
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDendronized delayed fluorescence emitters for non-doped, solution-processed organic light-emitting diodes with high efficiency and low efficiency roll-off simultaneously: two parallel emissive channels†
Yifan
Li‡
,
Guohua
Xie‡
 ,
Shaolong
Gong
,
Kailong
Wu
and
Chuluo
Yang
*
,
Shaolong
Gong
,
Kailong
Wu
and
Chuluo
Yang
*
Hubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Hubei Key Lab on Organic and Polymeric Optoelectronic Materials, Department of Chemistry, Wuhan University, Wuhan, 430072, People's Republic of China. E-mail: clyang@whu.edu.cn
First published on 26th April 2016
Abstract
We have developed two new carbazole-dendronized emitters based on a green emissive thermally activated delayed fluorescence (TADF) core. Both dendrimers possess excellent thermal stability, good solution processability, and an obvious TADF feature. Non-doped OLEDs based on the emitters formed by a solution process exhibit a maximum external quantum efficiency (EQE) of 13.8%. Remarkably, the EQE remains as high as 13.3% at the high luminance of 1000 cd m−2. To the best of our knowledge, this is one of the highest EQE values for dendrimer-based fluorescent OLEDs, which nearly harvest all of the generated excitons and exhibit a considerably low loss of EQE estimated from 1000 to 5000 cd m−2. Furthermore, we reveal a new emissive approach to utilize the excitons by a combination of both TADF and exciplex emission.
Introduction
Metal-free thermally activated delayed fluorescence (TADF) emitters have recently aroused considerable attention because they can harvest both singlet and triplet excitons through up-converting non-radiative triplet excitons to radiative singlet excitons and thereby approach a theoretical internal quantum efficiency (IQE) of 100%.1,2 Highly efficient OLEDs based on TADF emitters have achieved maximum external quantum efficiencies (EQEs) over 20%, which are comparable with heavy-metal based phosphorescent OLEDs. For example, the devices based on a blue emitter of a triazine/carbazole hybrid, and a green emitter of 2,4,5,6-tetra(carbazol-9-yl)-1,3-dicyanobenzene (4CzIPN) have achieved EQEs of 20.6% and 28.6%, respectively.1b,2b,3 In addition to the TADF emitters, the exciplex emission formed between electron-donating and electron-accepting molecules has recently been demonstrated to be promising for highly efficient fluorescent OLEDs.4 For example, the highest EQE of 15.4% for a fluorescent OLED employing an exciplex with a high photoluminescence quantum yield (PLQY) of 0.68 has been reported.5Despite the high efficiency of these two types of emissive approaches in fluorescent OLEDs, further utilization of these emitters in the solution process has been rarely explored.6 Generally, most highly efficient devices are fabricated via vacuum evaporation technology, which requires a complicated process, high costs and precise engineering. Comparatively, solution processes, such as spin-coating and ink-jet printing, represent low-cost methods to fabricate large-area OLEDs.7 In the past three years, OLEDs based on solution-processable TADF emitters have made some progress. For example, Lee et al. reported a solution-processed OLED with a maximum EQE of 18.3% by utilizing a green TADF emitter;8 Su et al. demonstrated a maximum EQE of 17.5% by employing an evaporation- and solution-process-feasible TADF emitter.9 Although these devices exhibited good performance, all of the emitting layers (EMLs) in the devices are host:dopant systems that require precise control and may result in phase separation. In these contexts, developing solution-processed non-doped emissive materials with a TADF feature is of practical and important significance.
Carbazole-dendronized materials are promising single-component functional materials for solution-processed OLEDs because of their good solubility in common solvents, good hole-transporting ability, amorphous film-forming property, and high thermal stability.10
Although some progress has been made on carbazole-dendronized phosphorescent emitters,11 much work remains to be done with the aim of developing metal-free emitters with a high EQE for solution-processed OLEDs. In this communication, we report two carbazole-dendronized TADF emitters by introducing carbazole dendrons into a TADF emissive core, bis(4-(9,9-dimethylacridin-10(9H)-yl)phenyl)methanone (DMAC-BP), which has been proven to be an excellent green TADF emitter peaking at 506 nm in neat film with a high PLQY of 0.85.12 We anticipate that the decoration of DMAC-BP with carbazole could not only improve hole injection and transport ability, but also minimize the exciton quenching approach, and consequently reduce efficiency roll-off at high luminance. Double-layer devices based on these emitters as the non-doped emissive layer exhibit a maximum EQE of 13.8%. Remarkably, the EQE remains at 13.3% at the high luminance of 1000 cd m−2. To the best of our knowledge, this is one of the highest EQE values for dendrimer-based fluorescent OLEDs. Furthermore, we reveal that the dendrimer-based devices display a dual-channel emissive feature, i.e., both TADF and interfacial exciplex emission.
Results and discussion
The chemical structures of two carbazole-dendronized emitters, named bis(4-(2,7-bis(3,6-di-tert-butyl-9H-carbazol-9-yl)-9,9-dimethylacridin-10(9H)-yl)phenyl)methanone (CDE1) and bis(4-(2,7-bis(3,3′′,6,6′′-tetra-tert-butyl-9′H-[9,3′′:6′,9′′-tercarbazole]-9′′-yl)-9,9-dimethylacridin-10(9H)-yl)phenyl)methanone (CDE2), are depicted in Fig. 1. Iodization of DMAC-BP with N-iodosuccinimide (NIS) produced the key intermediate, bis(4-(2,7-diiodo-9,9-dimethylacridin-10(9H)-yl)phenyl)methanone (4IDMACBP),13 which was coupled with the first and second generation carbazole dendrons through the Ullmann C–N coupling reaction to afford the target compounds CDE1 and CDE2, respectively (Scheme S1†).6a These compounds were characterized by 1H NMR and 13C NMR spectroscopies, mass spectrometry, and elemental analysis. The two compounds exhibit superior solubility in usual organic solvents, such as dichloromethane, tetrahydrofuran, toluene, and chlorobenzene, due to the peripheral tert-butyl groups. Their good thermal stability was indicated by the high decomposition temperatures (Td, corresponding to 5% weight loss) of 471 °C for CDE1 and 507 °C for CDE2 (Fig. S1†). Moreover, the glass transition temperatures (Tg) of these compounds reach up to 283 °C for CDE1 and 289 °C for CDE2 (Fig. S2†). To evaluate the morphological stability of these compounds, we utilized atomic force microscopy (AFM) to explore their surface images in neat films before and after annealing at 160 °C (Fig. S3†). Apparently, the surfaces of the neat films after annealing remained smooth and pinhole-free, almost the same as the unannealed surfaces. The good thermal and morphological stability of these materials guarantee homogeneous and stable amorphous thin films via solution processing, which is indispensable for the operation of OLEDs. | ||
| Fig. 1 Chemical structures of DMAC-BP, CDE1 and CDE2. | ||
Fig. 2 shows the absorption, photoluminescence (PL) and phosphorescence spectra of CDE1 and CDE2 in film. Both compounds exhibit similar absorption profiles with two types of absorption bands: the absorption bands at 289 and 298 nm, which can be attributed to the π–π* transition of the carbazole dendrons;11a the charge-transfer (CT) absorption bands at 348 and 349 nm, which can be assigned to the π–π* electron transition from 9,9-dimethyl-9,10-dihydroacridine (DMAC) and the carbazole to the benzophenone. In their PL spectra in neat film, the emission peaks are blue-shifted with the increasing generation of the carbazole dendrons, indicating that the donor–acceptor feature of CDE2 is weaker than that of CDE1, which can be verified by the TD-DFT calculation results (Fig. S6†).11b,14 In comparison with the neat films, the toluene solutions of the two dendrimers exhibit bathochromic shifts of 31 and 33 nm, respectively (Fig. S4†). This implies that the intermolecular π–π* interactions of the fluorescent core in the solid state are significantly weakened by the bulky molecular structure of the external carbazole dendrons.15 The energy gaps between the singlet and triplet state energy (ΔEST) are 0.11 eV for CDE1 and 0.15 eV for CDE2, which were estimated from the onsets of the fluorescence and phosphorescence spectra. The small ΔEST values suggest that triplet excitons can be easily harvested by T1 → S1 reverse intersystem crossing. The PLQYs of CDE1 and CDE2 in neat film were measured to be 0.77 and 0.75 under N2 conditions, respectively, showing good potential in OLEDs.
 | ||
| Fig. 2 The absorption and fluorescence spectra at room temperature, and phosphorescence spectra at 77 K of CDE1 (a) and CDE2 (b) in neat film. | ||
To confirm the existence of the TADF in CDE1 and CDE2, their transient PL spectra both in toluene and in neat film were measured. As shown in Fig. 3a and b, both compounds exhibit a second-order exponential decay in toluene under oxygen-free conditions with a delayed fluorescence (DF) decay of 523 ns (CDE1) and 627 ns (CDE2). Their prompt fluorescence decay lifetimes of 15 ns (CDE1) and 21 ns (CDE2) indicate the radiative decay from S1 to S0. After the exposure of the solution in ambient air for 30 minutes, the delayed component significantly decreases.1b,16 This implies that PL decay is sensitive to oxygen, which proves that the two compounds feature typical TADF behavior. Moreover, the temperature-dependence of the transient PL decay in film is consistent with the typical characteristics of TADF materials. The delayed component gradually increases when raising the temperature from 100 to 300 K (Fig. 3c and d).17
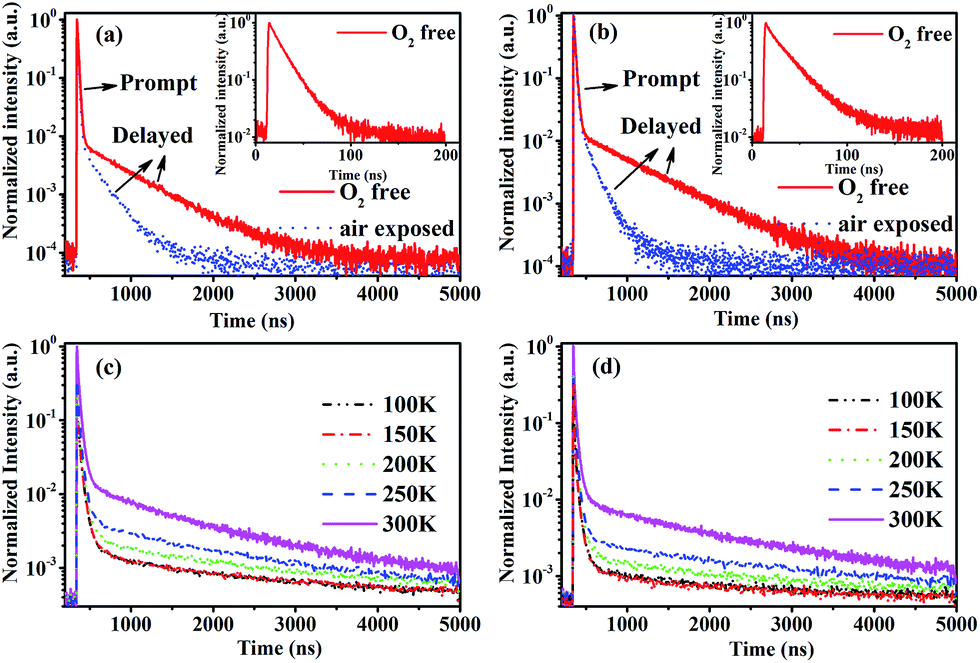 | ||
| Fig. 3 Transient decay curves of CDE1 (a) and CDE2 (b) in toluene without and with oxygen exposure for 30 min. Inset: prompt fluorescence. Temperature dependence of the transient decay for CDE1 (c) and CDE2 (d). | ||
Inspired by the highly thermal stability and good film forming ability of the two dendrimers, simple double-layer solution-processed OLEDs were fabricated with an architecture of ITO/PEDOT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PSS (30 nm)/CDE1 (Devices A1–A4) or CDE2 (Devices B1–B4) (70 nm)/TPBi (40 nm)/Liq (2 nm)/Al (Fig. 4), where poly(3,4-ethylenedioxythiophene) doped with poly(styrenesulfonate) (PEDOT:PSS) served as a hole-injecting layer and 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBi) acted as an electron transporting layer (ETL). Meanwhile, the EMLs were exposed to elevated temperatures at 80 (A2 and B2), 120 (A3 and B3) and 160 °C (A4 and B4), for comparison with those without thermal annealing (A1 and B1). The EML was spin-coated from chlorobenzene solution.
:PSS (30 nm)/CDE1 (Devices A1–A4) or CDE2 (Devices B1–B4) (70 nm)/TPBi (40 nm)/Liq (2 nm)/Al (Fig. 4), where poly(3,4-ethylenedioxythiophene) doped with poly(styrenesulfonate) (PEDOT:PSS) served as a hole-injecting layer and 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBi) acted as an electron transporting layer (ETL). Meanwhile, the EMLs were exposed to elevated temperatures at 80 (A2 and B2), 120 (A3 and B3) and 160 °C (A4 and B4), for comparison with those without thermal annealing (A1 and B1). The EML was spin-coated from chlorobenzene solution.
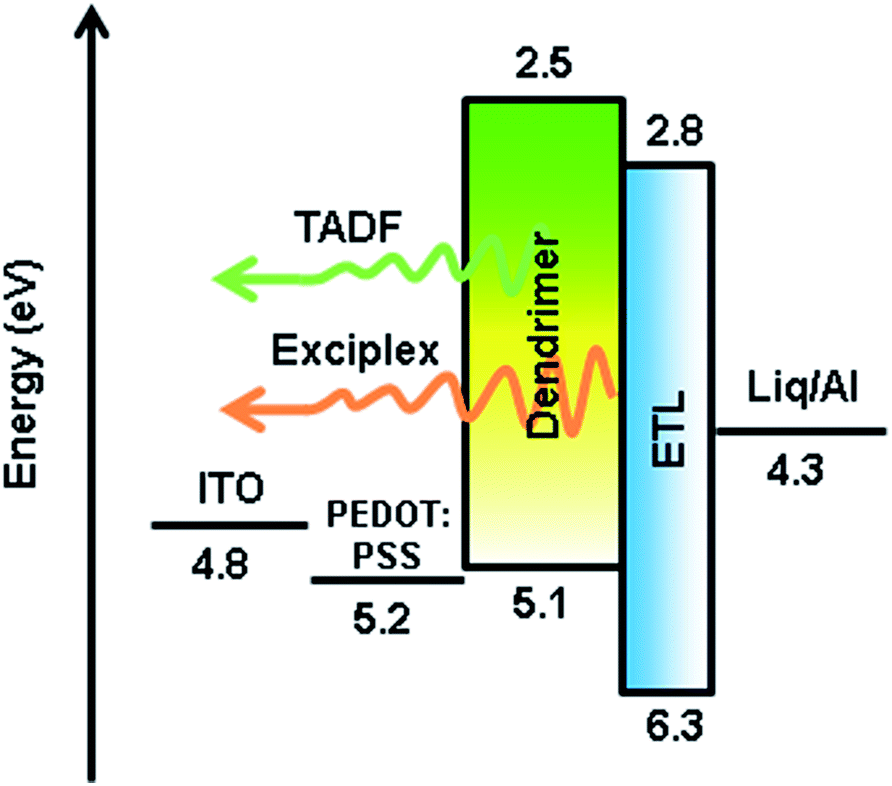 | ||
| Fig. 4 Schematic diagram of the energy levels of Devices A1–B4. | ||
Fig. 5 shows the EL spectra, the current density–voltage–luminance (J–V–L) characteristics, and the EQEs versus luminance curves of Devices A1–A4. The corresponding curves for Devices B1–B4 are shown in Fig. S8.† The characteristic data for all devices are summarized in Table 1. The driving voltages of Devices A1–A4 are significantly lower than the corresponding Devices B1–B4. For example, Device A2 exhibits driving voltage at 4.5 V that is lower than the 7.7 V of Device B2 at the same luminance of 10 cd m−2. The superior electrical performance of CDE1 is attributed to the better matched highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels that lower the hole and electron injection barriers of Device A. Among Series A, Device A2 exhibits considerably high luminance and current density (Fig. 5a) at the same bias. A maximum current efficiency (ηc,max) of 38.9 cd A−1 (Fig. S7a†), a maximum power efficiency (ηp,max) of 17.3 lm W−1 (Fig. S7b†), and a maximum external quantum efficiency (ηext,max) of 12.0% (Fig. 5b) were achieved. It is worth mentioning that these devices exhibit very high color stability at the elevated temperatures (see the inset of Fig. 5b) which is one of the prerequisites for practical application. Moreover, these devices show rather low efficiency roll-off values. At the luminance of 1000 cd m−2, the ηext of Device A2 is as high as 11.9%, and at the luminance of 5000 cd m−2, the ηext still remains at 10.3%.
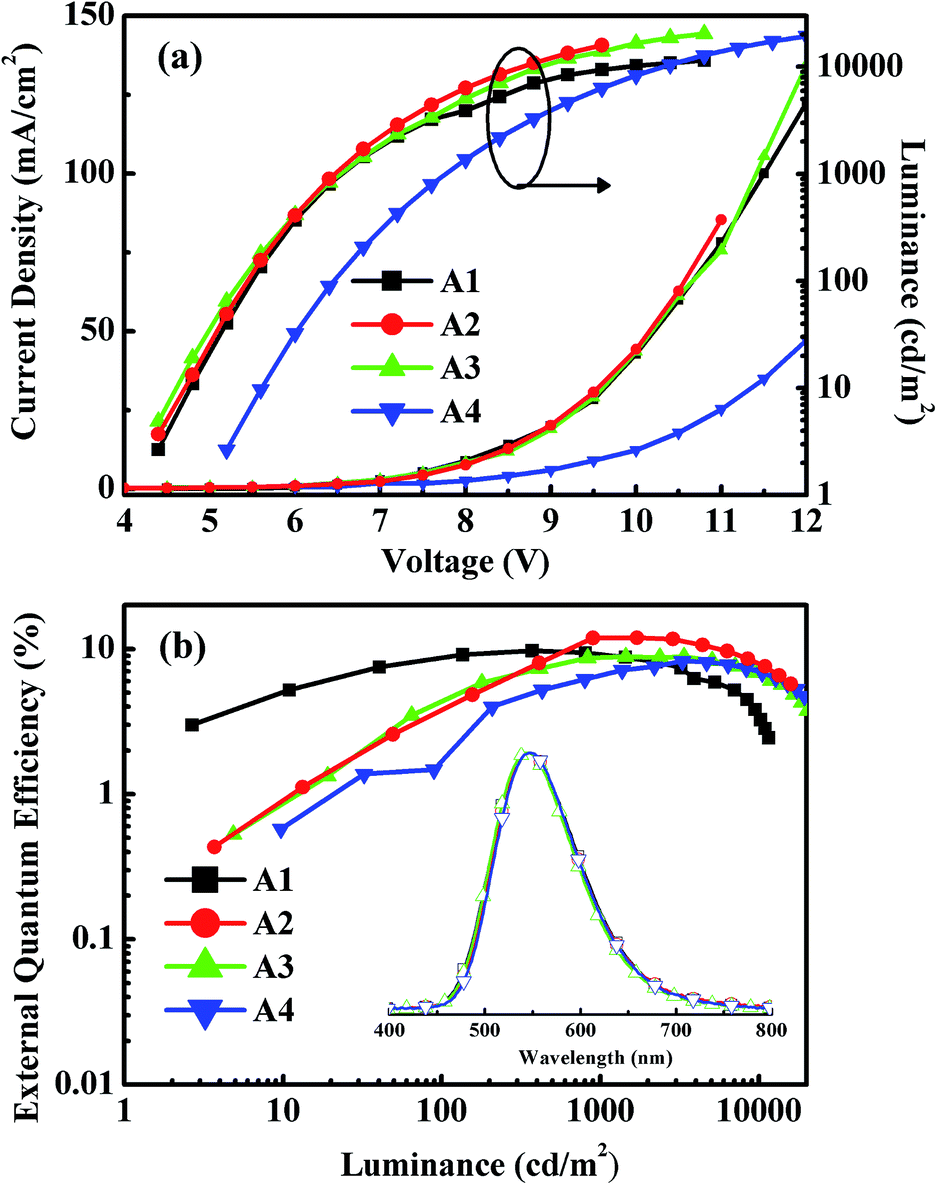 | ||
| Fig. 5 (a) Current density–voltage–luminance (J–V–L) characteristics. (b) External quantum efficiency versus luminance curves of Devices A1–A4 and the EL spectra of CDE1 annealed at different temperatures (inset). | ||
| Device | V 10 [V] | η ext,max [%] | η ext,1000 [%] | L max [cd m−2] | λ ems [nm] | FWHMe [nm] | CIE(x, y)f |
|---|---|---|---|---|---|---|---|
| a Voltage at 10 cd m−2. b Maximum EQE. c EQE at 1000 cd m−2. d Peak EL wavelength. e Full-width half maximum of the EL spectrum. f Commission Internationale de L'Eclairage (CIE) coordinates. | |||||||
| A1 | 4.9 | 9.7 | 9.0 | >10000 |
546 | 106 | (0.38, 0.55) |
| A2 | 4.8 | 12.0 | 11.9 | >10000 |
546 | 103 | (0.38, 0.56) |
| A3 | 4.7 | 8.8 | 8.7 | >10000 |
546 | 102 | (0.38, 0.56) |
| A4 | 6.0 | 8.3 | 6.7 | >10000 |
546 | 101 | (0.39, 0.55) |
| B1 | 9.4 | 3.3 | 2.0 | 1197 | 522 | 114 | (0.32, 0.52) |
| B2 | 7.7 | 5.2 | 4.1 | 2512 | 522 | 118 | (0.32, 0.51) |
| B3 | 8.1 | 5.2 | 4.1 | 2418 | 528 | 115 | (0.32, 0.52) |
| B4 | 9.3 | 4.7 | 4.3 | 2278 | 528 | 112 | (0.33, 0.53) |
| C3 | 4.4 | 13.8 | 13.3 | >10000 |
552 | 114 | (0.40, 0.54) |
Interestingly, we find significant red-shifts (26 nm for CDE1 and >23 nm for CDE2) of the EL spectra in comparison with their PL spectra (Table S1†). We suppose that the exciplex formed at the interface between the EML and the ETL contribute to the red-shift.4a To shed light on this hypothesis of exciplex existence, we fabricated devices based on CDE1 with four different ETLs including 4,7-diphenyl-1,10-phenanthroline (BPhen) for Device C1, 4,6-Bis(3,5-di-3-pyridylphenyl)-2-methylpyrimidine (B3PYMPM) for Device C2, 1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB) for Device C3 and TPBi for Device C4. Apparently, all the devices with different ETLs exhibit red-shifts in the EL spectra (Fig. S11†) compared to the so-called single layer (without ETL) device (peak at 528 nm shown in Fig. S12†) with a full width at half maximum (FWHM) of 95 nm. Judging from Fig. S11 and 12,† it is evident that CDE1 easily forms the interfacial exciplex with the ETLs through the external carbazole dendrons. Furthermore, we use a Gaussian function to fit the EL spectra curves and find that all of the EL spectra contain two main components which could be assigned to TADF emission (equivalent to that of Fig. S12†) and the exciplex emission which depends on the ETL (Fig. S13†).
Beyond the steady state study, to further prove the existence of the exciplex, we measured the transient decay of the mixtures of TmPyPB and CDE1. The steady state PL spectra of TmPyPB:CDE1 films show gradual red-shifts on increasing the ratio of TmPyPB from 10 wt% to 50 wt% (Fig. 6a).4a,18 Undoubtedly, the time-resolved PL spectroscopy data of the pristine CDE1 film and the mixed TmPyPB:CDE1 (1:1) film provide direct evidence of the existence of the exciplex. At the early delay stage (50 ns), the red-shift in the emission profile of the mixture is almost negligible compared to that of the neat film (Fig. 6b). In contrast, at the longer delayed time (at 1 μs), a distinguishable change is observed, which indicates that the exciplex contributes to red-shift of the spectra. The red-shifted delayed emission is quite similar to the spectral fitting (Fig. S13c†) of the device with TmPyPB as the ETL.4b,19 All of the aforementioned evidence sufficiently proves the existence of the two parallel emission mechanisms: intrinsic TADF from the dendrimer and the exciplex emission at the interface between the EML and the ETL. Among Device Series C, the Device C3 with TmPyPB as the ETL achieves a maximum EQE of up to 13.8% (Fig. S10b†), partially due to the high electron mobility and excellent hole blocking ability of TmPyPB. Table 2 summarizes the performances of the state-of-the-art OLEDs with TADF or exciplex emission reported in recent literature. It can be seen that very few emitters achieved high efficiencies at 5000 cd m−2. Basically, regardless of wet or dry processes, only the host-guest structure could maintain high efficiencies at high luminance. In contrast, the dendrimer-based non-doped double-layer architectures boost the EQE to 11.9% (TPBi as the ETL) and 13.3% (TmPyPB as the ETL) at a luminance of 1000 cd m−2. Impressively, the roll-offs of the EQEs from 1000 to 5000 cd m−2 are reasonably low compared to those of the counterpart devices, which could be ascribed to the self-host like nature of the dendrimer based emitters. To the best of our knowledge, the peak EQE and the luminance of CDE1 are some of the highest among the non-doped dendrimer-based OLEDs featuring a TADF mechanism.
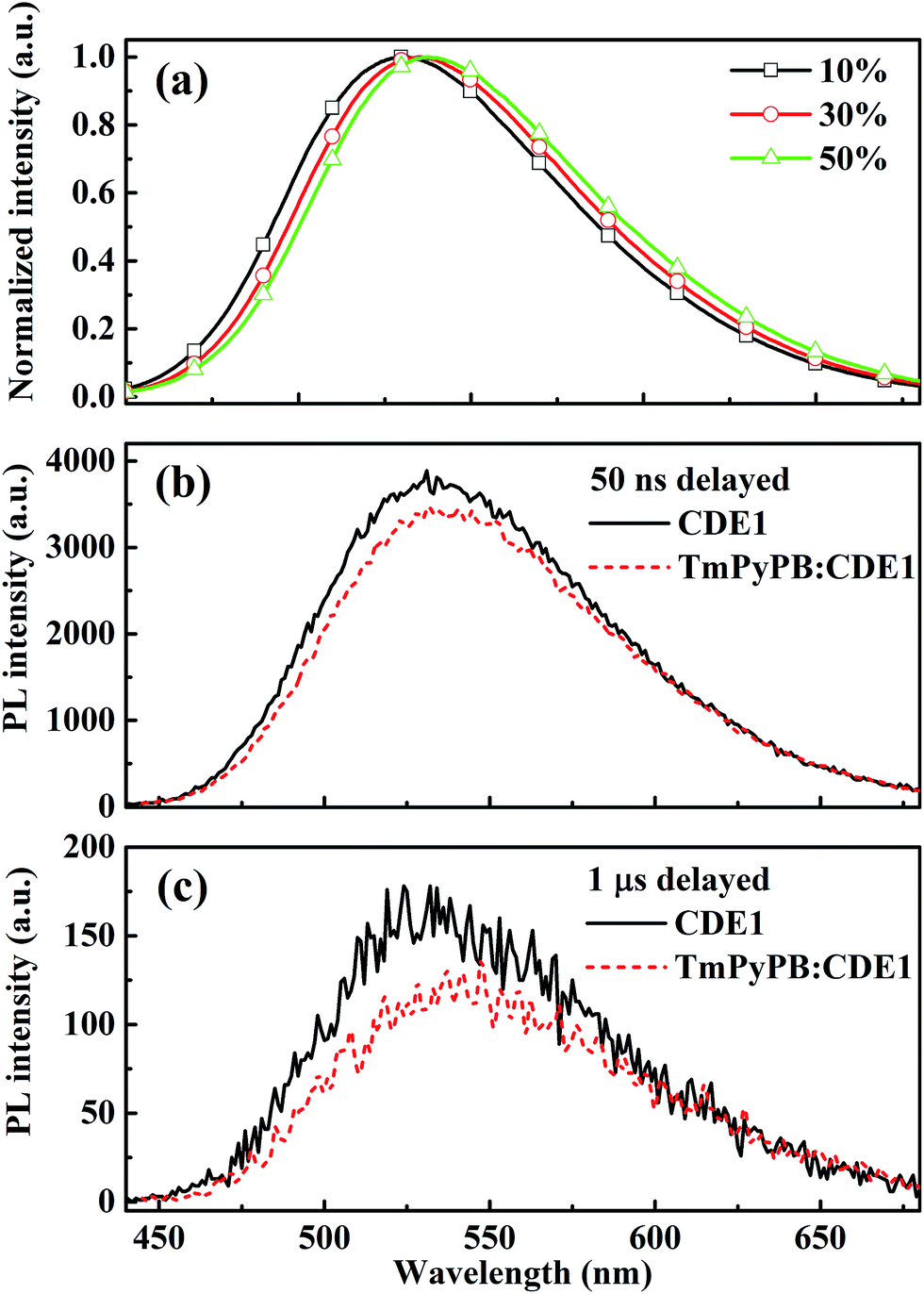 | ||
| Fig. 6 (a) Steady state PL spectra of CDE1 doped with 10, 30 and 50 wt% TmPyPB in film. Time-resolved transient fluorescence spectra delayed at 50 ns (b) and 1 μs (c) for CDE1 and TmPyPB:CDE1 (1:1) in film, respectively. | ||
| Process | Architecture | EML | Mechanism | η ext,max [%]a | η ext,1000 [%]b | η ext,5000 [%]c | Roll-off [%]d | |
|---|---|---|---|---|---|---|---|---|
| a Maximum EQE. b EQE at 1000 cd m−2. c Measured at 1000 cd m−2. d The roll-off of the EQE from 1000 to 5000 cd m−2. e Solution-processed device. f Double-layer device. g Device C4 with TBPi as the ETL. h Device C3 with TmPyPB as the ETL. i Not available. j Vacuum evaporated device. | ||||||||
| This work | Sole | Doublef | Non-doped | TADF and exciplex | 12.0g | 11.9 | 10.3 | 13.4 |
| 13.8h | 13.3 | 8.2 | 38.3 | |||||
| Ref. 6a | Sol | Double | Non-doped | TADF | 3.4 | 1.5 | —i | — |
| Ref. 9 | Sol | Double | Doped | TADF | 15.2 | 13.3 | 8.8 | 33.8 |
| Ref. 6b | Sol | Double | Doped | TADF | 11.3 | 10.6 | 6.9 | 34.9 |
| Ref. 8 | Sol | Four | Doped | TADF | 18.3 | 12.0 | 6.0 | 50 |
| Ref. 7a | Sol | Double | Doped | TADF | 18.6 | 13.0 | 8.0 | 38.5 |
| Ref. 20 | Sol | Double | Doped | TADF | 5.2 | 3.0 | — | — |
| Ref. 12 | Vacj | Double | Non-doped | TADF | 10.6 | 10.6 | 10.2 | 3.8 |
| Ref. 5 | Vac | Three | Doped | Exciplex | 15.4 | 8.6 | — | — |
| Ref. 18 | Vac | Four | Doped | Exciplex | >14 | 6.5 | — | — |
| Ref. 4a | Vac | Double | Doped | Exciplex | 12.2 | 6.0 | — | — |
Conclusions
In conclusion, we have developed two new carbazole-dendronized emitters based on a green emissive TADF core. Both dendrimers possess excellent thermal stability, good solution processability, and an obvious TADF feature accompanied by hidden exciplex emission. A non-doped OLED based on the dendrimer of CDE1 formed by a solution process exhibits a high EQE of 13.8% compared to recent simple dendrimer-based OLEDs, which is contributed to by two parallel emissive channels of TADF and exciplex emission fully harvesting all the generated excitons. This work presents a judicious design strategy for non-doped solution-processable TADF emitters, and reveals a new emissive approach by combining both TADF and exciplex emission. We believe that the strategy will pave the way to practical application of solution-processable fluorescent OLEDs for displays and lighting sources.Experimental section
General information
All reagents were purchased from commercial sources and used as received without further purification. Solvents for synthesis were purified according to standard procedures prior to use. 1H NMR and 13C NMR spectra were measured on a MERCURYVX300 or Bruker Advanced II (400 MHz) spectrometer. Elemental analysis of carbon, hydrogen, and nitrogen was obtained using a Vario EL III microanalyzer. MALDI-TOF (matrix-assisted laser-desorption/ionization time-of-flight) mass spectra were performed on a Bruker BIFLEX III TOF mass spectrometer. UV-vis absorption spectra were collected on a Shimadzu UV-2700 recording spectrophotometer. Photoluminescence (PL) spectra were collected on a Hitachi F-4600 fluorescence spectrophotometer. Absolute PLQYs were measured on a Quantaurus-QY measurement system (C11347-11, Hamamatsu Photonics) and all the samples were excited at 330 nm. Thermogravimetric analysis (TGA) was undertaken with a NEZSCH STA 449C instrument and the thermal stability of the samples under nitrogen flow was investigated at a heating rate of 15 °C min−1 from 25 °C to 800 °C. Differential scanning calorimetry (DSC) was performed using a NETZSCH DSC 200 PC unit at a heating rate of 10 °C min−1 from room temperature to 300 °C under a nitrogen flow. Cyclic voltammetry (CV) was measured on a CHI voltammetric analyzer with the conventional three-electrode configuration which consists of a platinum working electrode, a platinum wire auxiliary electrode, and an Ag wire pseudo reference electrode with ferrocenium-ferrocene (Fc+/Fc) as the internal standard. Tetrabutylammonium hexaflorophosphate (TBAPF6) (0.1 M) was used as the supporting electrolyte. Cyclic voltammograms were obtained at a scan rate of 100 mV s−1. The onset potential was determined from the intersection of two tangents drawn at the rising and background current of the cyclic voltammogram.Device fabrication and characterizations
Patterned indium tin oxide (ITO) coated glass substrates with a sheet resistance of 15–20 ohm/square underwent a wet-cleaning course in an ultrasonic bath, beginning with acetone, followed by ethanol. After UV-Ozone treatment for 20 min, a 30 nm thick PEDOT:PSS layer used as a hole-injecting layer was spin-coated on the ITO substrate and then baked inside the glove-box at 120 °C for 10 min. The EML was prepared by spin-coating from chlorobenzene solution on top of the PEDOT:PSS layer. The electron-transporting material was thermally evaporated onto the emitter layer in a vacuum chamber. Finally, Al was sequentially deposited through a shadow mask successively to define the size of the active area. Except for the spin-coating of the PEDOT:PSS layer, all the solution processes were carried out in the controlled atmosphere of a nitrogen dry-box containing less than 0.1 ppm oxygen and moisture. The devices were encapsulated with UV-curable epoxy before being taken out from the glove-box. All the measurements were carried out at room temperature under ambient conditions.
Synthesis
The solvents (1,4-dioxane, and chloroform) were purified by a conventional procedure and distilled under dry argon before using.:2 by vol.) as the eluent to give a green powder. Yield 92%. 1H NMR (CDCl3, 400 MHz): δ [ppm] 1.66 (s, 12H), 6.08 (d, J = 8.8 Hz, 4H), 7.29 (d, J = 8.8 Hz, 4H), 7.49 (d, J = 8.4 Hz, 4H), 7.71 (s, 4H), 8.18 (d, J = 8.4 Hz, 4H). 13C NMR (CDCl3, 100 MHz): δ [ppm] 194.5, 144.5, 139.8, 137.1, 135.3, 134.1, 132.9, 132.6, 131.0, 116.4, 83.9, 36.0, 31.0. MALDI-TOF (m/z): 1085.0 [M+ − CH3]. Anal. calcd for C43H32I4N2O (%): C, 46.94; H, 2.93; N, 2.55; O, 1.45. Found: C, 46.72; H, 3.02; N, 2.63.
:1 by vol.) as the eluent to give a green powder. Yield 73%. 1H NMR (CDCl3, 400 MHz): δ [ppm] 1.36 (s, 72H), 1.74 (s, 12H), 6.53 (d, J = 8.8 Hz, 4H), 7.24 (d, J = 8.8 Hz, 4H), 7.28 (dd, J = 8.8, 2.0 Hz, 8H), 7.42 (dd, J = 8.4, 2.0 Hz, 8H), 7.70 (d, J = 2.0 Hz, 4H), 7.87 (d, J = 8.4 Hz, 4H), 8.23 (d, J = 1.6 Hz, 8H), 8.26 (d, J = 8.4 Hz, 4H). 13C NMR (CDCl3, 100 MHz): δ [ppm] 194.7, 145.2, 142.5, 139.5, 139.1, 137.2, 133.0, 131.5, 131.4, 131.3, 125.2, 124.4, 123.5, 123.1, 116.3, 115.2, 109.4, 36.6, 34.7, 32.0, 31.6. MALDI-TOF (m/z): 1690.7 [M+ − CH3]. Anal. calcd for C123H128N6O (%): C, 86.58; H, 7.56; N, 4.93; O, 1.45. Found: C, 86.43; H, 7.66; N, 4.88.
Acknowledgements
We gratefully acknowledge financial support from the National Basic Research Program of China (973 Programs 2015CB655002 and 2013CB834805), the National Natural Science Foundation of China (Nos. 91433201, 61575146, 51573141 and 51125013), the Innovative Research Group of Hubei Province (No. 2015CFA014) and the Fundamental Research Funds for the Central Universities of China.Notes and references
- (a) K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253 CrossRef CAS; (b) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- (a) Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326 CrossRef CAS; (b) S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330 CrossRef CAS PubMed.
- B. S. Kim and J. Y. Lee, Adv. Funct. Mater., 2014, 24, 3970 CrossRef CAS.
- (a) D. Chen, G. Xie, X. Cai, M. Liu, Y. Cao and S. Su, Adv. Mater., 2016, 28, 239 CrossRef CAS PubMed; (b) D. Graves, V. Jankus, F. B. Dias and A. P. Monkman, Adv. Funct. Mater., 2014, 24, 2343 CrossRef CAS.
- X. Liu, Z. Chen, C. Zheng, C. Liu, C. S. Lee, F. Li, X. Ou and X. Zhang, Adv. Mater., 2015, 27, 2378 CrossRef CAS PubMed.
- (a) K. Albrecht, K. Matsuoka, K. Fujita and K. Yamamoto, Angew. Chem., Int. Ed., 2015, 54, 5677 CrossRef CAS PubMed; (b) C. Tang, T. Yang, X. Cao, Y. Tao, F. Wang, C. Zhong, Y. Qian, X. Zhang and W. Huang, Adv. Opt. Mater., 2015, 3, 786 CrossRef CAS.
- (a) Y. Wada, K. Shizu, S. Kubo, K. Suzuki, H. Tanaka, C. Adachi and H. Kaji, Appl. Phys. Lett., 2015, 107, 183303 CrossRef; (b) C. Liu, Y. Li, Y. Li, C. Yang, H. Wu, J. Qin and Y. Cao, Chem. Mater., 2013, 25, 3320 CrossRef CAS.
- Y. J. Cho, K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 6642 CrossRef CAS PubMed.
- G. Xie, X. Li, D. Chen, Z. Wang, X. Cai, D. Chen, Y. Li, K. Liu, Y. Cao and S. Su, Adv. Mater., 2016, 28, 181 CrossRef CAS PubMed.
- (a) J. Ding, B. Wang, Z. Yue, B. Yao, Z. Xie, Y. Cheng, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2009, 48, 6664 CrossRef CAS PubMed; (b) X. Wang, S. Wang, Z. Ma, J. Ding, L. Wang, X. Jing and F. Wang, Adv. Funct. Mater., 2014, 24, 3413 CrossRef CAS.
- (a) D. Xia, B. Wang, B. Chen, S. Wang, B. Zhang, J. Ding, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2014, 53, 1048 CrossRef CAS PubMed; (b) J. Ding, J. Lu, Y. Cheng, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Funct. Mater., 2008, 18, 2754 CrossRef CAS.
- Q. Zhang, D. Tsang, H. Kuwabara, Y. Hatae, B. Li, T. Takahashi, S. Y. Lee, T. Yasuda and C. Adachi, Adv. Mater., 2015, 27, 2096 CrossRef CAS PubMed.
- F. Schlutter, F. Rossel, M. Kivala, V. Enkelmann, J. P. Gisselbrecht, P. Ruffieux, R. Fasel and K. Mullen, J. Am. Chem. Soc., 2013, 135, 4550 CrossRef PubMed.
- T. Qin, J. Ding, L. Wang, M. Baumgarten, G. Zhou and K. Mullen, J. Am. Chem. Soc., 2009, 131, 14329 CrossRef CAS PubMed.
- (a) P. Moonsin, N. Prachumrak, S. Namuangruk, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, Chem. Commun., 2013, 49, 6388 RSC; (b) N. Prachumrak, S. Pojanasopa, S. Namuangruk, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, ACS Appl. Mater. Interfaces, 2013, 5, 8694 CrossRef CAS PubMed.
- (a) Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706 CrossRef CAS PubMed; (b) H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed.
- H. Wang, L. Xie, Q. Peng, L. Meng, Y. Wang, Y. Yi and P. Wang, Adv. Mater., 2014, 26, 5198 CrossRef CAS PubMed.
- V. Jankus, P. Data, D. Graves, C. McGuinness, J. Santos, M. Bryce, F. B. Dias and A. P. Monkman, Adv. Funct. Mater., 2014, 24, 6178 CrossRef CAS.
- G. Mallesham, C. Swetha, S. Niveditha, M. E. Mohanty, N. J. Babu, A. Kumar, K. Bhanuprakash and V. Rao, J. Mater. Chem. C, 2015, 3, 1208 RSC.
- L. Mei, J. Hu, X. Cao, F. Wang, C. Zheng, Y. Tao, X. Zhang and W. Huang, Chem. Commun., 2015, 51, 13024 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Thermal, photophysical, and electrochemical data of the materials, TGA curves, DSC curves, CV curves, AFM topographic images, DFT calculation, EL spectra of Devices A1–C4, various current densities and efficiency versus luminance curves for Devices A1–C4 and 1H NMR spectrum. See DOI: 10.1039/c6sc00943c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |