO III) which is finally converted to heme.6,7The last reaction in the alternative heme biosynthesis pathway, i.e. the conversion of Fe-COPRO III to heme, is catalyzed by the heme synthase AhbD (alternative heme biosynthesis enzyme D) which is, so far, scarcely characterized.9 During the reaction the two propionate side chains at positions C3 and C8 of Fe-COPRO III are oxidatively decarboxylated to the corresponding vinyl groups (Scheme 1).
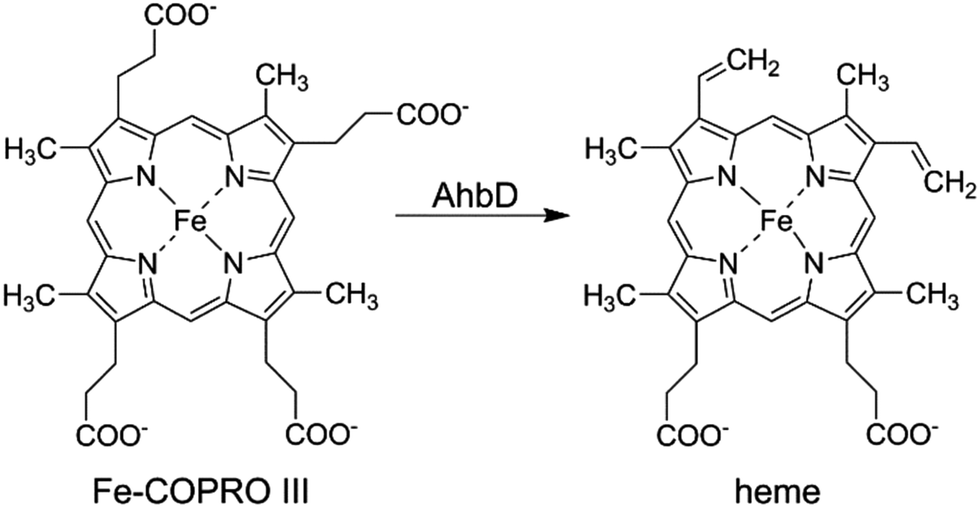 | ||
| Scheme 1 Reaction catalyzed by AhbD. | ||
AhbD belongs to the large family of Radical SAM (RS) enzymes. As such, the amino acid sequence of AhbD contains a typical cysteine-rich motif at the N-terminus (CX3CX2C) which provides three cysteine ligands for the coordination of a catalytically essential [4Fe–4S]2+/1+ cluster present in all RS enzymes (RS cluster).10 The fourth, non-cysteine ligated iron atom of this cluster is coordinated by one molecule of S-adenosyl-L-methionine (SAM) which is used to initiate radical catalysis.11 Thus, the common reaction steps employed by all members of the RS family consist of the initial reduction of the RS cluster to the [4Fe–4S]1+ state and subsequent electron transfer onto the bound SAM which results in the cleavage of the latter to methionine and a 5′-deoxyadenosyl radical (DOA˙). This radical then abstracts a hydrogen atom from the actual substrate which is unique for each RS enzyme.12 In the case of AhbD, it is assumed that the hydrogen atom abstraction occurs at the β-carbon of the propionate side chain of Fe-COPRO III leading to substrate radical formation in analogy to the formation of the substrate radical during HemN catalysis (see below).6,13 The substrate radical is then decarboxylated to form the vinyl group of the reaction product. Overall, this reaction takes place twice in order to obtain both vinyl groups at C-atoms C3 and C8 of the end product heme. Assuming that the decarboxylation step yields CO2, a terminal electron acceptor is required for the overall reaction (Scheme 2).
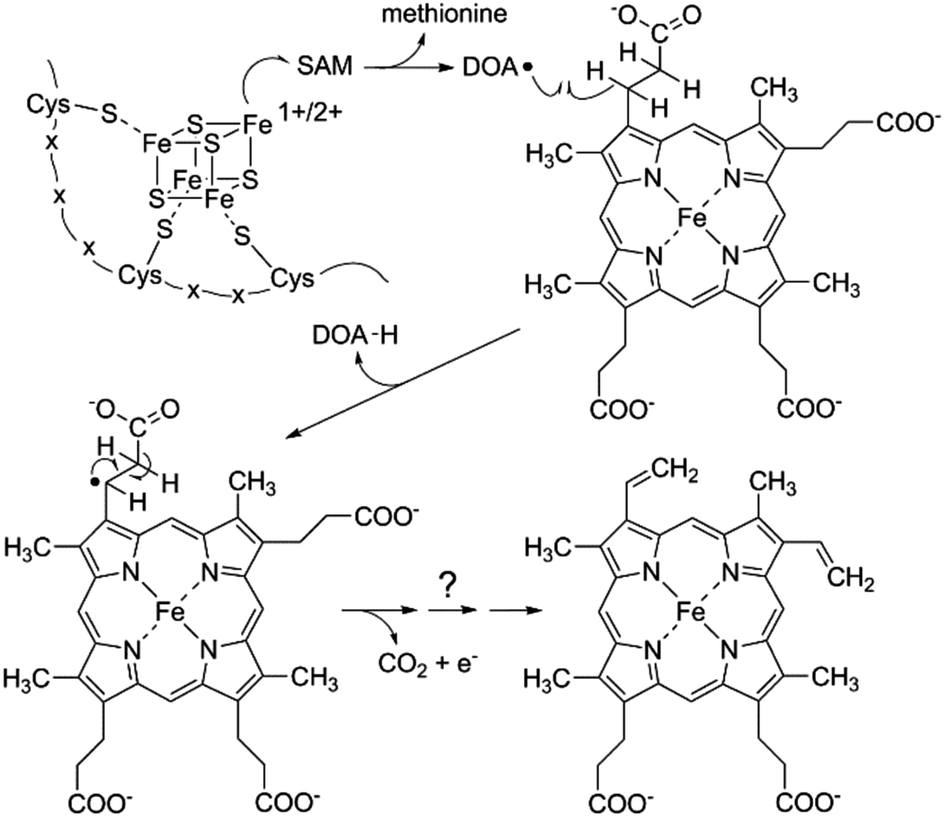 | ||
| Scheme 2 Initial reaction steps of AhbD catalysis. | ||
Interestingly, the coproporphyrinogen III dehydrogenase HemN, which also belongs to the RS enzyme family,10 performs exactly the same reaction during the classical heme biosynthesis pathway. However, HemN uses coproporphyrinogen III as its substrate which is converted to protoporphyrinogen IX.14 The three-dimensional structure of HemN was determined and the first steps of its catalytic mechanism up to substrate radical formation (as described above) were elucidated.11,13 In contrast, the final steps of HemN catalysis are still uncharacterized. AhbD from M. barkeri and HemN from Escherichia coli share only 17% amino acid sequence identity within the 349 amino acids of the AhbD sequence. As described above, the two enzymes catalyze the same reaction but use different substrates: an iron-porphyrin in the case of AhbD in contrast to a porphyrinogen in the case of HemN. Moreover, in contrast to HemN, AhbD was shown to contain a second [4Fe–4S] cluster in addition to the RS cluster.9 Based on amino acid sequence alignments, it was proposed that this auxiliary cluster might be coordinated by the cysteine residues present in an additional cysteine-rich motif (CX2CX5CX2CX17C) at the C-terminal end of AhbD.9 However, the function of the auxiliary cluster of AhbD during catalysis is not known. It might be involved in the binding or positioning of the substrate Fe-COPRO III. Alternatively, it might be required for a late step during catalysis such as electron transfer to a terminal electron acceptor. Therefore, the aims of this study were to obtain novel insights into the unknown function of the auxiliary [4Fe–4S] cluster of AhbD and to investigate the late steps of AhbD catalysis taking place after substrate radical formation.
Results and discussion
Production of single cluster AhbD variants
The first aim of this study was to analyze the properties of the two [4Fe–4S] clusters within AhbD from M. barkeri individually. For this purpose, two single cluster variants were created in which either the N-terminal RS cluster or the C-terminal auxiliary cluster was missing. For the deletion of the RS cluster the two cysteine residues Cys19 and Cys23 of the typical RS cysteine motif C19X3C23X2C26 (numbers corresponding to the AhbD sequence from M. barkeri) were exchanged against alanine by site directed mutagenesis. The resulting AhbD variant was named AhbD C19A/C23A. The auxiliary [4Fe–4S] cluster of AhbD was proposed to be ligated by three or four cysteine residues of the additional cysteine-rich motif C312X2C315X5C321X2C324X17C342.7,9 In order to delete this C-terminal cluster, the two cysteine residues Cys321 and Cys324 were exchanged against alanine resulting in the formation of the AhbD variant C321A/C324A. The recombinant wild type (wt) AhbD and the two single cluster variants were produced as fusion proteins carrying an N-terminal His6-tag and purified aerobically via immobilized metal ion affinity chromatography (IMAC). After the purification of the proteins, an in vitro iron–sulfur cluster reconstitution was performed under anaerobic conditions. The purified, reconstituted wt AhbD contained 7.6 mol iron and 7.5 mol sulfide per mol monomeric AhbD in agreement with the presence of two [4Fe–4S] clusters. In contrast, the iron and sulfide contents of the purified and reconstituted single cluster variant AhbD C19A/C23A were only 54% and 55%, respectively, compared to the wt protein. Similarly, the enzyme variant AhbD C321A/C324A exhibited iron and sulfide contents of 43% and 51%, respectively, compared to the wt protein. These results suggested that in each AhbD variant one [4Fe–4S] cluster binding site was successfully eliminated.Characterization of the individual [4Fe–4S] clusters by EPR spectroscopy
In order to further characterize the two [4Fe–4S] clusters individually and to verify their structural integrity in the single cluster variants, EPR spectroscopy was performed. After dithionite reduction, EPR spectra were recorded for wt AhbD and the two single cluster variants (Fig. 1). The measurement of each variant, AhbD C19A/C23A and AhbD C321A/C324A, showed a typical spectrum of a single [4Fe–4S]1+ cluster with g = 2.0525(5), 1.928(1), 1.903(1) for AhbD C19A/C23A and g = 2.039(1), 1.912(1), 1.896(1) for AhbD C321A/C324A. The sum of the two individual spectra of the AhbD variants equals the spectrum of wt AhbD confirming the presence of two distinct [4Fe–4S]2+/1+ clusters within AhbD, one located at the N-terminus and the other at the C-terminus of the enzyme.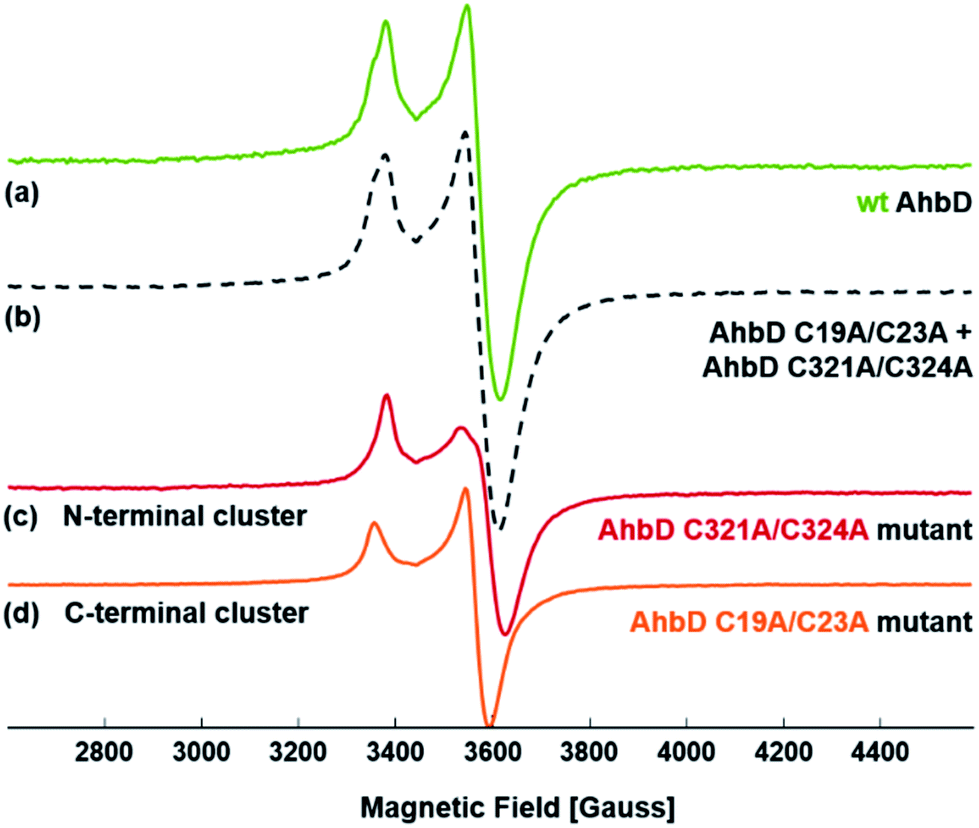 | ||
| Fig. 1 EPR spectra of the [4Fe–4S]1+ clusters in AhbD. The spectra were recorded at 10 K for (a) wt AhbD, (c) AhbD C321A/C324A and (d) AhbD C19A/C23A. The trace in (b) represents the sum of (c) and (d). Experimental parameters: field modulation 8 Gauss. Microwave frequency = 9.6428 GHz. Power 0.2 mW. | ||
Both [4Fe–4S] clusters of AhbD are essential for its heme synthase activity
The ability of purified, reconstituted wt AhbD and the two single cluster variants to convert Fe-COPRO III into heme was tested using an established in vitro enzyme activity assay with sodium dithionite (DT) as the reducing agent for the initial reduction of the RS cluster.7 After incubation of the assay mixtures their tetrapyrrole content was analyzed by HPLC. As shown in Fig. 2, heme was formed in the presence of wt AhbD in the activity assay. In this case, the substrate Fe-COPRO III was completely consumed after 24 h of incubation at 17 °C. In addition to the final reaction product heme, another iron-porphyrin was detected (retention time of about 30.1 min) most likely representing a monovinyl reaction intermediate (Fe-harderoporphyrin or Fe-isoharderoporphyrin) as observed previously.9 In contrast to the wt enzyme, both AhbD variants, C19A/C23A and C321A/C324A, were unable to convert Fe-COPRO III into detectable amounts of heme under identical reaction conditions (Fig. 2). Moreover, in both cases no monovinyl reaction intermediate was observed. Therefore, the deletion of either one of the two [4Fe–4S] clusters apparently abolished the ability of AhbD to decarboxylate the two propionate side chains of Fe-COPRO III to the corresponding vinyl groups. This result showed that both [4Fe–4S] clusters are essential for the overall heme synthase activity of AhbD from M. barkeri.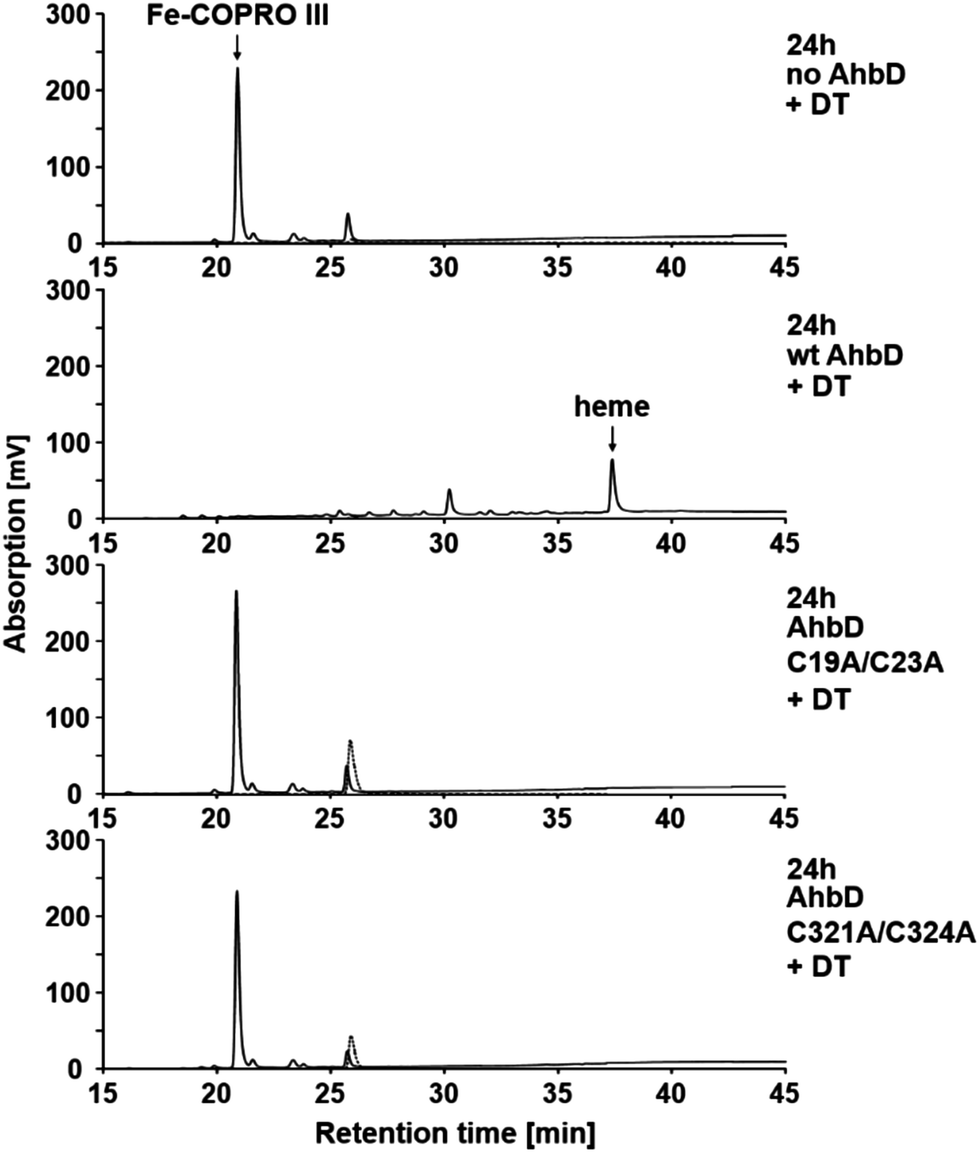 | ||
| Fig. 2 Heme synthase activity of wt AhbD and the AhbD variants. The tetrapyrrole content in the assay mixtures was analyzed after 24 h of incubation at 17 °C by HPLC. The elution of the tetrapyrroles was monitored by measuring the absorption at 400 nm (solid line) and the fluorescence (dotted line, excitation at 409 nm, emission at 630 nm). Fe-COPRO III eluted at 20.9 min, the potential monovinyl intermediate at 30.1 min and heme at 37.5 min. | ||
For the AhbD variant C19A/C23A this result was not surprising. This variant lacked the N-terminal RS cluster and was therefore unable to initiate SAM cleavage and radical catalysis. The reason for the lack of heme synthase activity of variant C321A/C324A was less obvious. Possibly, the C-terminal cluster might also be involved indirectly in SAM cleavage. Alternatively, it might be required for substrate binding or it could play an essential role for a late step during catalysis. These different hypotheses for the role of the auxiliary cluster of AhbD were tested and the obtained results are described in the following sections.
The auxiliary cluster of AhbD is not required for SAM cleavage
As described above, both [4Fe–4S] clusters of AhbD are required for the overall heme synthase activity of the enzyme. However, the reaction sequence of AhbD can be dissected into at least two observable parts: (i) the reductive cleavage of SAM and (ii) the overall formation of heme. The homolytic cleavage of SAM results in the formation of methionine and the 5′-deoxyadenosyl radical which is quenched by the abstraction of a hydrogen atom from the substrate. This first part of the AhbD reaction sequence can be followed by the HPLC analysis of the SAM cleavage product 5′-deoxyadenosine (DOA). In vitro, some Radical SAM enzymes such as RlmN and HemN require the presence of their substrate in order to be able to cleave SAM,15,16 whereas for others, like HydE and HydG, the substrate is not necessary for SAM cleavage.17 In the latter case, the 5′-deoxyadenosyl radical is probably quenched by hydrogen abstraction from the solvent or the enzyme itself.Here, we first tested whether wt AhbD from M. barkeri requires the presence of its substrate Fe-COPRO III for SAM cleavage. As shown in Fig. 3, no DOA was formed in the SAM cleavage assay conducted without Fe-COPRO III. In contrast, in the presence of Fe-COPRO III in the reaction mixture the formation of DOA was observed by HPLC analysis. Thus, in the case of AhbD from M. barkeri the substrate Fe-COPRO III is required for SAM cleavage in vitro. This observation is in line with the previous description of the SAM cleavage activity of the homologous AhbD protein from Desulfovibrio vulgaris. In this case, SAM cleavage was observed to take place to a small extent even without the substrate, but was enhanced about ten-fold in the presence of Fe-COPRO III.9 Since for AhbD from M. barkeri the formation of DOA is dependent on the presence of the substrate, the SAM cleavage assay can be used as an indirect indicator for the binding of Fe-COPRO III to the enzyme. Consequently, in the following, we assume that if DOA is formed, the substrate is bound in the active site of AhbD.
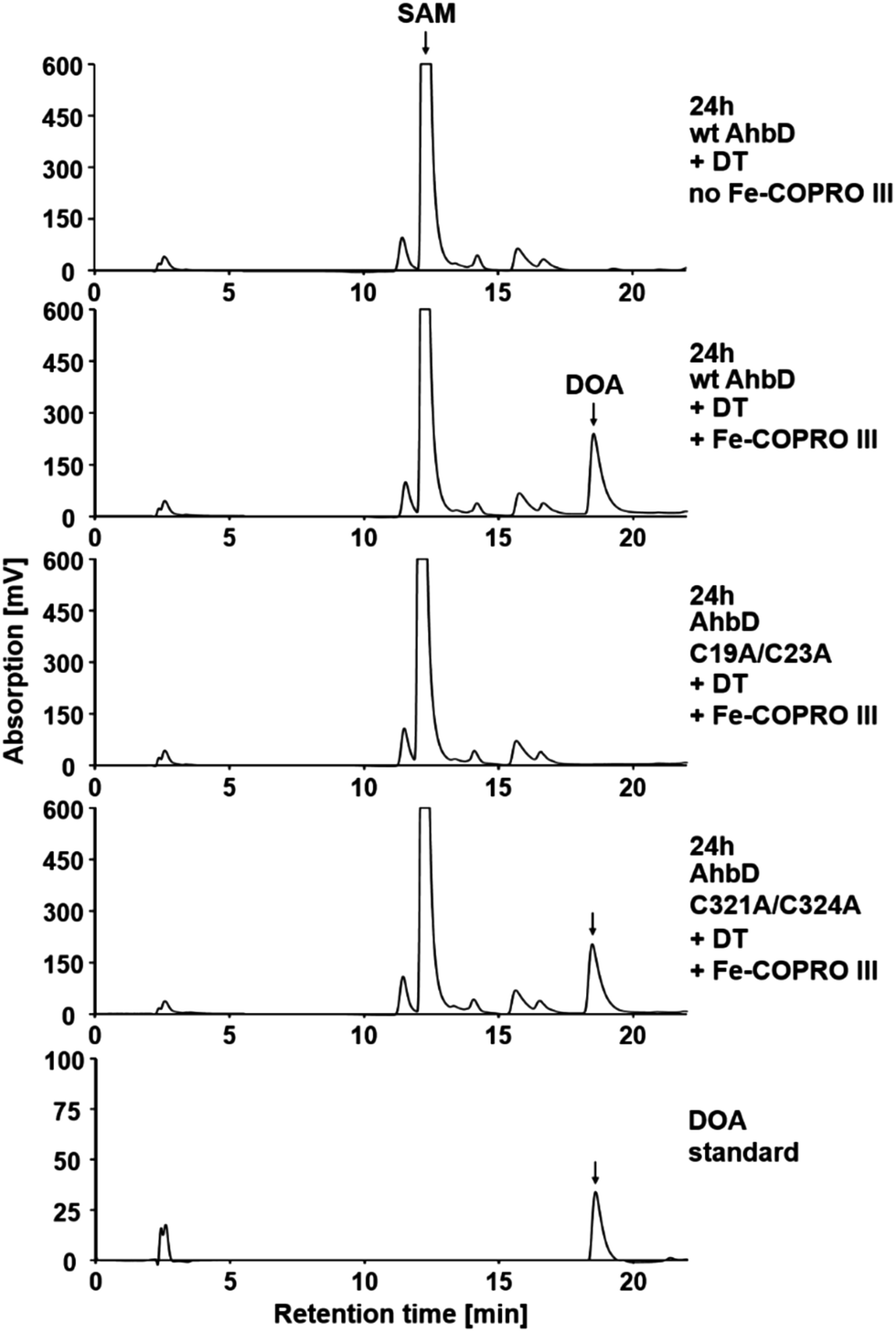 | ||
| Fig. 3 SAM cleavage activity of wt AhbD and the AhbD variants. The formation of the SAM cleavage product DOA was followed by HPLC analysis. The elution of SAM and DOA was monitored by measuring the absorption at 254 nm. SAM eluted at 12.3 min and DOA at 18.6 min. | ||
Next, the two single cluster variants of AhbD were tested for their ability to cleave SAM in the presence of Fe-COPRO III. As expected, the AhbD variant C19A/C23A was unable to cleave SAM confirming the role of the N-terminal [4Fe–4S] cluster as the RS cluster of AhbD. In contrast, the AhbD variant C321A/C324A lacking the C-terminal cluster was able to cleave SAM comparable to the wt enzyme (Fig. 3). Therefore, the auxiliary C-terminal [4Fe–4S] cluster is apparently not required for the first part of the AhbD reaction sequence up to the step of substrate radical formation. Instead, it must fulfil an essential function during the late steps of AhbD catalysis since it is absolutely required for overall heme formation (see above). Alternatively, the C-terminal cluster might also be required for substrate binding. However, this possibility seems less likely since our SAM cleavage assay which also serves as an indicator for substrate binding was positive for the enzyme variant C321A/C324A. Nevertheless, in order to further characterize the ability of the two single cluster variants to bind Fe-COPRO III, a substrate binding assay using UV-visible absorption spectroscopy was performed.
Neither of the iron–sulfur clusters is required for substrate binding
For the substrate binding assay an Fe(III)-COPRO III solution was mixed with either wt AhbD, AhbD C19A/C23A or AhbD C321A/C324A, and UV-visible absorption spectra were recorded immediately after mixing and after 270 s incubation time. Additionally, the Fe(III)-COPRO III solution was also mixed with either buffer alone or alcohol dehydrogenase (Sigma-Aldrich) as a control, and UV-visible absorption spectra were recorded after the same time intervals as for the mixtures containing AhbD (Fig. 4). The Soret band of monomeric Fe(III)-COPRO III in buffer was recorded at 382 nm while the second peak at about 350 nm indicated that the substrate solution also contained a fraction of dimeric Fe(III)-COPRO III.18 When no protein was added to this substrate solution (buffer control) the absorption spectrum of Fe(III)-COPRO III remained unchanged within the time frame of the measurements and even after prolonged incubation for 24 h. In contrast, the addition of wt AhbD led to an immediate shift of the Soret band of the monomeric form of the substrate to 390 nm indicating the binding of Fe(III)-COPRO III to the enzyme. After 270 s of incubation the peak was further shifted to 392 nm. Finally, after 24 h of incubation the Soret band of the enzyme-bound substrate displayed its maximum at 396 nm.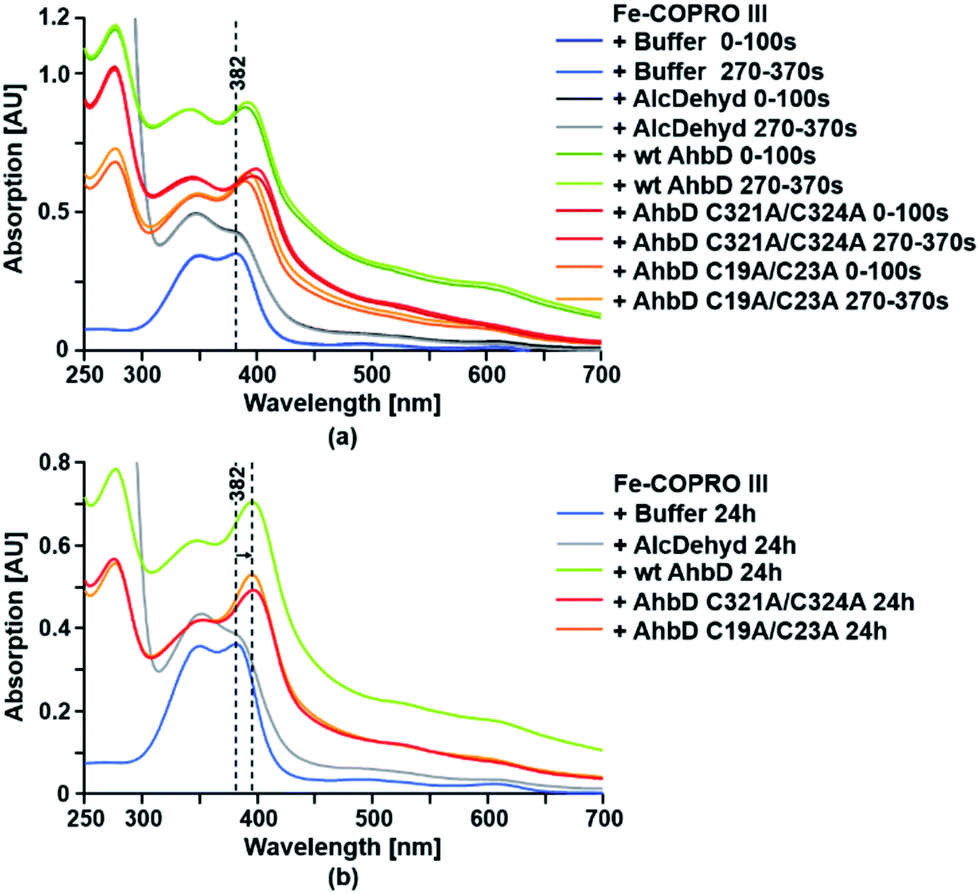 | ||
| Fig. 4 Substrate binding assay for wt AhbD and the AhbD variants. The binding of Fe-COPRO III to wt AhbD and the AhbD variants was followed by UV-visible absorption spectroscopy. Fe-COPRO III was mixed with either buffer or the indicated proteins and spectra were recorded (a) directly after mixing (0–100 s), after 270 s (270–370 s) and (b) after 24 h. The observed shift of the Soret band upon addition of AhbD is indicated. An UV-visible absorption spectrum of purified wt AhbD alone is shown in ESI Fig. A† for comparison. | ||
Essentially the same spectral changes were observed after the addition of either AhbD C19A/C23A or AhbD C321A/C324A to the substrate solution with only insignificant variations of the initial wavelength shifts. Additionally, the absorption spectra measured after 24 h of incubation clearly showed that both AhbD variants were able to bind the substrate comparable to the wt enzyme. In contrast, no shift of the Soret band of monomeric Fe(III)-COPRO III was observed after the addition of alcohol dehydrogenase to the substrate solution even after 24 h of incubation.
In order to quantify the binding of the Fe(III)-COPRO III to wt AhbD and the two AhbD cluster variants, the dissociation constants (Kd) were determined by the measurement of the quenched tryptophan fluorescence of AhbD upon substrate binding (ESI Fig. B†). Using this method, a Kd value of 14.2 ± 1.9 μM was determined for the wt AhbD. Similar Kd values of 15.3 ± 1.8 μM for AhbD C19A/C23A and 11.2 ± 1.2 μM for AhbD C321A/C324A were obtained. Thus, all three AhbD proteins bind the substrate with similar affinity.
Altogether, these experiments demonstrate that Fe(III)-COPRO III binds specifically to AhbD from M. barkeri and that both single cluster variants are still able to bind the substrate with affinities comparable to that of the wt protein. Therefore, concerning the role of the auxiliary C-terminal [4Fe–4S] cluster we conclude that it is not required for substrate binding as already suggested by the results of the SAM cleavage assay described above. Since the auxiliary cluster of AhbD is not required for substrate binding and SAM cleavage, it might be involved in a late step of the AhbD reaction sequence such as the transfer of an electron from the substrate radical onto a terminal electron acceptor. In order to investigate whether such a role in electron transfer is feasible, the redox potentials of the individual iron–sulfur clusters within AhbD were measured by cyclic voltammetry.
The redox potential of the auxiliary [4Fe–4S] cluster of AhbD is compatible with a role in electron transfer
The redox potentials of the two [4Fe–4S] clusters within AhbD were determined for the wt protein and the two variants C19A/C23A and C321A/C324A either in the absence or presence of SAM and the substrate Fe-COPRO III. All electrochemical measurements of the proteins showed a quasi-reversible reduction of the [4Fe–4S]2+/1+ clusters with half-wave potentials in the range of −340 to −400 mV (ESI Fig. C†), which is common for proteins containing such clusters.19,20 The half-wave potentials of the individual clusters were determined to be at −344 mV for the RS cluster (AhbD C321A/C324A) and at −359 mV for the auxiliary cluster (AhbD C19A/C23A) in the absence of SAM. For both clusters these potentials were not significantly altered in the presence of SAM (−343 mV for the RS cluster and −366 mV for the auxiliary cluster). The cyclic voltammogram of the wt AhbD, which contains both iron–sulfur clusters, showed one single reduction process at −371 mV in the absence of SAM. This finding indicates that there is no electronic interaction between the two iron–sulfur clusters and such an electrochemical behavior was also described for other proteins, which contain two cuboidal [4Fe–4S] units.21 Moreover, this observation is in agreement with the reported reduction potential determined for AhbD from D. vulgaris, for which also one single reduction potential at −390 mV was measured.9 Further measurements of wt AhbD in the presence of added SAM did not result in a significantly shifted potential (−396 mV). Also, cyclic voltammograms of AhbD in the presence of Fe-COPRO III displayed traces representing the sum of the single compounds without any noticeable shifts in the potentials of the iron–sulfur clusters (ESI Fig. C†). In summary, these experiments showed that not only the RS cluster is redox active, but also the C-terminal auxiliary cluster supporting its putative function in electron transfer during AhbD catalysis.One possible scenario for the electron transfer events during the AhbD catalytic cycle is depicted in Scheme 3 and ESI Fig. D.† After the formation of the substrate radical, the single electron is delocalized over the iron-porphyrin ring system and the central metal ion facilitating the decarboxylation of the propionate side chain. Assuming that CO2 is the product of the decarboxylation event, the vinyl group formation is accompanied by the reduction of the central iron ion to the Fe(II) state. Then, the Fe(II)-harderoporphyrin might transfer one electron to the auxiliary iron–sulfur cluster which in turn would pass the electron further on to an external, terminal electron acceptor. The resulting Fe(III)-harderoporphyrin could then serve as the substrate for the second decarboxylation reaction proceeding by a mechanism identical to the one for the first decarboxylation after release of methionine and DOA and rebinding of a new SAM. Thus, the role of the C-terminal [4Fe–4S] cluster would be to accept an electron from the reduced reaction intermediate and to pass it on to a terminal, so far unknown electron acceptor.
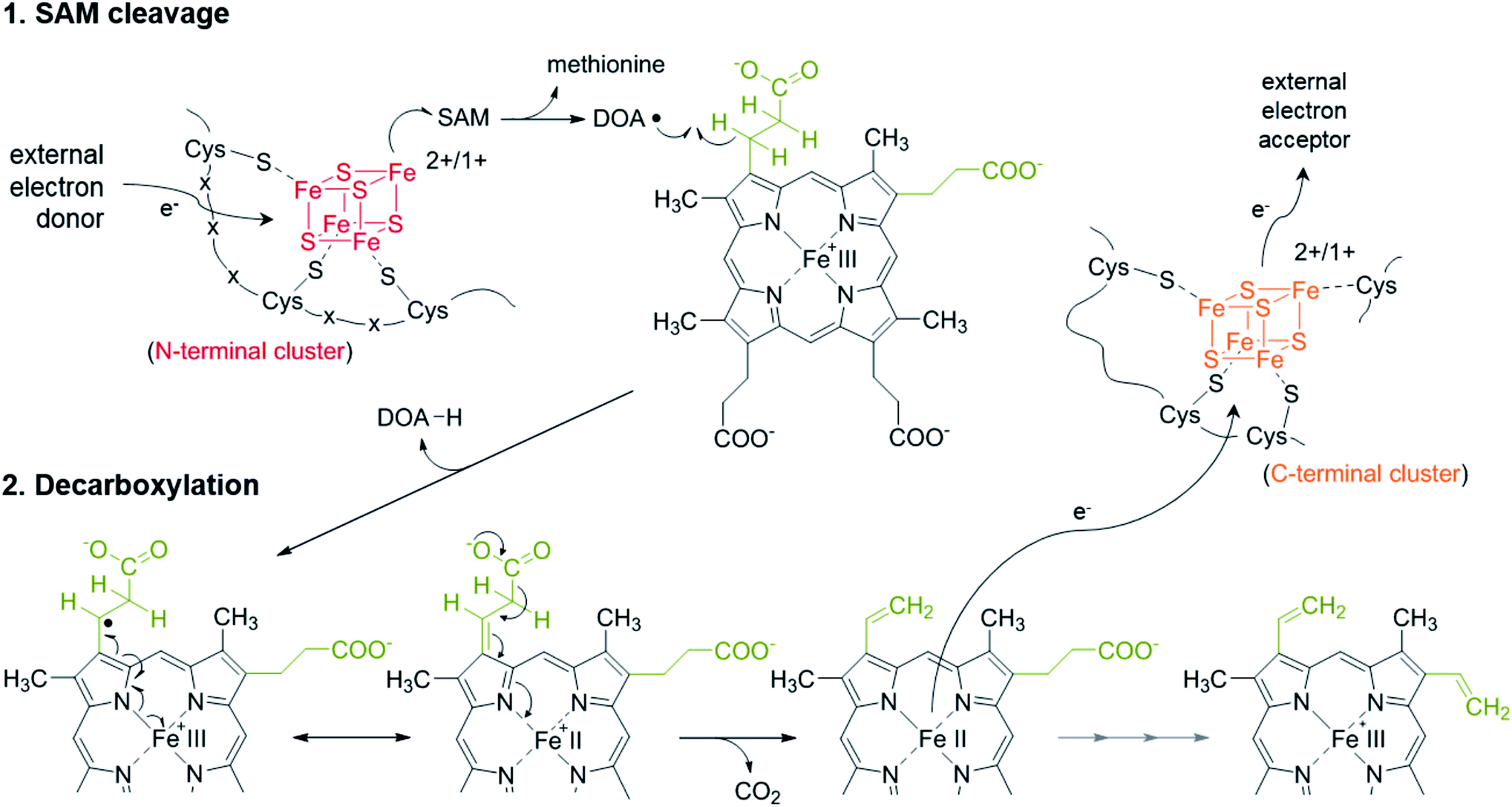 | ||
| Scheme 3 Proposed reaction mechanism for AhbD. | ||
In order to test whether an electron transfer from a reduced iron-porphyrin to the C-terminal cluster (−359 mV) is feasible, we also determined the half-wave potential of Fe-COPRO III and heme (haemin) by cyclic voltammetry. It can be assumed that the redox potential of Fe-harderoporphyrin, which was not available for measurements, lies between those of Fe-COPRO III and heme. In the absence or presence of AhbD half-wave potentials in the range of −650 mV to −659 mV were determined for Fe-COPRO III. The half-wave potential of haemin in buffer (ESI Fig. E†) was determined at −609 mV. Therefore, assuming that the redox potential of Fe-harderoporphyrin lies between −659 mV and −609 mV, the proposed electron transfer from the monovinyl Fe(II)-porphyrin intermediate to the auxiliary cluster is possible.
For the proposed reaction mechanism including the reversible reduction/oxidation of the central metal ion of the porphyrin substrate/intermediate, the central iron plays a crucial role. In order to further substantiate the proposed mechanism and to investigate the role of the central iron ion, alternative substrates were used for AhbD activity assays. These substrate analogs contained either no metal at all (coproporphyrin III (COPRO III)) or Cu (Cu-COPRO III) or Zn (Zn-COPRO III). Before testing the AhbD activity with these substrates, we tested whether the enzyme was able to bind the substrate analogs.
AhbD binds the substrate analogs COPRO III, Cu-COPRO III and Zn-COPRO III
The binding of the substrate analogs to AhbD was tested under identical conditions as for the true substrate Fe-COPRO III. A shift of the Soret band to longer wavelengths upon addition of the enzyme was taken as an indication for the formation of an enzyme–substrate complex. As shown in Fig. 5 and ESI Fig. F,† such a shift of the Soret band was observed for all three substrate analogs. For the metal-free COPRO III the Soret band was shifted from 397 nm for the unbound form to 406 nm for the AhbD-bound form. For the Cu-COPRO III the shift of the Soret band upon AhbD addition was from 379 nm to 398 nm. Finally, the Soret band of free Zn-COPRO III at 403 nm was shifted to 410 nm in the presence of AhbD.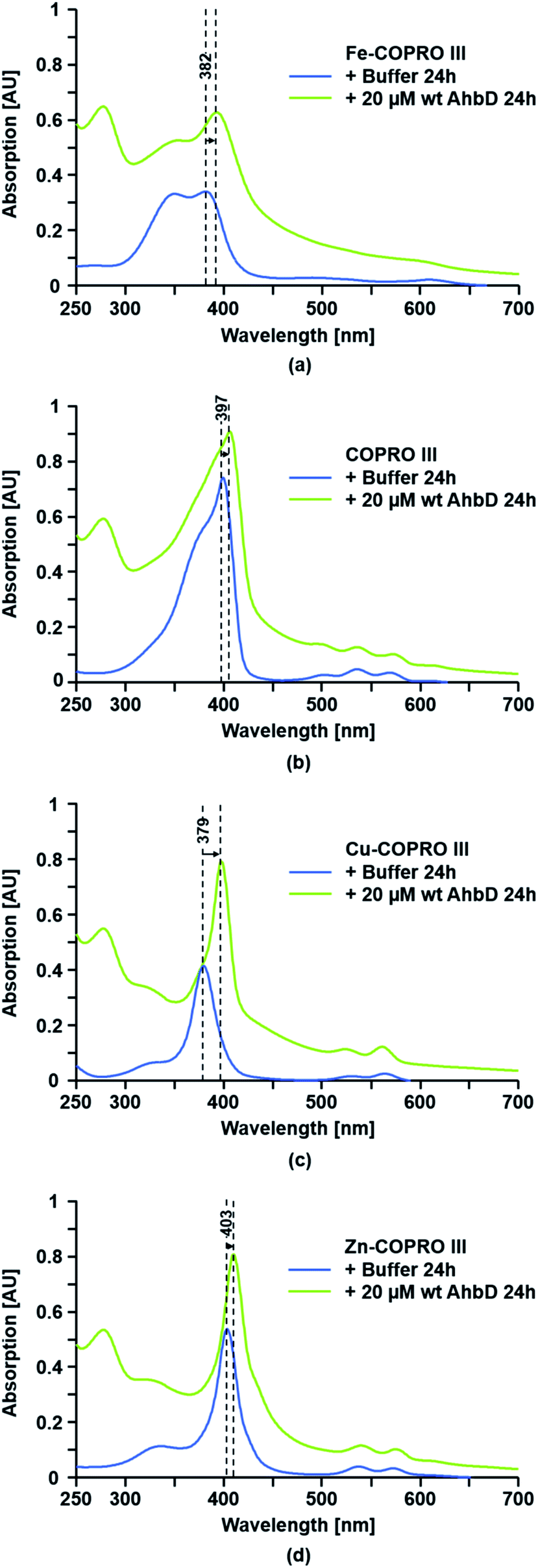 | ||
| Fig. 5 Binding assay for wt AhbD using different substrate analogs. The binding of (a) Fe-COPRO III, (b) COPRO III, (c) Cu-COPRO III and (d) Zn-COPRO III to wt AhbD was followed by UV-visible absorption spectroscopy. The spectra were recorded after 24 h of incubation after mixing the protein with the respective substrate analog. The shift of the Soret bands upon AhbD addition is indicated. | ||
In addition to the binding assays using UV-visible absorption spectroscopy, we also performed SAM-cleavage assays of AhbD in the presence of the substrate analogs to test their binding to the enzyme. As described above, the cleavage of SAM only occurs when the substrate is bound to AhbD. In the presence of all three substrate analogs SAM was cleaved and DOA was detected by HPLC analysis (ESI Fig. G†). However, for COPRO III and Cu-COPRO III the SAM cleavage activity seemed to be a little bit lower than with Fe-COPRO III and the SAM cleavage activity in the presence of Zn-COPRO III was even more impaired, although still detectable.
In order to test, whether these slight differences in SAM cleavage in the presence of the substrate analogs as compared to the SAM cleavage in the presence of the true substrate were due to differences in the binding affinities, the Kd values for the substrate analogs and wt AhbD were determined by the measurement of tryptophan fluorescence quenching (ESI Fig. H†). For COPRO III a Kd of 33.9 ± 3.9 μM, for Cu-COPRO III a Kd of 56 ± 3.5 μM and for Zn-COPRO III a Kd of 38.8 ± 2.4 μM was determined. Thus, all three substrate analogs bind with slightly lower affinity to the wt AhbD than the true substrate according to the two-fold higher Kd values for COPRO III and Zn-COPRO III and the four-fold higher Kd for Cu-COPRO III. The somewhat higher Kd values might explain the slightly lower SAM cleavage activity in the presence of COPRO III and Cu-COPRO III, but do not explain the low SAM cleavage activity in the presence of Zn-COPRO III. Here, a slightly different orientation of the substrate analog within the active site might be responsible for the inefficient SAM cleavage.
Nevertheless, all three methods, UV-visible absorption spectroscopy, SAM-cleavage assays and fluorescence quenching, showed that AhbD was able to bind all three artificial substrates and, therefore, enzyme activity assays employing these molecules were performed.
AhbD displays very low decarboxylase activity with the substrate analogs COPRO III, Cu-COPRO III and Zn-COPRO III
To investigate the influence of the central iron ion of Fe-COPRO III on the activity of AhbD, an enzyme activity assay using COPRO III instead of Fe-COPRO III was performed. For comparison, the AhbD assay was performed under the same assay conditions using the true substrate Fe-COPRO III. As already described above, for the assay containing Fe-COPRO III the substrate was completely consumed after 24 h of incubation and converted to heme or at least the monovinyl intermediate (Fig. 6 and 2). In contrast, in the assay mixture containing COPRO III there was still a high amount of COPRO III (retention time around 25.6 min) present after the same incubation time. However, minor amounts of PROTO IX (retention time 40.5 min) and a potential monovinyl intermediate (retention time 34.5 min) were also detected. These reaction products were not found in a control assay mixture lacking AhbD. Therefore, AhbD was able to convert COPRO III to the corresponding monovinyl intermediate and the product PROTO IX; however, the activity of AhbD with the artificial substrate was very low compared to its activity in the presence of the true substrate Fe-COPRO III.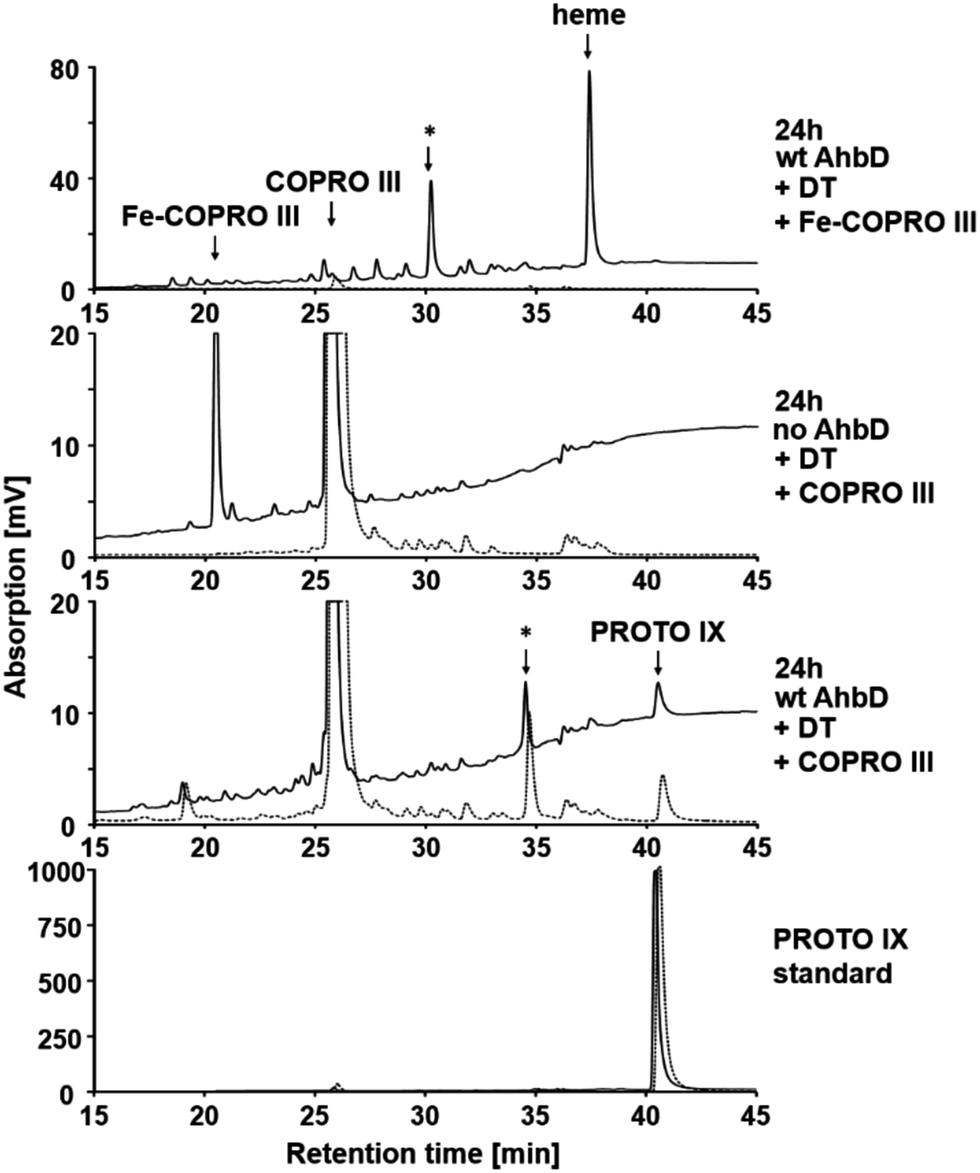 | ||
| Fig. 6 Decarboxylase activity of wt AhbD with COPRO III as the substrate. The tetrapyrrole content in the assay mixtures was analyzed after 24 h of incubation at 17 °C by HPLC. The elution of the tetrapyrroles was monitored by measuring the absorption at 400 nm (solid line) and the fluorescence (dotted line, excitation at 409 nm, emission at 630 nm). Fe-COPRO III eluted at 20.9 min, the potential monovinyl intermediate at 30.1 min and heme at 37.5 min (top panel). COPRO III eluted at 25.7 min, the corresponding monovinyl intermediate at 34.5 min and PROTO IX at 40.5 min (lower panels). The respective monovinyl intermediates are highlighted by an asterisk. | ||
Since the binding affinity of COPRO III to AhbD is not drastically different from that of Fe-COPRO III (only two-fold higher Kd) and SAM cleavage is clearly taking place in the presence of COPRO III, this result indicates that the central iron ion of the porphyrin substrate indeed plays a crucial role for the efficient conversion of the propionate side chains to the corresponding vinyl side chains by AhbD.
In order to further substantiate this idea, the same AhbD activity assay was also performed using Cu-COPRO III and Zn-COPRO III. As shown in ESI Fig. I† similar results were obtained as in the assay with COPRO III. In both cases a very low decarboxylase activity of AhbD was observed since low amounts of monovinyl intermediates and the reaction products Cu-PROTO IX and Zn-PROTO IX (detected as PROTO IX due to extraction conditions) were detected by HPLC analysis. These reaction products were not found in the respective assay mixtures lacking AhbD. However, as in the case for the assay with COPRO III and in contrast to the assay with Fe-COPRO III, the major amount of the two artificial substrates was not consumed after 24 h showing that they served only as very poor substrates for AhbD. In the case of the Zn-COPRO III the product formation is comparable to that of the assay with COPRO III despite the seemingly lower SAM cleavage activity. For Cu-COPRO III product formation is hardly detectable at all. Again, these very low decarboxylase activities of AhbD in presence of the substrate analogs cannot be explained by their lower binding affinity alone.
Overall, the experiments with the substrate analogs support the hypothesis that the central iron ion of the substrate Fe-COPRO III participates in catalysis most likely as an electron relay ultimately transferring the electron from the reaction intermediate to the auxiliary [4Fe–4S] cluster. In our current activity assay, the reductant for the RS cluster in order to initiate catalysis is sodium dithionite which is commonly used for activity assays of Radical SAM enzymes. However, in the presence of sodium dithionite the auxiliary cluster as well as the substrate itself are most likely also present in their reduced forms. Considering our proposed reaction mechanism this might be a reason why the reaction catalyzed by AhbD is quite slow in vitro. Therefore, in the particular case of AhbD, sodium dithionite should be replaced by a physiological electron donor such as flavodoxin or ferredoxin in future experiments.
Conclusions
All RS enzymes contain at least one [4Fe–4S] cluster coordinated by three cysteine residues which is able to cleave SAM into a 5′-deoxyadenosyl radical and methionine thereby initiating radical catalysis. In AhbD from M. barkeri a second, auxiliary [4Fe–4S] cluster is present, coordinated via cysteine residues at the C-terminal end of the protein. Like for several other RS enzymes containing auxiliary [4Fe–4S] clusters, the overall enzyme activity of AhbD is lost upon deletion of the auxiliary cluster. The functions of the auxiliary clusters in the various RS SAM enzymes are diverse and include roles in substrate binding and orientation, sulfur donation and electron transfer.22 In the case of AhbD, a role in sulfur donation is irrelevant and a role in substrate binding could be excluded by the experiments in this study. However, based on the results described here, we propose a role of the auxiliary [4Fe–4S] cluster in electron transfer from the reaction intermediate to a terminal, so far unknown electron acceptor. Furthermore, we observed that the central iron ion of the Fe-COPRO III substrate of AhbD participates in this electron transfer process. Therefore, we obtained new insights into the reaction steps that follow the initial formation of the substrate radical during AhbD catalysis. Obviously, these steps differ from those (still uncharacterized) occurring during HemN catalysis which catalyzes the identical reaction but uses a different substrate (porphyrinogen without metal ion) and does not contain an auxiliary iron–sulfur cluster. Nevertheless, both enzymes require a terminal electron acceptor for the oxidative decarboxylation of the propionate side chains which in both cases also remains to be identified in the future.Experimental section
Chemicals
All chemicals and reagents were obtained from Sigma-Aldrich (Taufkirchen, Germany), Carl Roth (Karlsruhe, Germany), Gerbu (Heidelberg, Germany), Merck (Darmstadt, Germany). DNA polymerases, restriction enzymes, PCR requisites as well as the Q5 Site-Directed Mutagenesis Kit were purchased from New England Biolabs (Frankfurt a. M., Germany). Oligonucleotide primers and plasmid-miniprep Kits were purchased from Metabion (Martinsried, Germany). PCR purification and gel extraction Kits were obtained from Qiagen (Hilden, Germany). Ni-NTA agarose was purchased from Macherey-Nagel (Düren, Germany). Porphyrins were all purchased from Frontier Scientific (Logan, UT, USA) except of Zn-coproporphyrin III. The for E. coli codon-optimized gene Mbar_A1458 from M. barkeri was synthesized by Life Technologies GmbH (Darmstadt, Germany).Construction of plasmids
For the construction of plasmid pETDuetahbD/HIS the codon-optimized gene Mbar_A1458 (encoding AhbD) was PCR amplified using the primers NirJ1_Duet_BamHI_fw (![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) G ATG ATT GCC ATG ACC) and NirJ1_Duet_HindIII_bw (G TT ATT TTT TAC CCG GAC GGT) containing BamHI and HindIII restriction sites (underlined) and first cloned into the pJET1.2/blunt cloning vector (Thermo Scientific) according to the manufacturer's instructions. Mbar_A1458 was then cut from this plasmid with BamHI and HindIII endonucleases and the resulting DNA fragment was ligated into the correspondingly digested vector pETDuet-1 (Novagen) generating pETDuetahbD/HIS.
G ATG ATT GCC ATG ACC) and NirJ1_Duet_HindIII_bw (G TT ATT TTT TAC CCG GAC GGT) containing BamHI and HindIII restriction sites (underlined) and first cloned into the pJET1.2/blunt cloning vector (Thermo Scientific) according to the manufacturer's instructions. Mbar_A1458 was then cut from this plasmid with BamHI and HindIII endonucleases and the resulting DNA fragment was ligated into the correspondingly digested vector pETDuet-1 (Novagen) generating pETDuetahbD/HIS.
For the construction of plasmid pETDuetahbDp.C19A/C23A/HIS plasmid pETDuetahbD/HIS was used as template for mutagenesis with the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) using the primers nebN_NirJ1 (CAT CGT GGT GCA AGC ACC AG) and nebN_NirJ1_anti (AAC ATT CAG ATT ACA ACC TGC GGT C) (introduced mutations underlined).
For the construction of plasmid pETDuetahbDp.C321A/C324A/HIS plasmid pETDuetahbD/HIS was used as template for mutagenesis with the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) using the primers nebC_NirJ1 (GGT CGT GCA CGT GCA TAT GC) and nebC_NirJ1_anti (TGC AAC TTT TTT ATA CTC GCA GAT GC) (introduced mutations underlined).
Bacterial strains and growth conditions
E. coli DH10B was used as the host for cloning. For production of recombinant proteins, the E. coli strain BL21(DE3) was used. For recombinant protein production the E. coli strains carrying the corresponding vectors were grown at 37 °C in LB-medium (aerobic growth conditions) supplemented with appropriate antibiotics. Protein production was induced by adding 500 μM IPTG (isopropyl-β-D-thiogalactopyranoside) to the cultures as soon as an optical density at 600 nm of 0.6 was reached. After induction the cells were further cultivated at 25 °C overnight and then harvested by centrifugation. Cell pellets were stored at −20 °C.Purification of enzymes
Recombinant wild type AhbD (wt AhbD) from M. barkeri and the AhbD variants (AhbD C19A/C23A and AhbD C321A/C324A) were purified by IMAC. The cell pellet was resuspended in buffer A (50 mM Tris/HCl, pH 7.5, 300 mM NaCl, 20 mM imidazole) and the cells were disrupted using a French Press system (1000 psi). The soluble protein fraction was obtained by ultracentrifugation (60 min, 175![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 4 °C). The supernatant was sonicated twice using a Sonopuls HD 2070 (Bandelin, Berlin, Germany) equipped with a KE 76 tip (1 min, 4 × 10%) and afterwards loaded on a Ni-NTA column by gravity flow. The column was washed with 10 column volumes (CV) of buffer A before eluting the bound proteins with 6 CV of buffer A containing 300 mM imidazole. The protein content of the elution fractions was analyzed by SDS-polyacrylamide gel electrophoresis and the fractions containing recombinant AhbD were pooled. The final buffer exchange against buffer A without imidazole (= buffer B) was performed with a NAP-25 Sephadex column (Illustra NAP-25, GE Healthcare). The purified proteins were stored at 4 °C.
000 × g, 4 °C). The supernatant was sonicated twice using a Sonopuls HD 2070 (Bandelin, Berlin, Germany) equipped with a KE 76 tip (1 min, 4 × 10%) and afterwards loaded on a Ni-NTA column by gravity flow. The column was washed with 10 column volumes (CV) of buffer A before eluting the bound proteins with 6 CV of buffer A containing 300 mM imidazole. The protein content of the elution fractions was analyzed by SDS-polyacrylamide gel electrophoresis and the fractions containing recombinant AhbD were pooled. The final buffer exchange against buffer A without imidazole (= buffer B) was performed with a NAP-25 Sephadex column (Illustra NAP-25, GE Healthcare). The purified proteins were stored at 4 °C.
Determination of protein concentration
For the determination of protein concentrations the Bradford Reagent (Sigma-Aldrich) was used according to the manufacturer's instructions with BSA as the protein standard.In vitro iron–sulfur cluster reconstitution
The in vitro reconstitution of iron–sulfur clusters was performed under anaerobic conditions in an anaerobic chamber (Coy Laboratories, Grass Lake, MI, USA) as previously described.23 All required solutions (1 M DTT, 20 mM ammonium iron citrate, 20 mM lithium sulfide) were freshly prepared in buffer B. 10 equiv. (relative to the protein concentration) of DTT were added to the protein solution and incubated for 1 h before the addition of 10 equiv. of ammonium iron citrate. After incubation for 5 min, 10 equiv. of lithium sulfide were added slowly and the mixture was incubated for 35 min. The excess of iron and sulfide was removed by centrifugation and subsequent passage of the protein solution through a NAP-25 column (GE Healthcare) which was used according to the manufacturer's instructions.Determination of iron and sulfide contents
The iron content of purified proteins was determined according to Fish24 after denaturation of the protein with 1 M perchloric acid and bathophenanthroline as the chelating reagent. The sulfide content was determined as previously described.25Molecular mass determination
The native molecular mass of purified proteins was estimated by gel permeation chromatography as previously described.5In vitro enzyme activity assay for AhbD
The assay was performed under anaerobic conditions in a glove box (Coy Laboratories). Stock solutions of 500 μM Fe-COPRO III, COPRO III and Cu-COPRO III were generated according to Chim et al.26 For Zn-COPRO III a stock solution of about 200 μM was prepared. The enzyme activity assay mixture contained either 20 μM of substrate or 20 μM of substrate analog, 500 μM SAM, 1 mM sodium dithionite (DT) and 5 μM purified, reconstituted AhbD in buffer B. The reaction mixtures were incubated for 24 h at 17 °C. To stop the enzymatic reaction 5% (v/v) of concentrated HCl were added to the mixture which was then stored at −20 °C until further use.SAM cleavage assay
The conditions of the SAM cleavage assay were identical to those of the enzyme activity assay described above except the concentration of SAM which was used at a final concentration of 1 mM.Preparation of HPLC samples
HPLC samples for the analysis of tetrapyrroles within enzyme activity assay mixtures and the corresponding HPLC standards were prepared as described previously.7 For the preparation of HPLC samples of SAM cleavage assays, the assay mixture containing 5% (v/v) of concentrated HCl was mixed using a Vortex-Genie 2 mixer (Scientific Industries Inc., Bohemia, NY, USA) for 1 min followed by centrifugation (10 min, 16100 × g).
High performance liquid chromatography of tetrapyrroles and SAM
Tetrapyrrole extracts were analyzed using a JASCO HPLC 2000 series system (Jasco, Gross-Umstadt, Germany) as previously described.7 The chromatographic separation of samples (20 μL injection volume) containing SAM and 5′-deoxyadenosine (DOA) was performed on a HYPERCARB column (Thermo Scientific, Dreieich, Germany) with 5 μm particle size and 100 mm × 2.1 mm column dimensions at 25 °C. The substances were eluted at a flow rate of 0.2 mL min−1 using a gradient system. Solvent A consisted of 0.1% trifluoroacetic acid (for UV-spectroscopy, Fluka) and solvent B of acetonitrile (HPLC grade, Sigma-Aldrich) containing 0.08% trifluoroacetic acid (HPLC grade, Sigma-Aldrich). At the time of sample injection the mobile phase consisted of 100% solvent A. Within 25 min the content of solvent B was increased to 100% with concomitant decrease of solvent A. Then, the content of solvent B was held for 5 min before returning to the initial conditions. SAM and DOA were detected by photometric diode array analysis at 200–650 nm.EPR-spectroscopy
EPR samples were prepared under anaerobic conditions in a glove box (Coy Laboratories). 400 μM of purified AhbD in buffer B was mixed with 10 mM sodium dithionite in a total volume of 300 μL. This solution was transferred to a 5 mm od quartz EPR tube and frozen in liquid nitrogen after 15 min of incubation. CW EPR spectra were recorded on a Bruker Elexsys E 500 X-band EPR spectrometer equipped with an Oxford ESR900 helium flow cryostat. The EPR samples were accommodated in a dual mode rectangular resonator operating in the perpendicular excitation mode. The EPR spectra were processed using home-written Matlab scripts and analyzed using the Easyspin Matlab package.27 The g-tensor values of the single cluster mutants were evaluated using the least-squares fitting procedure “esfit” including the line-shape and g-strain models supported by Easyspin.Fe-COPRO III binding assay
The assay was performed under anaerobic conditions in a glove box (Coy Laboratories). Either purified AhbD in buffer B or buffer B alone as a control was directly added to a diluted Fe-COPRO III solution in a quartz SUPRASIL precision cell with 10 mm light path (Hellma Analytics, Müllheim, Germany) and mixed by pipetting. The resulting solution contained a total amount of 5 μM Fe-COPRO III and 10 μM AhbD or only 5 μM Fe-COPRO III without AhbD as a control. UV-visible absorption spectra from 250–700 nm were recorded using a V-650 spectrophotometer (Jasco, Gross-Umstadt, Germany) immediately after mixing (0–100 s), after 270 s (270–370 s) and after 24 h.Synthesis of Zn-COPRO III (zincphyrin28)
COPRO III dihydrochloride (Frontier Scientific, 9.0 mg, 12.3 μmol, 1.0 equiv.) was solved in 0.1 M NaOH (5 mL). Zinc acetate dihydrate (11 mg, 50 μmol, 4.0 equiv.) was added and the mixture was stirred at room temperature for 30 min. The precipitated brown Zn-COPRO carboxylate zinc salt was collected by centrifugation and washed multiple times with water. The residue was again solved in 0.1 M NaOH. During that process zinc hydroxide precipitated out and was removed by centrifugation. To prepare the protonated Zn-COPRO III, the solution was acidified with 0.1 M HCl. The precipitate was collected by centrifugation, dissolved in methanol, filtered over Na2SO4 and the solvent was evaporated to yield Zn-COPRO III as a purple solid (6.5 mg, 9 μmol, 74%). 1H NMR (600 MHz, (CD3)2SO): δ 12.35 (br. s, 4H), 10.17 (s, 1H), 10.10 (s, 2H), 10.08 (s, 1H), 4.36–4.29 (m, 8H), 3.62 (s, 12H), 3.18 (m, 8H). 13C NMR (150 MHz, (CD3)2SO): δ 174.4 (2C), 174.2 (2C), 147.7, 147.6, 147.6, 147.5, 146.9 (2C), 146.8, 146.7, 139.6, 139.3, 136.7, 136.6, 136.5, 136.5, 97.2, 97.1 (2C), 97.0, 37.6 (2C), 37.4 (2C), 21.7 (2C), 21.6 (2C), 11.5 (4C). HRMS (ESI): m/z = 716.1822 [M]+ calcd for C36H36N4O8Zn+: 716.1819 (error = 0.4 ppm). UV-Vis (MeOH): λmax/nm (ε[rel]) = 406 (1), 538 (0.1), 574 (0.1). See also ESI Fig. J–M.†Binding assay of different substrate analogs
The assay was performed as described above. The resulting solutions contained a total amount of either 5 μM Fe-COPRO III, 5 μM COPRO III, 5 μM Cu-COPRO III, or 5 μM Zn-COPRO III and 20 μM wt AhbD, respectively. Mixtures containing the respective substrate analog but without wt AhbD served as a control. Stock solutions of Cu-COPRO III and Zn-COPRO III were prepared according to Chim et al.26Quantification of substrate binding by fluorescence quenching
Dissociation constants (Kd) for the affinity of Fe-COPRO III to wt AhbD and the AhbD cluster variants C19A/C23A and C321A/C324A as well as for COPRO III, Zn-COPRO III and Cu-COPRO III to wt AhbD were determined by fluorescence spectroscopy. The quenching of the intrinsic tryptophan fluorescence of AhbD upon substrate or substrate analog addition was followed using a JASCO spectrofluorimeter FP-8500 (Jasco) with excitation at 295 nm and emission at 337 nm. The samples contained 1 μM AhbD and increasing amounts of the substrate or substrate analogs (0–300 μM) in a total volume of 50 μL of buffer B. The protein and the substrate or substrate analog were mixed and incubated at room temperature overnight prior to the fluorescence measurements. The fluorescence values at 337 nm for the samples with substrate/substrate analog were corrected by the subtraction of the fluorescence values for the corresponding samples containing only substrate/substrate analog. The difference in fluorescence between the sample containing no substrate and the corrected values for the samples containing increasing amounts of substrate was plotted against the substrate concentration (see ESI Fig. B and H†). The Kd values were then determined using equation29and the software OriginPro 8G (Originlab Corporation, Northampton, MA, USA). [E] = AhbD concentration, [S] = substrate concentration, ΔF = difference in fluorescence at [S], ΔF0 = difference in fluorescence at [S] = 0, ΔFmax = difference in fluorescence at substrate saturation.
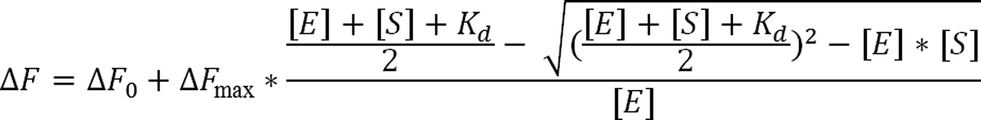
Cyclic voltammetry
Cyclic voltammetry measurements were performed using an Ametek Versastat 3. The measurements were carried out in a self-made anaerobic three-electrode electrochemical cell flushed with nitrogen. As the reference, a silver/silver-chloride electrode was used (3 mol L−1 KCl). All potentials in the text and figures are given vs. NHE (+210 mV). A platinum wire was used as the counter electrode, with glassy carbon as the working electrode. Before each measurement the platinum wire was annealed in a natural gas flame and the glassy carbon electrode was pretreated in nitric acid, neutralized, polished with 0.05 μm alumina and annealed in a natural gas flame as described before.30,31 For each experiment 20 cycles were recorded. The potential slightly drifted only over the first 10 cycles and stabilized thereafter. In this work, only the stabilized potential is discussed. The cycles were recorded with a scan rate of 1 V s−1. All electrochemical experiments were carried out at ambient temperature in a 100 μL drop. Samples were prepared under anaerobic conditions in a glove box (Coy Laboratories) and transferred into HPLC vials before injecting into the CV chamber directly on the glassy carbon electrode. Samples contained 120 μM AhbD, 500 μM SAM, 40 μM Fe-COPRO III, 40 μM Zn-COPRO III, Cu-COPRO III or 40 μM heme in diverse combinations in buffer B.Acknowledgements
The authors would like to thank Prof. Dr Dieter Jahn and Dr Jürgen Moser (Technische Universität Braunschweig, Germany) for helpful discussions. This work was supported by grants from the Deutsche Forschungsgemeinschaft to MB (BR 2010/12-1) and GL (LA 2412/5-1).References
- H. Akutsu, J.-S. Park and S. Sano, J. Am. Chem. Soc., 1993, 115, 12185–12186 CrossRef CAS.
- T. Ishida, L. Yu, H. Akutsu, K. Ozawa, S. Kawanishi, A. Seto, T. Inubushi and S. Sano, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4853–4858 CrossRef CAS.
- H. Panek and M. R. O'Brian, Microbiology, 2002, 148, 2273–2282 CrossRef CAS PubMed.
- B. Buchenau, J. Kahnt, I. U. Heinemann, D. Jahn and R. K. Thauer, J. Bacteriol., 2006, 188, 8666–8668 CrossRef CAS PubMed.
- S. Storbeck, S. Rolfes, E. Raux-Deery, M. J. Warren, D. Jahn and G. Layer, Archaea, 2010, 2010, 175050 CrossRef PubMed.
- S. Bali, A. D. Lawrence, S. A. Lobo, L. M. Saraiva, B. T. Golding, D. J. Palmer, M. J. Howard, S. J. Ferguson and M. J. Warren, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18260–18265 CrossRef CAS PubMed.
- M. Kühner, K. Haufschildt, A. Neumann, S. Storbeck, J. Streif and G. Layer, Archaea, 2014, 2014, 327637 CrossRef PubMed.
- G. Layer, J. Reichelt, D. Jahn and D. W. Heinz, Protein Sci., 2010, 19, 1137–1161 CrossRef CAS PubMed.
- S. A. Lobo, A. D. Lawrence, C. V. Romao, M. J. Warren, M. Teixeira and L. M. Saraiva, Biochim. Biophys. Acta, Proteins Proteomics, 2014, 1844, 1238–1247 CrossRef CAS PubMed.
- H. J. Sofia, G. Chen, B. G. Hetzler, J. F. Reyes-Spindola and N. E. Miller, Nucleic Acids Res., 2001, 29, 1097–1106 CrossRef CAS PubMed.
- G. Layer, J. Moser, D. W. Heinz, D. Jahn and W. D. Schubert, EMBO J., 2003, 22, 6214–6224 CrossRef CAS PubMed.
- P. A. Frey, A. D. Hegeman and F. J. Ruzicka, Crit. Rev. Biochem. Mol. Biol., 2008, 43, 63–88 CrossRef CAS PubMed.
- G. Layer, A. J. Pierik, M. Trost, S. E. Rigby, H. K. Leech, K. Grage, D. Breckau, I. Astner, L. Jänsch, P. Heathcote, M. J. Warren, D. W. Heinz and D. Jahn, J. Biol. Chem., 2006, 281, 15727–15734 CrossRef CAS PubMed.
- G. Layer, K. Verfürth, E. Mahlitz and D. Jahn, J. Biol. Chem., 2002, 277, 34136–34142 CrossRef CAS PubMed.
- F. Yan, J. M. LaMarre, R. Röhrich, J. Wiesner, H. Jomaa, A. S. Mankin and D. G. Fujimori, J. Am. Chem. Soc., 2010, 132, 3953–3964 CrossRef CAS PubMed.
- G. Layer, K. Grage, T. Teschner, V. Schünemann, D. Breckau, A. Masoumi, M. Jahn, P. Heathcote, A. X. Trautwein and D. Jahn, J. Biol. Chem., 2005, 280, 29038–29046 CrossRef CAS PubMed.
- J. K. Rubach, X. Brazzolotto, J. Gaillard and M. Fontecave, FEBS Lett., 2005, 579, 5055–5060 CrossRef CAS PubMed.
- E. Tipping, B. Ketterer and P. Koskelo, Biochem. J., 1978, 169, 509–516 CrossRef CAS PubMed.
- F. Capozzi, S. Ciurli and C. Luchinat, Struct. Bonding, 1998, 90, 127–160 CrossRef CAS.
- J. M. Blamey, M. Chiong, C. Lopez and E. T. Smith, Anaerobe, 2000, 6, 285–290 CrossRef CAS.
- F. A. Armstrong, H. A. O. Hill and N. J. Walton, Acc. Chem. Res., 1988, 21, 407–413 CrossRef CAS.
- N. D. Lanz and S. J. Booker, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824, 1196–1212 CrossRef CAS PubMed.
- L. Flühe, T. A. Knappe, M. J. Gattner, A. Schäfer, O. Burghaus, U. Linne and M. A. Marahiel, Nat. Chem. Biol., 2012, 8, 350–357 CrossRef PubMed.
- W. W. Fish, Methods Enzymol., 1988, 158, 357–364 CAS.
- H. Beinert, Anal. Biochem., 1983, 131, 373–378 CrossRef CAS PubMed.
- N. Chim, A. Iniguez, T. Q. Nguyen and C. W. Goulding, J. Mol. Biol., 2010, 395, 595–608 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- M. Toriya, S. Yaginuma, S. Murofushi, K. Ogawa, N. Muto, M. Hayashi and K. Matsumoto, J. Antibiot., 1993, 46, 196–200 CrossRef CAS PubMed.
- Fluorescent methods for molecular motors, ed. C. P. Toseland and N. Fili, Springer, Basel, 2014 Search PubMed.
- W. R. Hagen, Eur. J. Biochem., 1989, 182, 523–530 CrossRef CAS PubMed.
- A. Aliverti, W. R. Hagen and G. Zanetti, FEBS Lett., 1995, 368, 220–224 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The cyclic voltammetry experiments, the binding assays with the substrate analogs, the determination of Kd values, the SAM cleavage assays with the substrate analogs, the decarboxylase activity assays with the substrate analogs and the synthesis and characterization of Zn-COPRO III. See DOI: 10.1039/c6sc01140c |
| This journal is © The Royal Society of Chemistry 2016 |