
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWell-structured bimetallic surface capable of molecular recognition for chemoselective nitroarene hydrogenation†
Shinya
Furukawa
*,
Katsuya
Takahashi
and
Takayuki
Komatsu
*
Department of Chemistry, Tokyo Institute of Technology, 2-12-1-E1-10 Ookayama, Meguro-ku, Tokyo 152-8551, Japan. E-mail: furukawa.s.af@m.titech.ac.jp; komatsu.t.ad@m.titech.ac.jp; tanakat@molemg.kyoto-u.ac.jp; Fax: +81-3-5734-2758; Tel: +81-3-5734-2602 Tel: +81-3-5734-3532
First published on 29th March 2016
Abstract
Unprecedented molecular recognition ability governed by a simple bimetallic surface is reported. A series of Rh-based ordered alloys supported on silica gel (RhxMy/SiO2, where M is Bi, Fe, Ga, Ge, In, Ni, Pb, Sb, Sn, or Zn) were tested in the hydrogenation of nitrostyrene to form aminostyrene. RhIn/SiO2 showed remarkably high catalytic activity and good selectivity under 1 atm H2 at room temperature. Moreover, various other nitroarenes containing carbonyl, cyano, or halo moieties were selectively hydrogenated into the corresponding amino derivatives using RhIn/SiO2. Kinetic study and density functional theory (DFT) calculations revealed that the high selectivity originates from RhIn/SiO2 adsorbing nitro groups much more favorably than vinyl groups. In addition, the DFT calculations indicated that the RhIn ordered alloy presents concave Rh rows and convex In rows on its surface, which are able to capture the nitro group with end-on geometry while effectively minimizing vinyl-π adsorption. Thus, the specific and highly ordered surface structure of RhIn enables the chemoselective molecular recognition of nitro groups over vinyl groups through geometric and chemical effects.
Molecular recognition is an important methodology used to achieve selective chemical transformations in fields such as organic chemistry, pharmacology, enzymology, and industrial chemistry. Molecular recognition typically requires sophisticated reaction environments, such as those found in the active centers of enzymes or metal complexes bearing tailor-made ligands.1,2 In contrast, heterogeneous inorganic materials, such as supported catalysts or amorphous oxides, do not typically provide well-structured reaction environments, but instead offer advantages in terms of practical use, such as durability, separability, and reusability. Although mesoporous and microporous materials, such as zeolites, have been used for shape-selective reactions,3–6 their molecular recognition relies on physical restriction by pore size, which is typically on the nanoscale level. However, a reaction site displaying regularity at the Angstrom-scale that achieves accurate molecular recognition through chemical interactions is more appealing in terms of selectivity than the sites found in porous materials.
Ordered alloys, also known as intermetallic compounds, exhibit specific crystalline structures and well-defined atomic arrangements of their metal components,7 making them attractive candidates for simple and ordered inorganic materials. Their highly ordered bimetallic surfaces may provide appropriate reaction environments for chemoselective molecular recognition. Moreover, strong chemical interactions between their surfaces and adsorbate molecules can be expected when a catalytically active metal element is used as a component. Our research group has previously reported unique catalytic properties of ordered alloys8,9 and the involvement of their specific surface structures in stereoselective conversions.10
In this study, we applied ordered alloys to the development of well-structured reaction sites capable of chemoselective molecular recognition. As a target reaction, we focused on the chemoselective hydrogenation of nitrostyrenes to form aminostyrenes. Functionalized anilines, including aminostyrenes, are valuable synthetic intermediates in the synthesis of pharmaceuticals, polymers, herbicides, and other fine chemicals.11,12 The use of conventional Pt-group metal catalysts (i.e., Pt, Pd, and Rh) typically results in undesired C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond hydrogenation13 because this reaction is kinetically and thermodynamically more favored than nitro hydrogenation. Au-14,15 and Ag-based16,17 catalysts have been reported to show high selectivity for nitro hydrogenation; however, these catalysts require high reaction temperatures and H2 pressures, typically higher than 100 °C and 6 atm, respectively, hampering their widespread application. This is partly due to the low hydrogenation activity of Au and Ag. Although highly dispersed Pt on reducible oxides has recently been reported to work well at lower temperatures,18,19 high H2 pressures remain a necessity. In this context, achieving the selective nitro hydrogenation of nitrostyrenes at room temperature under atmospheric H2 pressure is a highly attractive challenge. In this study, we focused on Rh, which has been reported to be very active in nitro hydrogenation,20 as the main metallic component of ordered alloys. The use of Rh-based ordered alloys that are capable of chemoselective molecular recognition shows enormous promise for attaining both high catalytic activity and selectivity.
C bond hydrogenation13 because this reaction is kinetically and thermodynamically more favored than nitro hydrogenation. Au-14,15 and Ag-based16,17 catalysts have been reported to show high selectivity for nitro hydrogenation; however, these catalysts require high reaction temperatures and H2 pressures, typically higher than 100 °C and 6 atm, respectively, hampering their widespread application. This is partly due to the low hydrogenation activity of Au and Ag. Although highly dispersed Pt on reducible oxides has recently been reported to work well at lower temperatures,18,19 high H2 pressures remain a necessity. In this context, achieving the selective nitro hydrogenation of nitrostyrenes at room temperature under atmospheric H2 pressure is a highly attractive challenge. In this study, we focused on Rh, which has been reported to be very active in nitro hydrogenation,20 as the main metallic component of ordered alloys. The use of Rh-based ordered alloys that are capable of chemoselective molecular recognition shows enormous promise for attaining both high catalytic activity and selectivity.
In the present study, we prepared a series of Rh-based ordered alloys supported on silica (RhxMy/SiO2; M = Bi, Fe, Ga, Ge, In, Ni, Pb, Sb, Sn, or Zn), which was tested in the hydrogenation of 4-nitrostyrene. The catalytic performances observed were rationalized based on a combination of kinetic study and density functional theory (DFT) calculations. Herein, we report the first demonstration of chemoselective molecular recognition governed by a bimetallic surface ordered at an atomic level and its application to highly efficient hydrogenation catalysis.
Results and discussion
Rh-based bimetallic alloys supported on silica (RhM/SiO2; M = Bi, Fe, Ga, In, Ni, Sb, Sn, or Zn) were prepared by successive impregnations of Rh/SiO2. Rh–Pb/SiO2 catalysts (Rh3Pb2, RhPb, and RhPb2) were prepared by a co-impregnation method. The crystalline phases of the prepared Rh-based catalysts were analyzed by X-ray diffraction (XRD), as shown in Fig. 1. The desired alloy or intermetallic phase was successfully formed with high phase purity for each catalyst. For Rh–Fe and Rh–Ni, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solid solution alloy phase with face-centered cubic structure is formed, whereas for other combinations, intermetallic phases exhibiting different crystalline forms are produced as follows: RhGa, RhIn, and RhZn: cubic Pm
:1 solid solution alloy phase with face-centered cubic structure is formed, whereas for other combinations, intermetallic phases exhibiting different crystalline forms are produced as follows: RhGa, RhIn, and RhZn: cubic Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, CsCl structure; RhSn: cubic P213; RhGe: orthorhombic Pnma; Rh3Pb2 and RhBi: hexagonal P63/mmc; RhPb: hexagonal P6/mmm; and RhPb2: tetragonal I4/mcm. The crystallite size of the metallic nanoparticles was estimated using the Scherrer equation, as applied to the most intense peaks following deconvolution. The crystallite sizes were typically 2–3 nm, which was close to that of the parent alloy Rh/SiO2 (1.6 nm). For In-, Bi-, and Pb-containing phases, larger sizes were observed, which is partially due to the incorporation of larger atoms (the atomic radii of Rh, In, Bi, and Pb are 135, 155, 160, and 180 pm, respectively).21 The difference in the preparation method for Rh–Pb catalysts may also be involved in the size difference. We also performed Fourier transform infrared (FT-IR) experiments with CO adsorption to characterize the surface of the prepared catalysts (Fig. S1†). Rh/SiO2 showed peaks at 2057 and 1880 cm−1, which are assigned to the stretching vibration of CO adsorbed linearly and bridged, respectively. Other Rh-based bimetallic alloys exclusively showed peaks assignable to linearly adsorbed CO, with no peaks at 2057 cm−1 nor bridged species being observed, demonstrating the absence of monometallic Rh phase at the surface of these alloys. Chemical analysis was also performed for the prepared catalysts containing metals with low melting (Ga; 30 °C, In; 157 °C) or boiling (Zn; 907 °C) points, using inductively coupled plasma atomic emission spectroscopy (ICP-AES). The composition ratio was almost 1:1 for RhGa and RhIn (1.00:1.03 and 1.00:1.06, respectively). However, for RhZn, a small amount of Zn was lost (1.00:0.88), probably because of its high vapor pressure and the high reduction temperature used (600 °C). The loss of Zn from an alloy at high temperature has been previously reported for the PdZn system.22 According to the XRD and FT-IR results, the small excess of Rh may be present as tiny clusters in the cores of the RhZn particles.
m, CsCl structure; RhSn: cubic P213; RhGe: orthorhombic Pnma; Rh3Pb2 and RhBi: hexagonal P63/mmc; RhPb: hexagonal P6/mmm; and RhPb2: tetragonal I4/mcm. The crystallite size of the metallic nanoparticles was estimated using the Scherrer equation, as applied to the most intense peaks following deconvolution. The crystallite sizes were typically 2–3 nm, which was close to that of the parent alloy Rh/SiO2 (1.6 nm). For In-, Bi-, and Pb-containing phases, larger sizes were observed, which is partially due to the incorporation of larger atoms (the atomic radii of Rh, In, Bi, and Pb are 135, 155, 160, and 180 pm, respectively).21 The difference in the preparation method for Rh–Pb catalysts may also be involved in the size difference. We also performed Fourier transform infrared (FT-IR) experiments with CO adsorption to characterize the surface of the prepared catalysts (Fig. S1†). Rh/SiO2 showed peaks at 2057 and 1880 cm−1, which are assigned to the stretching vibration of CO adsorbed linearly and bridged, respectively. Other Rh-based bimetallic alloys exclusively showed peaks assignable to linearly adsorbed CO, with no peaks at 2057 cm−1 nor bridged species being observed, demonstrating the absence of monometallic Rh phase at the surface of these alloys. Chemical analysis was also performed for the prepared catalysts containing metals with low melting (Ga; 30 °C, In; 157 °C) or boiling (Zn; 907 °C) points, using inductively coupled plasma atomic emission spectroscopy (ICP-AES). The composition ratio was almost 1:1 for RhGa and RhIn (1.00:1.03 and 1.00:1.06, respectively). However, for RhZn, a small amount of Zn was lost (1.00:0.88), probably because of its high vapor pressure and the high reduction temperature used (600 °C). The loss of Zn from an alloy at high temperature has been previously reported for the PdZn system.22 According to the XRD and FT-IR results, the small excess of Rh may be present as tiny clusters in the cores of the RhZn particles.
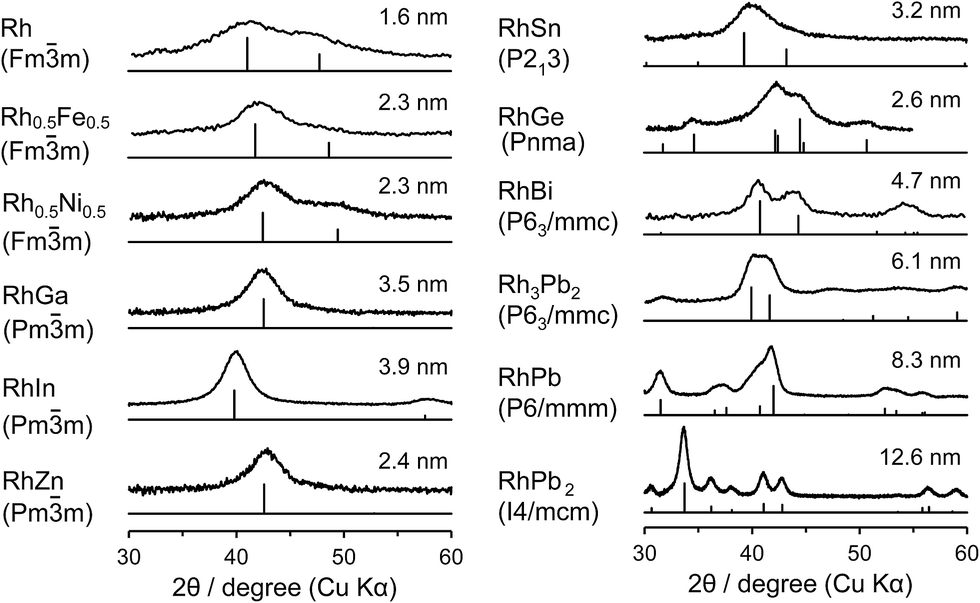 | ||
| Fig. 1 XRD patterns of RhxMy/SiO2 and corresponding references (ICDD-PDF, vertical lines). Values indicate the crystallite sizes estimated using the Scherrer equation. | ||
Fig. 2a shows the transmission electron microscopy (TEM) images of RhIn/SiO2. The particle sizes were observed to be 2–5 nm, which is consistent with the crystallite size estimated by XRD. A high-resolution TEM image of a single RhIn nanoparticle is shown in Fig. 2b. Lattice fringes with 2.24 Å spacing can be clearly observed, which is consistent with the spacing of RhIn (110) planes (2.25 Å). Neither amorphous nor other crystal phases were observed at the surface region, suggesting that the surface is terminated by the intermetallic RhIn structure. Note that the TEM sample underwent treatment in carbon tetrachloride in air for dispersion on a grid prior to the observation. Therefore, this result demonstrates the stability of the RhIn surface even in an organic solvent. The fast Fourier transform of this nanoparticle image (Fig. 2c) showed a set of diffraction spots assignable to a single RhIn crystal oriented along the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1] direction. The RhIn crystalline structure viewed along with this direction exhibits good agreement with the magnified TEM image (Fig. 2d). A model structure for the RhIn nanocrystal with {110} facets is illustrated in Fig. 2e.
1] direction. The RhIn crystalline structure viewed along with this direction exhibits good agreement with the magnified TEM image (Fig. 2d). A model structure for the RhIn nanocrystal with {110} facets is illustrated in Fig. 2e.
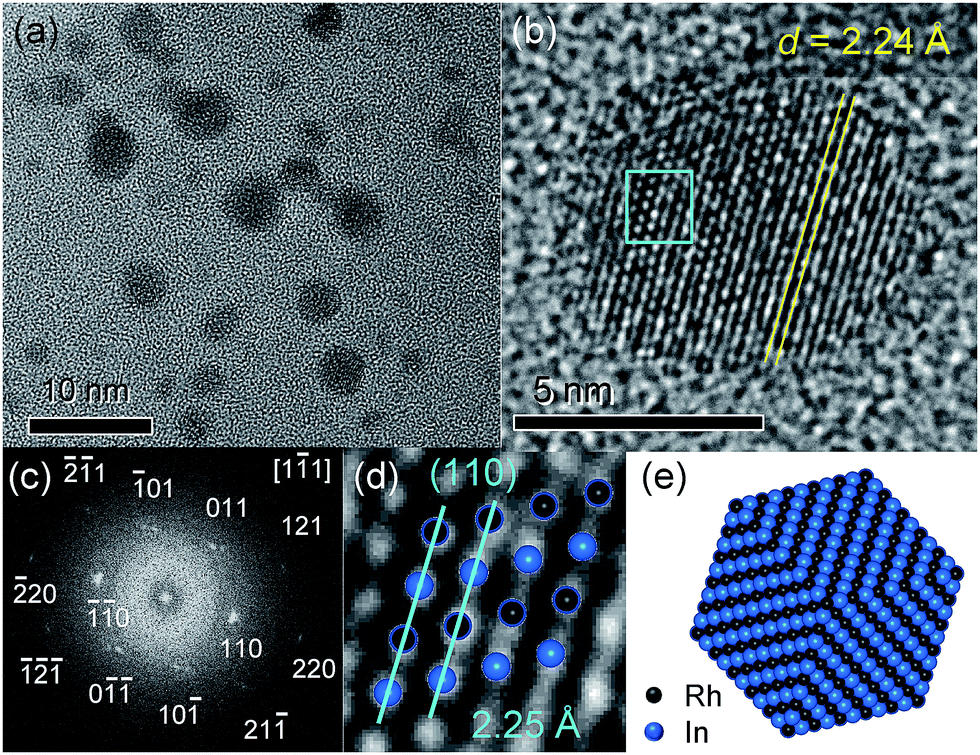 | ||
| Fig. 2 (a) TEM image of RhIn/SiO2. (b) High-resolution TEM image of a single RhIn nanoparticle. (c) Fast Fourier transform of the TEM image shown in (b). (d) Magnification of the region designated by the light-blue square in (b). RhIn crystalline structure oriented along the [11] direction is overlapped. (e) Model structure of a RhIn nanocrystal with {110} facets. | ||
The prepared Rh-based catalysts were trialed in the hydrogenation of 4-nitrostyrene (NS). Fig. 3 shows the NS conversion at 0.5 h and product distribution at ca. 50% conversion for each catalyst. Although monometallic Rh showed high conversion, the undesired product 4-ethylnitrobenzene (EN) was afforded with 94% selectivity. This result reflects the intrinsic difficulty in nitro-selective hydrogenation of NS using monometallic catalysts. Conversely, the Rh-based bimetallic catalysts showed higher selectivities for the desired product 4-aminostyrene (AS) than monometallic Rh. The product distribution differed depending on the second metal, and the highest AS selectivity (97%) was obtained with RhIn. 4-Ethylaniline (EA) was also formed as an over-hydrogenated by-product. However, in most cases, EA levels were very low (typically <15%), indicating that AS selectivity depends mainly on the relative rate of the initial hydrogenation (i.e., NS → AS vs. NS → EN).
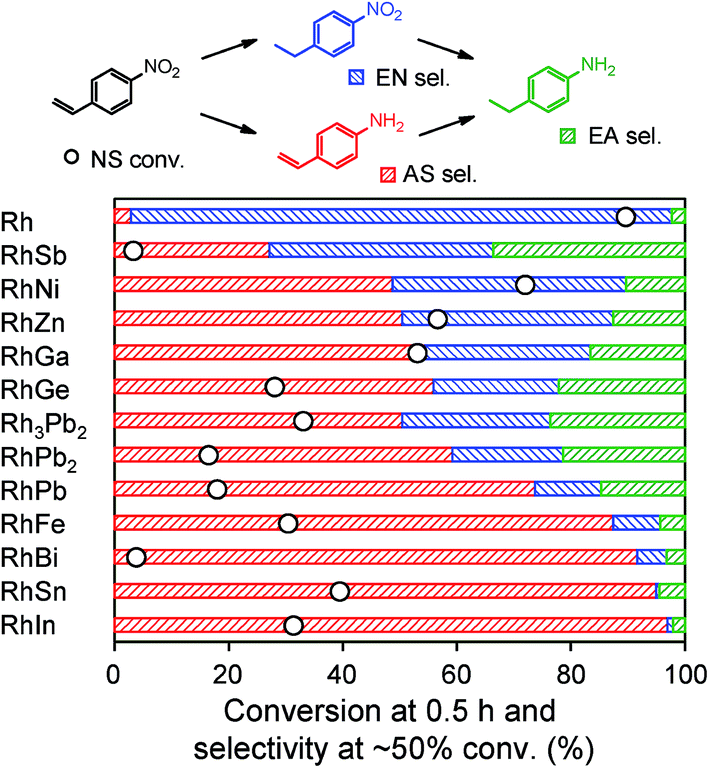 | ||
| Fig. 3 Conversion and product distribution in NS hydrogenation over various Rh-based alloys supported on SiO2. Conversion at 0.5 h and selectivity at approximately 50% conversion are shown. Reaction conditions: catalyst, 50 mg (Rh: 3 wt%); NS, 0.5 mmol; solvent, 5 mL ethyl acetate; hydrogen pressure, 1 atm; temperature, 75 °C. | ||
We subsequently performed the hydrogenation of various nitroarenes under 1 atm H2 at 75 °C using RhIn/SiO2 as the nitro hydrogenation catalyst. 3-Nitrostyrene and 4-nitrostilbene were also hydrogenated to the corresponding aminoarenes in excellent yields (Table 1, entries 3 and 4). Moreover, nitroarenes containing cyano, acetoxyl, acetyl, and halo groups were selectively hydrogenated (Table 1, entries 5, 7, 8, and 12–14). Note that this catalyst worked well under 1 atm H2, even at room temperature (25 °C): various nitroarenes, including NS, were converted to the amino derivatives within a few hours (Table 1, entries 2, 6, 9, and 11). To the best of our knowledge, this is the first example of such in inorganic heterogeneous catalytic systems. In addition, we performed a gram-scale reaction with 4-acetylnitrobenzene, which resulted in almost quantitative conversion to the amino product with 97% isolated yield (Table 1, entry 10) and a turnover number of 36000. The RhIn/SiO2 catalyst could be separated by simple decantation and reused at least four times without significant decrease in its catalytic performance (Fig. S2†). Thus, a remarkably efficient catalytic system for nitroarene hydrogenation using the RhIn/SiO2 catalyst was developed.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Temp./°C | Time/h | Conv. (%) | Yield (%) |
| a Reaction conditions: RhIn/SiO2, 50 mg (Rh: 3 wt%); substrate, 0.5 mmol; solvent, 5 mL ethyl acetate; hydrogen pressure, 1 atm. b Solvent, 10 mL. c Substrate 6 mmol (1.0 g). d Isolated yield. e Substrate, 0.25 mmol; in the dark. f Catalyst, 100 mg. | |||||
| 1 | 4-Vinyl | 75 | 2 | 99 | 93 |
| 2 | 25 | 2 | 99 | 84 | |
| 3 | 3-Vinyl | 75 | 2 | 99 | 91 |
| 4b | 4-Styryl | 75 | 2.5 | 99 | 93 |
| 5 | 4-Cyano | 75 | 4 | 100 | >99 |
| 6 | 25 | 4 | 100 | >99 | |
| 7 | 4-Acetoxy | 75 | 1.5 | 100 | >99 |
| 8 | 4-Acetyl | 75 | 2 | 100 | >99 |
| 9 | 25 | 1.5 | 100 | >99 | |
| 10c | 25 | 22 | 100 | >99(97)d | |
| 11b,e | 4-Formyl | 25 | 1 | 100 | 83 |
| 12 | 4-Chloro | 75 | 1.5 | 100 | >99 |
| 13 | 4-Bromo | 75 | 1.6 | 100 | >99 |
| 14f | 4-Iodo | 75 | 1.8 | 100 | 96 |
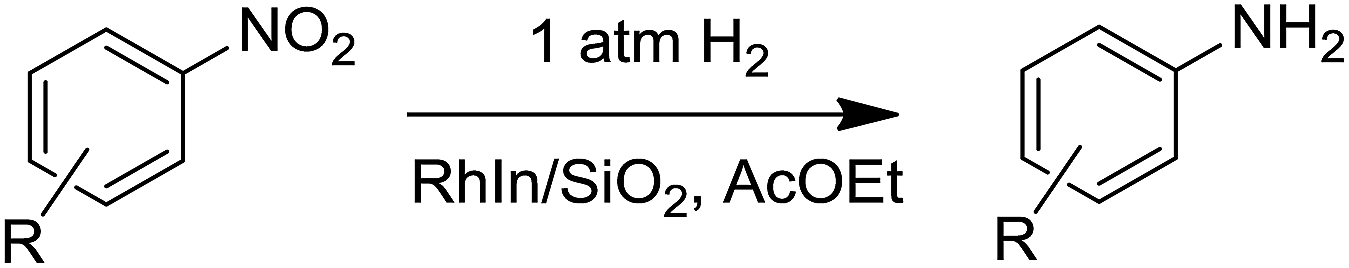
Next, the origin of the chemoselectivity and high catalytic activity were investigated in detail. Kinetic studies on NS hydrogenation over Rh/SiO2 and RhIn/SiO2 at 75 °C and 25 °C revealed a zero-order dependence of the NS conversion rate on the NS concentration ([NS] = 62–390 mM) and a first-order relationship with hydrogen pressure (PH2 = 0.25–1.00 atm) (Fig. S3†). Moreover, when D2 was used instead of H2, a small kinetic isotope effect value was observed (kH/kD = 1.2 for RhIn/SiO2 at 75 °C). These results indicate saturation coverage by NS of the catalyst surface and indicate that hydrogen dissolution or diffusion is the rate-limiting step. The Arrhenius-type plot obtained from NS hydrogenation over RhIn/SiO2 showed a linear correlation below 60 °C, affording an apparent activation energy of 20 kJ mol−1 (Fig. S4†). This low activation energy is consistent with the rate-determining step being hydrogen dissolution or diffusion rather than being a chemical reaction. The reaction rate began to decrease above 70 °C (Fig. S4†), which may be due to the boiling of the ethyl acetate solvent (boiling point, 77 °C) inhibiting H2 dissolution. Consequently, comparable reaction rates were obtained at 75 °C and 25 °C. Considering the rate-determining step being H2 supply, the catalytic activity based on H2 consumption should not depend on the catalyst. Indeed, most Rh-based catalysts tested in this study showed similar H2 consumption (ca. 0.5 mmol at 0.5 h, estimated as 1 × EN + 3 × AS + 4 × EA, as shown in Fig. S5†). Therefore, the difference in NS conversion observed in Fig. 3 can be attributed to the functional group being hydrogenated. However, in the case of the catalysts with very low catalytic activities, such as RhSb and RhBi, the rate-determining step may be a surface reaction.
We also performed a control experiment using nitrobenzene (NB) and styrene (ST) as the substrates. Fig. 4 shows the time courses of NB and ST conversion when these substrates were used alone or together. When the substrates were used alone (hereafter termed “alone mode”) with Rh/SiO2, ST hydrogenation was much faster than NB hydrogenation, and the initial ST conversion rate (rST) was 9.5 times higher than that of NB (rNB) (Table 2). Although a similar trend was also observed in the presence of both substrates (hereafter termed “together mode”), the rate ratio was slightly higher (rST/rNB = 13), indicating that the adsorption of ST onto monometallic Rh is preferred to that of NB. For RhIn/SiO2, a different situation was observed. ST and NB were hydrogenated at similar rates (rST/rNB = 1.9) in alone mode. However, in together mode, ST hydrogenation was significantly inhibited, even though NB hydrogenation took place as well as in the alone mode (rST/rNB = 0.04). Note that rST recovered the level observed in alone mode after the complete consumption of NB. These results suggest that, on RhIn, NB is much more strongly adsorbed than ST, which causes the chemoselective adsorption of NB. Moreover, we performed a similar experiment with RhZn, which exhibited moderate selectivity (Fig. S6†), with a result between those of Rh and RhIn being observed (rST/rNB = 4.8 and 1.1 for alone and together modes, respectively, as shown in Table 2). The selectivity for NB hydrogenation in the together mode, rNB/(rST + rNB), can be represented using rST/rNB, as listed in Table 2. These values are consistent with the AS selectivity observed in NS hydrogenation. Based on these results, we conclude that the product selectivity in NS hydrogenation depends strongly on the relative adsorption affinity of the nitro and vinyl groups of NS. Considering that the rate-determining step is H2 supply, the ratio of nitro adsorption and vinyl adsorption should be under thermodynamic control.
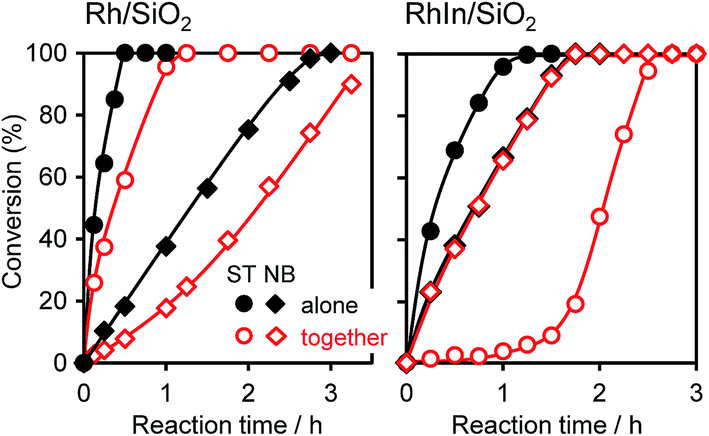 | ||
| Fig. 4 Time courses of ST and NB conversion when these substrates were used alone or together. The reaction conditions were identical to that for Table 1 except for the catalyst amount; Rh/SiO2, 30 mg; RhIn/SiO2, 50 mg. | ||
To evaluate the individual energies of the nitro and vinyl adsorptions of NS, we carried out DFT calculations using NS-adsorbed slab models of some of the catalysts. First, we selected the model planes that were expected to be exposed, stable, and active for hydrogenation by considering the following criteria: (1) low Miller index, (2) low surface energy, and (3) large Rh–Rh coordination numbers at the outermost surface (i.e., Rh–Rh rows or ensembles) for H2 activation and H diffusion (e.g. Rh(111), RhIn(110), and RhPb2(100); other planes are listed in Table S1†). For each selected surface, the vibrational frequency of CO adsorbed on a Rh top site was calculated (Table S2†), and compared with the experimental value obtained by the FT-IR experiment (Fig. S1 and Table S2†). Good agreement between the theoretical and experimental values was obtained, thus indicating that the selected surfaces are indeed exposed as main facets. However, the theoretical values for other low-Miller-index surfaces differed from the experimental values.
Then, the configuration of the adsorbed NS molecules was optimized. In general, there are two adsorption modes for an alkenyl moiety on a metal surface, i.e. di-σ and π modes. The surface science literature concerning Pt(111)23 suggests that π alkene adsorption is energetically favored for hydrogenation, whereas di-σ alkene adsorption is not. Moreover, our previous DFT study of hydrogenation over Rh-based intermetallic compounds has shown that π-alkene was much more easily hydrogenated than di-σ alkene.10 Therefore, in the present work, we studied π modes for vinyl adsorption. We also compared the monodentate and bidentate ligation modes of the nitro moieties, which revealed that the bidentate modes are energetically unfavored, probably because inappropriate N–O–Rh angles are forced (Fig. S7†). The optimized structures of NS bound to the Rh(111) and RhIn(110) surfaces are shown in Fig. 5 (structures on other surfaces are shown in Fig. S8†). For vinyl adsorption on Rh(111), not only the vinyl moiety but also the aromatic ring were bound to Rh, affording a significantly negative adsorption energy (Ead = −62.4 kJ mol−1). Interestingly, sp3-like structures were observed for the aromatic C–H bonds, which clearly show the formation of Rh–C bonds. A similar result has also been reported for benzene adsorption on Ni(111).24 In contrast, only the vinyl moiety was bound to the Rh on RhIn(110), in which the π plane of NS was bent, and Ead was positive (63.1 kJ mol−1). This is most likely because the large In atoms (155 pm)21 adjacent to Rh (135 pm) sterically hinder the access of the aromatic ring to the Rh atoms. Note that In atoms are slightly upshifted from their original positions in the bulk-terminated structure during geometrical optimization (Fig. S9†), which enhances the steric effect. Such surface relaxation was also observed with other alloys possessing large second metals, such as Bi (160 pm) and Pb (180 pm)21 (Fig. S9†). For nitro adsorption, the adsorption geometries on both planes were almost identical, yielding similarly negative Ead values (−27.4 and −41.0 kJ mol−1 for the Rh(111) and RhIn(110) planes, respectively). This may be due to the lower steric hindrance to the nitro adsorption geometry on the surface.
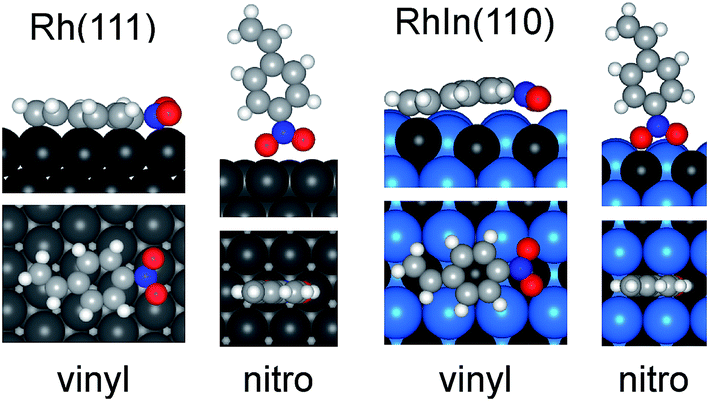 | ||
| Fig. 5 Optimized structure of NS adsorbed on Rh(111) and RhIn(110) surface with vinyl and nitro moieties. Side (top) and top (bottom) views are shown. | ||
The calculated Ead values for various bimetallic planes in relation to nitro hydrogenation selectivity are summarized in Fig. 6. As a scale of the selectivity, the ratio of nitro hydrogenation rate to vinyl hydrogenation rate (rNO2/rCC) was set as the y-axis. The Ead of vinyl adsorption differed drastically depending on the surface (i.e., from −62.4 to 63.1 kJ mol−1) with a positive correlation to selectivity. Conversely, for nitro adsorption, no correlation was observed between Ead and the selectivity. Moreover, the Ead values were mostly within a small range (i.e., from −41.0 to −27.4 kJ mol−1), except for those of RhBi and RhPb (−12.8 and 1.8 kJ mol−1, respectively). These slightly higher values may be due to the steric repulsion from large Bi and Pb atoms. Thus, the results show that the selectivity varies depending on the favorability of vinyl adsorption rather than of nitro adsorption. Note that vinyl adsorption is favored over nitro adsorption on Rh(111), whereas this situation is reversed on the bimetallic surfaces. This is consistent with the results of ST–NB hydrogenation in Fig. 4 and the product distribution in Fig. 3, i.e., the Rh surface predominantly affords EN, but the bimetallic catalysts yield AS as the main product. This is also reflected in the rNO2/rCC values, which are lower and higher than unity for the Rh surface and the bimetallic surfaces, respectively (Fig. 6). This can be rationalized by the aforementioned steric effect. For monometallic Rh, vinyl adsorption is significantly favored by multiple coordination to Rh ensembles, as clearly reflected in the aromatic C–H bonds with sp3-like conformations. However, for bimetallic surfaces, this coordination effect is diminished significantly by inert second metal atoms adjacent to Rh atoms. The aromatic C–H bonds show sp2-like conformations (e.g., RhZn and RhPb2), indicating very weak adsorption and resulting in increased Ead. When the Rh sites are concave (e.g., RhIn and RhBi), vinyl adsorption requires bending of the π plane because of the significant steric hindrance between the second metal and the phenyl ring. This deformation causes a further increase in Ead. Conversely, nitro adsorption may be free from such steric hindrance because the nitro adsorption has end-on geometry, thus fitting within the concave Rh sites. Consequently, the concave Rh structure as observed for RhIn(110) is capable of chemoselective molecular recognition. The larger size of the second metal seems to favor the formation of such convex sites. However, note that not only the size of the second metal but also the original atom positions in the bulk-terminated structure contribute to the formation of concave sites. For example, the RhPb2(100) facet presents a flat surface despite the large size of the Pb atoms and their upshifting. This is because the original position of the Rh atoms in the RhPb2(100) facet is slightly higher than that of the Pb atoms (Fig. S9†). Moreover, the density of the second atom at the surface makes an important contribution to the steric effect. The indium atoms on the RhIn(110) surface are densely aligned (6.62 atoms per nm2) compared with Bi (4.84 atoms per nm2) and Pb (3.40 atom per nm2) on the RhBi(110) and RhPb(002) surfaces, respectively (Fig. S8,† top views). Therefore, the steric effect and ECC on RhIn(110) are greater than those on RhBi(110) and RhPb(002), and the RhIn(110) surface provides an ideal structure for molecular recognition.
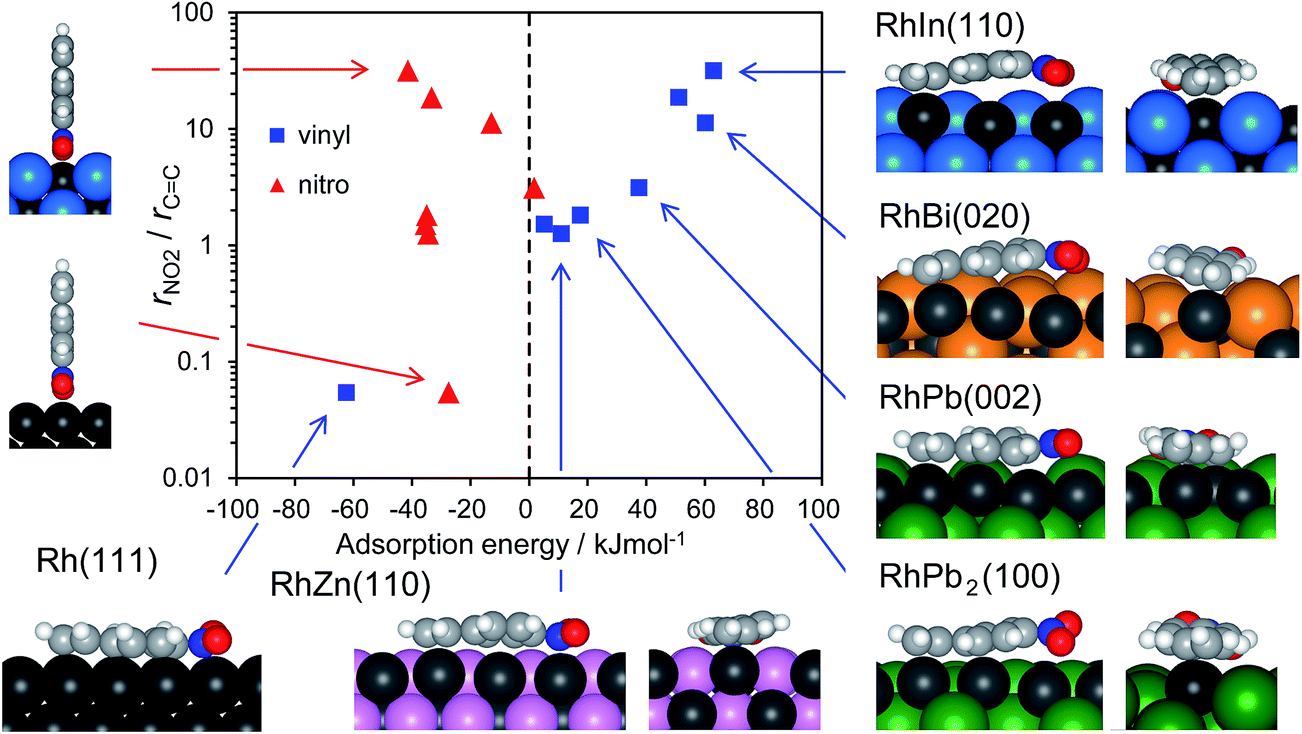 | ||
| Fig. 6 Relationship between the nitro hydrogenation selectivity (rNO2/rCC) and the adsorption energy of NS on slab models with vinyl (square) or nitro (triangle) moiety. Several NS structures are shown. | ||
The physical meaning of the relationship between selectivity and Ead is represented in eqn (1):
 | (1) |
C) are the hydrogenation rate of x, adsorbed NS with x, unadsorbed NS, and adsorption energy of NS with x, respectively. The first equation indicates that the hydrogenation rate ratio depends on the ratio of adsorbate concentration. Note that rNO2 is tripled because nitro hydrogenation requires three times the amount of hydrogen compared with vinyl hydrogenation. The third equation represents the ratio of adsorbed and unadsorbed NS using the Boltzmann distribution with the adsorption energy. Taking the logs of both sides yields a simple proportional relationship between ln(3rNO2/rCC) and ΔE. As shown in Fig. 7, the plot obtained using the experimental hydrogenation rates and calculated adsorption energy difference shows a good linear relationship through the origin. This validates our calculation method and our proposed rationale for the chemoselectivity of the hydrogenation.
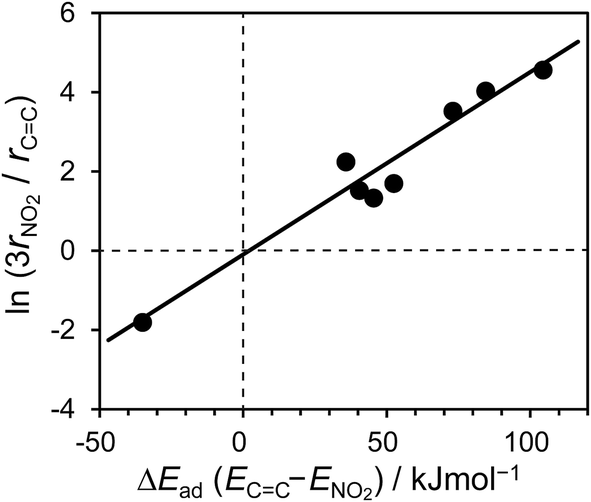 | ||
| Fig. 7 Relationship between ln(3rNO2/rCC) and ΔEad (ECC − ENO2) for RhM/SiO2. | ||
We also investigated the effect of the electronic state of Rh on the catalytic performances. The vibrational frequency of linearly adsorbed CO on Rh (Fig. S1 and Table S2†) and calculated partial charge of Rh were employed as indicators of the electron density of Rh. For most Rh-based alloys, the atomic charges of the surface Rh were slightly more negative than those of bulk Rh (Fig. 8a), most likely due to the presence of dangling bonds. A strong correlation was observed between the vibrational frequencies and the Rh atomic charges (Fig. 8a), showing that the former indicates the electron density of Rh well. However, no apparent correlation was observed between the selectivity and these electronic factors (Fig. 8b), excluding the electronic effects on the observed molecular recognition properties. We also studied the size effect on the selectivity in NS hydrogenation using RhIn/SiO2 with various crystallite sizes. The AS selectivity decreased with increasing size of the RhIn crystallites (Fig. S10†). This might be due to the contribution of minor facets allowing vinyl hydrogenation, such as Rh-terminated RhIn(100) and (111), becoming more significant for large crystallites. For sufficiently small nanoparticles (<4 nm), if any such minor facets appear they are unlikely to be large enough to work as vinyl hydrogenation sites.
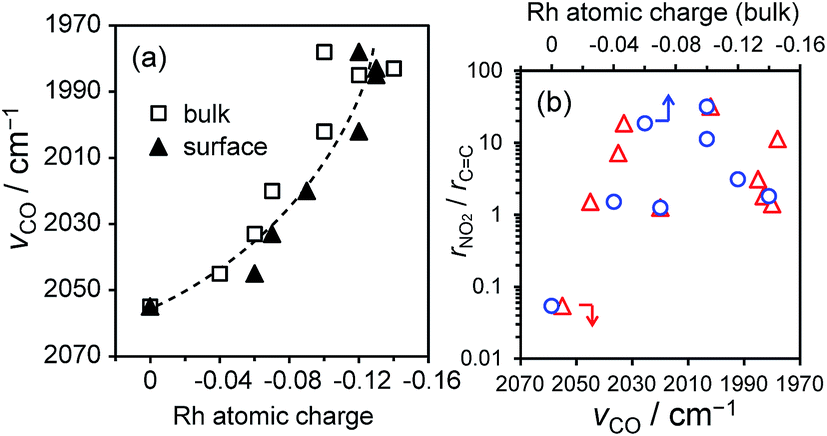 | ||
| Fig. 8 (a) Relationship between the vibrational frequencies of linearly adsorbed CO on Rh-based catalysts and the atomic charges (Hirshfeld charge) of the bulk or surface Rh. (b) Relationship between the electronic factors show in (a) and the nitro-hydrogenation selectivity (rNO2/rCC). | ||
Based on these results, we concluded that the chemoselectivity for the nitro hydrogenation of NS is due to the preferential adsorption of the nitro group over the vinyl group. This selectivity originates from the concave nature of the Rh site, as observed for the RhIn(110) surface, which can accurately distinguish the shape and geometry of nitro and vinyl adsorption. The combination of steric hindrance from the inert In atoms and the chemical interaction between Rh and the functional groups plays a significant role on this molecular recognition. It should be noted that the molecular recognition does not depend on the crystal structure, but on the individual surface structure determined by the nature of the second metal. For example, although the RhZn catalyst has a crystal structure identical to that of RhIn (CsCl-type structure) and a similar crystallite size, it shows much lower selectivity than RhIn. This is because the RhZn(110) surface is flat rather than concave, as shown in Fig. 6. Although we focused on the adsorption geometries of the nitro and vinyl groups of NS, a similar rationale can be applied for the other nitro arenes tested in Table 1. As well as the vinyl group, other unsaturated functional groups (CO and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) and carbon–halogen moieties (C–X) generally need to adsorb in a side-on fashion for the group to be activated toward hydrogenation (CO25 and CN)26,27 or dissociation (C–X).28,29 In contrast, end-on geometry is allowed for nitro group hydrogenation.30 This unique adsorption geometry of the nitro group may also contribute to the observed molecular recognition ability and chemoselectivity.
N) and carbon–halogen moieties (C–X) generally need to adsorb in a side-on fashion for the group to be activated toward hydrogenation (CO25 and CN)26,27 or dissociation (C–X).28,29 In contrast, end-on geometry is allowed for nitro group hydrogenation.30 This unique adsorption geometry of the nitro group may also contribute to the observed molecular recognition ability and chemoselectivity.
Recently, we reported a catalytic system for nitrostyrene hydrogenation using Rh-based ordered alloys and methanol as a hydrogen source.31 Although that catalytic system looks similar to the one reported in the present study, we emphasize that the reaction mechanism and the origin of the chemoselectivity are completely different. In the case of the methanol system, selectivity for nitro hydrogenation is always >99% regardless of the second metal because the methanol-derived hydrogen species is almost inactive for vinyl hydrogenation. The electron density of Rh varies depending on the second metal, which changes the methanol activation ability and the overall reaction rate. Thus, for the methanol system, there is only an electronic effect on the catalytic activity.
Conclusion
In this study, a series of Rh-based ordered alloys were tested in the hydrogenation of nitrostyrene. Of the tested catalysts, RhIn/SiO2 showed the highest selectivity for nitro hydrogenation. A high aminostyrene yield was obtained within a few hours at room temperature under atmospheric H2 pressure, which had never been previously attained using an inorganic heterogeneous catalyst. Moreover, RhIn/SiO2 exhibited excellent catalytic performance in the nitro hydrogenation of nitroarenes containing vinyl, cyano, carbonyl, and halo moieties. The selectivity of these catalysts varies significantly depending on the difference in the adsorption energies of the two functional groups on the Rh sites. RhIn presents concave Rh sites on its surface, which are able to capture the nitro group with end-on geometry while inhibiting vinylic π adsorption. Thus, the specific and highly ordered surface structure of RhIn enables the chemoselective molecular recognition of nitro and vinyl groups through a combination of geometric effects and chemical interactions between Rh and C or O atoms. This is the first demonstration of molecular recognition properties being governed by an ordered bimetallic surface. The obtained results provide not only a highly efficient catalytic system, but also important insight into the design and construction of all-inorganic reaction environments for molecular recognition.Methods
Catalyst preparation
Silica-supported monometallic Rh (Rh/SiO2) was prepared by pore-filling impregnation. Aqueous solution of Rh(NO3)3 was added to dry silica gel (Cariact G-6, Fuji Silysia Ltd.). The solution volume was calculated to fill silica gel pores and achieve 3 wt% Rh loading. The mixture was sealed overnight at room temperature using a piece of plastic film, dried over a hot plate at 100 °C and calcined under dry air at 500 °C for 2 h. The catalyst was subsequently reduced under H2 flow (60 mL min−1, 99.9995%) for 2 h at 400 °C. Rh-based bimetallic alloys supported on silica (RhM/SiO2; M = Bi, Fe, Ga, In, Ni, Sb, Sn, and Zn) were prepared by successive impregnations with Rh/SiO2. An aqueous solution of the second metal (typically nitrates, excepting (NH4)2GeF6, InCl3, SbCl3, and (NH4)2SnCl6) was added to Rh/SiO2 so that the Rh/M atomic ratio was adjusted to unity. Rh–Pb/SiO2 catalysts (Rh3Pb2, RhPb, and RhPb2) were prepared by co-impregnation using a mixed aqueous solution of Rh(NO3)3 and Pb(NO3)2 possessing Rh/Pb ratios at 1.5, 1, and 0.5, respectively. For all bimetallic catalysts, the impregnated mixture was kept overnight and dried in a similar way for Rh/SiO2, followed by reduction under H2 flow at 800 °C for 1 h.Catalytic hydrogenation
Hydrogenation of nitroarenes was conducted in a 50 mL, three-necked, round-bottom flask equipped with a reflux condenser, gas balloon and stopcock possessing a silicone septum. Prior to the reaction, catalyst (50 mg) was reduced in the reactor for 30 min under H2 gas flow (60 mL min−1) at 400 °C using a mantle heater. After the reduction pre-treatment, the reactor was cooled to a reaction temperature (25–75 °C) under H2 gas flow. An ethyl acetate solution (5 mL) of nitroarene (0.5 mmol) and n-dodecane as an internal standard was added into the reactor through the septum. The reaction was initiated by stirring at a constant rate (400 rpm). The products in the liquid phase were identified and quantitated by flame-ionization gas-chromatogram (Shimadzu, GC-14B) equipped with a capillary column (GL Science, TC-17, 0.25 mm × 30 m). The partial pressure of H2 was adjusted using He as a diluent for the kinetic study. The hydrogenation rates of the nitro (rNO2) and vinyl (rCC) groups in NS were determined as formation rates (mmol min−1) of AS and EN, respectively.
Characterization
The crystalline structures of the catalysts were examined by powder XRD with a Rigaku RINT2400 diffractometer using a Cu-Kα X-ray source. TEM was conducted by a JEOL JEM-2010F microscope at an accelerating voltage of 200 kV. To prepare the TEM specimens, all samples were sonicated in carbon tetrachloride and dispersed on copper grids supported by an ultra-thin carbon film. CO pulse chemisorption was conducted at room temperature to estimate the Rh dispersion. The FT-IR spectra of adsorbed CO were measured with a JASCO FT/IR-430 spectrometer in transmission mode. A self-supporting wafer (20 mg cm−2) of the catalyst was placed in a quartz cell with CaF2 windows and attached to a glass circulation system. The catalyst was reduced at 450 °C for 30 min under H2 gas flow (60 mL min−1). Then, the catalyst was evacuated at the same temperature for 30 min, followed by cooling to room temperature. After the introduction of 5 Pa CO, spectra were repeatedly recorded at 1 cm−1 of resolution until the peak growth was saturated. ICP-AES analysis of the catalysts was performed using Shimadzu ICPE-9000 spectrometer. The catalysts were dissolved using aqua regia and a HF solution.Computational details
Periodic DFT calculations except vibrational calculations were performed using the CASTEP code32 with Vanderbilt-type ultra-soft pseudopotentials33 and a revised Perdew–Burke–Ernzerhof exchange–correlation functional (RPBE)34,35 based on the generalised gradient approximation. The plane wave basis set was truncated at a kinetic energy of 370 eV. A Fermi smearing of 0.1 eV was utilised. The reciprocal space was sampled using a k-point mesh with a typical spacing of 0.04 Å−1 generated by the Monkhorst–Pack scheme.36 Convergence criteria comprised (a) a self-consistent field (SCF) tolerance of 1.0 × 10−6 eV per atom, (b) an energy tolerance of 1.0 × 10−5 eV per atom, (c) a maximum force tolerance of 0.05 eV Å−1, and (d) a maximum displacement tolerance of 1.0 × 10−3 Å for structural optimization and energy calculation. Atomic coordinates were fully relaxed, whereas lattice constants were fixed. Surface energies of bimetallic plane were calculated using eqn (2):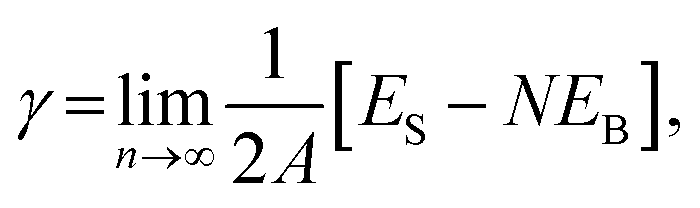 | (2) |
m (CsCl-type) structures, the Rh-terminated (or Rh-rich) plane was chosen as an active surface for hydrogenation. The surface energies of such planes were estimated using a procedure reported elsewhere.10,37 For the geometry optimization of adsorbed CO, (2 × 2) structures were considered for all Rh-based bimetallic surfaces, so that CO coverage on Rh was ≤1/4. Slabs were modelled typically with 4-atomic-layer-thickness and a 10 Å vacuum spacing between the two surfaces. For a monometallic Rh(111) surface, various adsorption geometries of CO were considered: (3 × 3), (2 × 2), 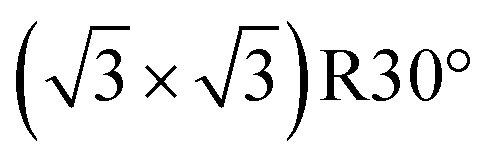 , c(4 × 2)-2CO, and (2 × 2)-3CO structures for 1/9, 1/4, 1/3, 1/2, and 3/4 full coverages, respectively. For the geometry optimization of NS adsorbed on the Rh-based planes, cut-off energy, k-point mesh spacing and vacuum spacing were set to 340 eV, 0.06 Å−1, and 17 Å, respectively. Surfaces were modelled using (4 × 4), (3 × 4), (2 × 2), and (2 × 2) unit cells for cubic Fmm (Rh, (111)), Pmm (RhGa, RhIn, RhZn, (110)), P213 (RhSn, (012)), hexagonal (P63/mmc; RhBi (110), P6/mmm; RhPb (002)), and tetragonal I4/mcm (RhPb2, (100)) structures, respectively. The adsorption energy was defined with eqn (3)
, c(4 × 2)-2CO, and (2 × 2)-3CO structures for 1/9, 1/4, 1/3, 1/2, and 3/4 full coverages, respectively. For the geometry optimization of NS adsorbed on the Rh-based planes, cut-off energy, k-point mesh spacing and vacuum spacing were set to 340 eV, 0.06 Å−1, and 17 Å, respectively. Surfaces were modelled using (4 × 4), (3 × 4), (2 × 2), and (2 × 2) unit cells for cubic Fmm (Rh, (111)), Pmm (RhGa, RhIn, RhZn, (110)), P213 (RhSn, (012)), hexagonal (P63/mmc; RhBi (110), P6/mmm; RhPb (002)), and tetragonal I4/mcm (RhPb2, (100)) structures, respectively. The adsorption energy was defined with eqn (3)| ΔEad = EA–S − (ES + EA), | (3) |
The frequency calculations of adsorbed CO molecules were conducted using the DMol3 code.38 These calculations involved the RPBE functional, a double-numeric quality basis set with polarization functions (DNP, comparable to Gaussian 6-311G**)39 associated with a real-space cut-off of 4.2 Å, DFT semi-core pseudopotential core treatment40 and Fermi smearing of 0.1 eV. The SCF convergence was accelerated using the iterative scheme proposed by Kresse and Furthmüller.41 The partial Hessian matrix, including C and O atoms, was computed to evaluate the harmonic frequencies of the adsorbed CO. All computed harmonic frequencies were scaled by an empirical factor of 1.0386, which corresponds to the ratio between the experimental42 and computed values for gas-phase CO (2143 cm−1/2063.4 cm−1).
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 26820350. We deeply thank the Center for Advanced Materials Analysis Tokyo Institute of Technology for aid with TEM observation.References
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS PubMed.
- R. E. Babine and S. L. Bender, Chem. Rev., 1997, 97, 1359–1472 CrossRef CAS PubMed.
- S. S. Kim, T. J. Pinnavaia and R. Damavarapu, J. Catal., 2008, 253, 289–294 CrossRef CAS.
- K. Smith, S. D. Roberts and G. A. Ei-Hiti, Org. Biomol. Chem., 2003, 1, 1552–1559 CAS.
- E. J. Creyghton, S. D. Ganeshie, R. S. Downing and H. vanBekkum, J. Mol. Catal. A: Chem., 1997, 115, 457–472 CrossRef CAS.
- E. J. Creyghton, S. D. Ganeshie, R. S. Downing and H. Vanbekkum, J. Chem. Soc., Chem. Commun., 1995, 1859–1860 RSC.
- J. H. Westbrook and R. L. Fleischer, Magnetic, Electrical and Optical Properties and Applications of Intermetallic Compounds, Wiley, 2000 Search PubMed.
- T. Komatsu and S. Furukawa, Mater. Trans., 2015, 56, 460–467 CrossRef CAS.
- T. Komatsu and A. Onda, Catal. Surv. Asia, 2008, 12, 6–15 CrossRef CAS.
- S. Furukawa, K. Ochi, H. Luo, M. Miyazaki and T. Komatsu, ChemCatChem, 2015, 7, 3472–3479 CrossRef CAS.
- R. S. Downing, P. J. Kunkeler and H. vanBekkum, Catal. Today, 1997, 37, 121–136 CrossRef CAS.
- M. Suchy, P. Winternitz and M. Zeller, World Patent 91/00278, 1991.
- P. N. Rylander, Catalytic Hydrogenation in Organic Synthesis, Academic Press, New York, 1979 Search PubMed.
- K. Shimizu, Y. Miyamoto, T. Kawasaki, T. Tanji, Y. Tai and A. Satsuma, J. Phys. Chem. C, 2009, 113, 17803–17810 CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- T. Mitsudome, Y. Mikami, M. Matoba, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2012, 51, 136–139 CrossRef CAS PubMed.
- K. Shimizu, Y. Miyamoto and A. Satsuma, J. Catal., 2010, 270, 86–94 CrossRef CAS.
- H. S. Wei, X. Y. Liu, A. Q. Wang, L. L. Zhang, B. T. Qiao, X. F. Yang, Y. Q. Huang, S. Miao, J. Y. Liu and T. Zhang, Nat. Commun., 2014, 5, 6634 Search PubMed.
- A. Corma, P. Serna, P. Concepcion and J. J. Calvino, J. Am. Chem. Soc., 2008, 130, 8748–8753 CrossRef CAS PubMed.
- R. A. W. Johnstone, A. H. Wilby and I. D. Entwistle, Chem. Rev., 1985, 85, 129–170 CrossRef CAS.
- J. C. Slater, J. Chem. Phys., 1964, 41, 3199–3204 CrossRef CAS.
- M. Armbruster, M. Behrens, K. Fottinger, M. Friedrich, E. Gaudry, S. K. Matam and H. R. Sharma, Catal. Rev., 2013, 55, 289–367 Search PubMed.
- P. S. Cremer, X. C. Su, Y. R. Shen and G. A. Somorjai, J. Am. Chem. Soc., 1996, 118, 2942–2949 CrossRef CAS.
- F. Mittendorfer and J. Hafner, J. Phys. Chem. B, 2002, 106, 13299–13305 CrossRef CAS.
- A. Vargas, T. B. Burgi and A. Baiker, J. Catal., 2004, 222, 439–449 CrossRef CAS.
- C. Oliva, C. van den Berg, M. H. Niemantsverdriet and D. Curulla-Ferre, J. Catal., 2007, 248, 38–45 CrossRef CAS.
- C. Oliva, C. van den Berg, J. W. H. Niemantsverdriet and D. Curulla-Ferre, J. Catal., 2007, 245, 436–445 CrossRef CAS.
- J. H. Lyu, J. G. Wang, C. S. Lu, L. Ma, Q. F. Zhang, X. B. He and X. N. Li, J. Phys. Chem. C, 2014, 118, 2594–2601 CAS.
- S. Iihama, S. Furukawa and T. Komatsu, ACS Catal., 2016, 6, 742–746 CrossRef CAS.
- L. Y. Zhang, J. H. Jiang, W. Shi, S. J. Xia, Z. M. Ni and X. C. Xiao, RSC Adv., 2015, 5, 34319–34326 RSC.
- S. Furukawa, Y. Yoshida and T. Komatsu, ACS Catal., 2014, 4, 1441–1450 CrossRef CAS.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- B. Hammer, L. B. Hansen and J. K. Norskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- Y. K. Zhang and W. T. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- A. Dannenberg, M. E. Gruner, A. Hucht and P. Entel, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 245438 CrossRef.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- Y. Inada and H. Orita, J. Comput. Chem., 2008, 29, 225–232 CrossRef CAS PubMed.
- B. Delley, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- D. Scarano, S. Bordiga, C. Lamberti, G. Spoto, G. Ricchiardi, A. Zecchina and C. O. Arean, Surf. Sci., 1998, 411, 272–285 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR spectra, recycling test, kinetic studies, and DFT calculations. See DOI: 10.1039/c6sc00817h |
| This journal is © The Royal Society of Chemistry 2016 |