
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomic-level organization of vicinal acid–base pairs through the chemisorption of aniline and derivatives onto mesoporous SBA15†
Bilel
Hamzaoui‡
a,
Anissa
Bendjeriou-Sedjerari‡
a,
Eva
Pump
a,
Edy
Abou-Hamad
a,
Rachid
Sougrat
a,
Andrei
Gurinov
a,
Kuo-Wei
Huang
a,
David
Gajan
b,
Anne
Lesage
b,
Lyndon
Emsley
c and
Jean-Marie
Basset
*a
aKing Abdullah University of Science and Technology (KAUST), KAUST Catalysis Center (KCC), Thuwal, 23955-6900, Saudi Arabia. E-mail: jeanmarie.basset@kaust.edu.sa
bUniversité de Lyon, Institut de Sciences Analytiques (CNRS/ENS-Lyon/UCB-Lyon 1), Centre de RMN à Très Hauts Champs, 69100 Villeurbanne, France
cInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
First published on 9th June 2016
Abstract
The design of novel heterogeneous catalysts with multiple adjacent functionalities is of high interest to heterogeneous catalysis. Herein, we report a method to obtain a majority of bifunctional acid–base pairs on SBA15. Aniline reacts with SBA15 by opening siloxane bridges leading to N-phenylsilanamine–silanol pairs. In contrast with ammonia treated surfaces, the material is stable under air/moisture. Advanced solid state MAS NMR (2D 1H–1H double-quantum, 1H–13C HETCOR) experiments and dynamic nuclear polarization enhanced 29Si and 15N spectra demonstrate both the close proximity between the two moieties and the formation of a covalent Si–N surface bond and confirm the design of vicinal acid–base pairs. This approach was successfully applied to the design of a series of aniline derivatives of bifunctional SBA15. A correlation between the substituent effects on the aromatic ring (Hammett parameters) with the kinetics of a model Knoevenagel reaction is observed.
Introduction
One of the major current challenges in heterogeneous catalysis is the ability to develop multifunctional catalyst systems where each active site plays a distinct role in the overall catalytic process (cascade approach). To date, two main approaches to introduce functionalities into mesoporous materials exist: the soft templating strategy for the synthesis of organic–inorganic hybrid materials1–6 and the surface organometallic chemistry (SOMC) methodology for the generation of well-defined surface species.7–11In the soft templating method, the inorganic materials provide the surface area and the porosity. The organic active site, linked to the surface via an alkyl spacer, can be randomly distributed or organized.12–21 However, the resulting materials are composed of complex mixtures of surface species statistically and randomly spread on the surface which are consequently difficult to characterize. Accordingly, the structure of their active site is generally not known at the molecular level.
In the SOMC methodology,7 the generation of well-defined surface species is achieved by understanding the reaction of organometallic complexes with the inorganic materials which act as a rigid ligand. This approach presents the advantage of establishing a structure–activity relationship, and provides molecular-level insight for the design and prediction of new catalysts for new reactions.22,23 Indeed, SOMC has been successful in designing “multifunctional” single site catalysts that are able to perform alkane metathesis via a multistep mechanism. However, the surface–complex bond is usually a σ-bonded oxygen ligand, e.g., siloxy [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O–)MLn], with M = metal and L = ligands, in the primary coordination sphere and the requirement for oxygen limits the development of SOMC methods. It would then be highly desirable to tune the coordination sphere of the metal center by designing surface ligands in close proximity to the surface to preserve the rigidity of the ensemble “surface ligand/complex”. By tuning the electronic and/or steric properties of the surface ligand, new catalysts and new reactions will be discovered.
Si–O–)MLn], with M = metal and L = ligands, in the primary coordination sphere and the requirement for oxygen limits the development of SOMC methods. It would then be highly desirable to tune the coordination sphere of the metal center by designing surface ligands in close proximity to the surface to preserve the rigidity of the ensemble “surface ligand/complex”. By tuning the electronic and/or steric properties of the surface ligand, new catalysts and new reactions will be discovered.
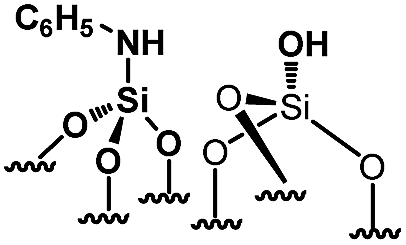
In 2013, our group developed a new strategy to create an N-donor surface pincer ligand where the functional groups are well-organized into silylamine and silanol pairs on mesoporous SBA15 materials, named [N,O]SBA15. This was achieved by opening strained siloxane bridges at 200 °C via treatment with ammonia, by analogy with the ring opening of epoxides by ammonia.24–26 By reaction with an organometallic complex (zirconium tetraneopentyl), the expected bipodal(siloxy-)(amido-)zirconium bis neopentyl was obtained.27–29 However, the successful formation of [Si–NH2][Si–OH] surface groups is associated with experimental, economical and safety disadvantages, such as the need for a high flow rate (200 mL min−1) of pure, expensive and corrosive ammonia, and the resulting materials being moisture sensitive. Aside from that, further chemical modifications of [N,O]SBA15 to offer opportunities to provide tunable steric and electronic properties were impossible without affecting the structural parameters of the materials. To face these issues, we investigate an alternative approach based on the chemisorption of dry aniline onto highly dehydroxylated SBA15 (1100 °C), which has never been reported (Scheme 1). The resulting new materials are inexpensive, easy to prepare and moisture stable and their performance as acid–base catalysts is evaluated in the Knoevenagel condensation where a controllable distance between two antagonist functionalities is known to be an influential factor in the enhancement of the reactivity.12,14,19,30–35
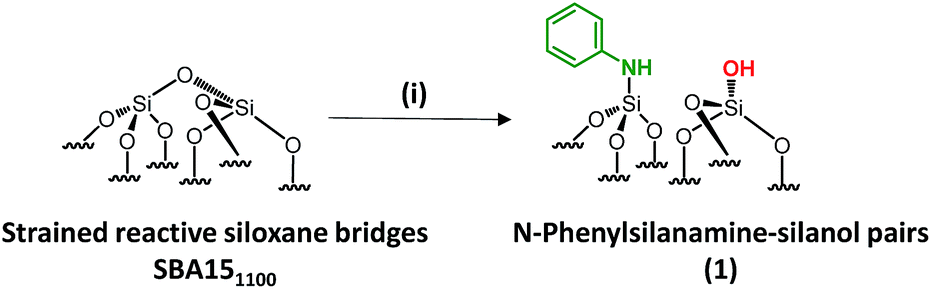 | ||
| Scheme 1 Synthesis of paired N-phenylsilanamine/silanols 1via the chemisorption of dry aniline on SBA151100 in toluene at 80 °C for 20 h. | ||
Results and discussion
Well-ordered hexagonal mesoporous silica, SBA15, was chosen as a support because of its high thermal stability (up to 1200 °C), its high surface area (700 m2 g−1) and its large and uniform pore diameter (6 nm) and relatively thick walls (3 to 6 nm).36 Thermal treatment at 1100 °C under vacuum (10−5 mbar) yields the condensation of adjacent silanols and generates a support that contains a surface of mainly strained siloxanes along with a small amount of isolated silanols (<0.4 OH per nm2).37–39 As described in Scheme 1, the reaction of SBA151100 with dry aniline was performed in a solution in toluene at 80 °C for 20 h. The resulting material 1 was evacuated at room temperature overnight under high vacuum (10−5 mbar) and characterized by FT-IR spectroscopy. Comparison of the FT-IR spectra of SBA151100 and 1 (Fig. 1) reveals a slight increase in intensity of the characteristic [νs(OH)] band with a red shift from 3748 to 3745 cm−1. Additionally, the typical single sharp infrared bands characteristic of secondary amines,40 here N-phenylsilylamine appears at 3435 and 1500 cm−1. They correspond to the [ν(NH)] and [δ(NH)], respectively. Vibrational bands of the aromatic group are clearly visible at 3089–3023 cm−1 [ν(CH)], at 1606 and 1500 cm−1 [δ(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)] (overlap a NH band). Finally, the shoulder in the range of 3690–3585 cm−1 is assigned to electronic interactions of the π system of the aromatic group with the newly formed adjacent silanol (π–OH interactions).41,42 It is important to mention that no reversible adsorption aniline (physisorbed) remains on the support after the reaction. Indeed, the adsorption of aniline, which we do not see, would lead to a decrease of the isolated silanol band due to the hydrogen bonding of aniline to surface silanol, and the appearance of two bands at 3395 and 3300 cm−1 assigned to the N–H stretching vibration of physisorbed aniline.41,42
C)] (overlap a NH band). Finally, the shoulder in the range of 3690–3585 cm−1 is assigned to electronic interactions of the π system of the aromatic group with the newly formed adjacent silanol (π–OH interactions).41,42 It is important to mention that no reversible adsorption aniline (physisorbed) remains on the support after the reaction. Indeed, the adsorption of aniline, which we do not see, would lead to a decrease of the isolated silanol band due to the hydrogen bonding of aniline to surface silanol, and the appearance of two bands at 3395 and 3300 cm−1 assigned to the N–H stretching vibration of physisorbed aniline.41,42
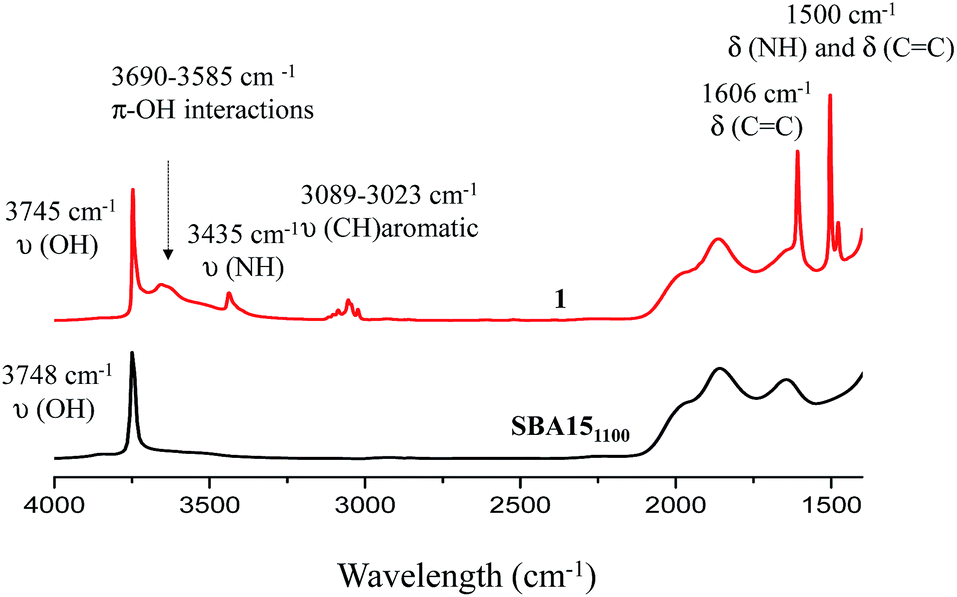 | ||
| Fig. 1 FT-IR spectra of SBA151100 (black) and 1 (red). | ||
Solid state NMR was used to characterize the pairwise nature of the atomic-level organization of the supported organic functionalities, N-phenylsilanamine and silanol. The 1H magic-angle-spinning (MAS) spectrum (Fig. 2A) shows four clear resonances at 1.9, 3.4, 6.6 and 7 ppm. The chemical shift at 1.9 ppm is assigned to the SiOH proton. Its value appears slightly downfield compared to the chemical shift of SiOH (1.7 ppm) generated by treatment with ammonia.27 This shift might be due to the proximity of the protons of the aromatic ring which induces π–OH interactions. The chemical shift at 3.4 ppm is attributed to the proton of the Si–NH–Ph.43
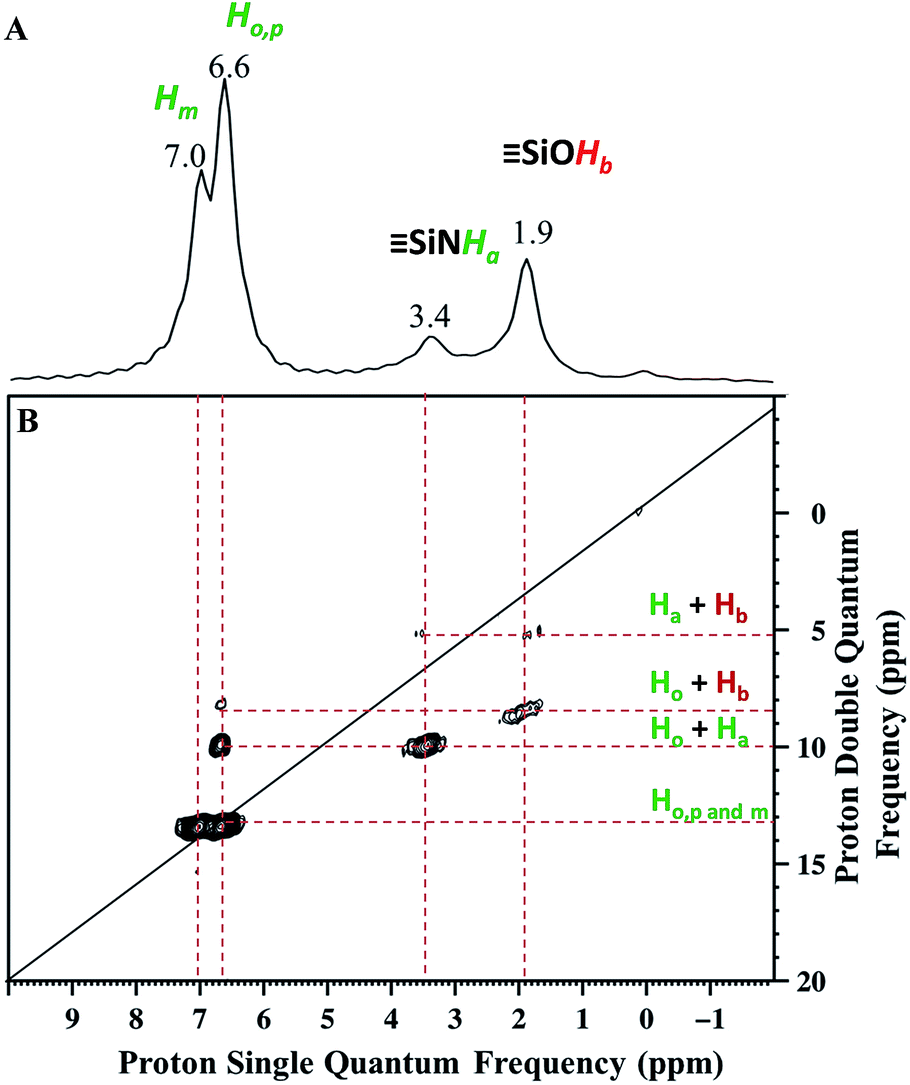 | ||
| Fig. 2 (A) 1H MAS NMR spectrum of 1. (B) DQ rotor-synchronized 2D 1H MAS NMR spectrum of 1 (see ESI† for details). | ||
The two intense signals at 6.6 ppm correspond to the protons of the aromatic ring in ortho (Ho) and para (Hp) positions. The proton in the meta position (Hm) appears at 7 ppm (Fig. 2A). To confirm these proton assignments, the synthesis of a model molecular silsesquioxane bearing a N-phenylsilanamine group was carried out (see ESI, Fig. S1†). The 1H liquid-state NMR spectrum of the resulting silsesquioxane is in agreement with the 1H MAS spectrum of 1 and confirms the formation of a σ bond between the nitrogen of aniline and the SBA15 surface.
Further information about the presence of vicinal functionalities are obtained from the 2D 1H–1H double quantum (DQ) NMR spectrum (Fig. 2). The 2D DQ spectrum of 1 shows a weak correlation between the proton resonance of SiOH at 1.9 ppm and the proton resonance of SiNHPh at 3.4 ppm [5.3 ppm in F1: δH(OH) + δH(NH) = 1.9 + 3.4] (Scheme 2.1), indicating that the two protons are in close proximity, typically <5 Å.
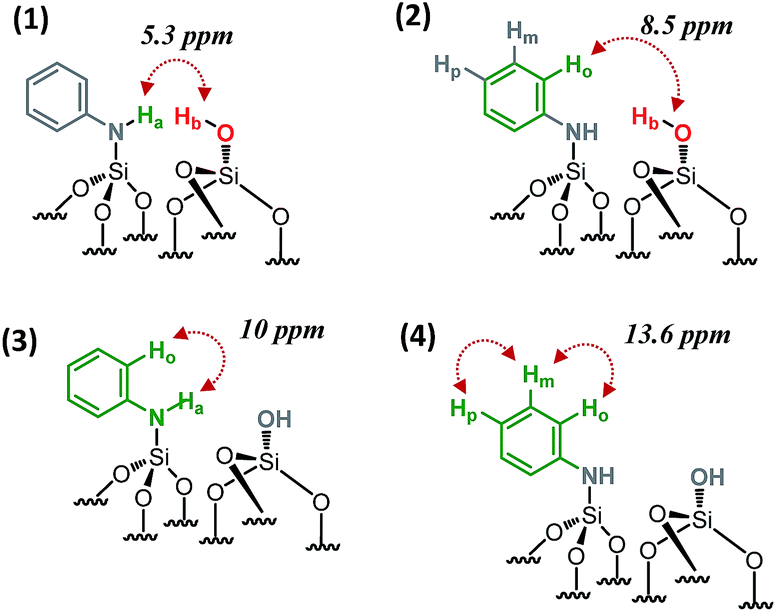 | ||
| Scheme 2 Schematic showing the observed proximities of the paired N-phenylsilanamine/silanol groups in 1 from the 2D 1H–1H DQ NMR spectrum. | ||
Moreover, the paired organization is reinforced by the presence of a correlation between the proton resonance of SiOH and the aromatic proton resonances of the neighboring SiNHPh at 6.6 ppm [8.5 ppm in F1: δH(OH) + δH(Ho) = 1.9 + 6.6] (Scheme 2.2). The correlation most likely arises from ortho position Ho. Note that Hm does not correlate with the silanol indicating that the aromatic ring is oriented in such a way that Ho is closest to the silanol. An additional correlation is observed between the proton resonance of SiNHPh at 3.4 ppm and the proton of the aromatic ring in ortho position Ho at 6.6 ppm [10 ppm in F1: δH(OH) + δH(Ho) = 3.4 + 6.6] (Scheme 2.3). Finally, strong correlation peaks are observed between all the protons of the aromatic ring at 13.6 ppm (Scheme 2.4).
Interestingly, the fact that no correlations between two silanols [SiOH] [SiOH] (1.9 × 2 = 3.8 ppm in F1) as well as between two [SiNHPh] [SiNHPh] (2 × 3.4 = 6.8 ppm in F1) are observed clearly demonstrates that the majority of sites are isolated “acid–base” pairs [SiNHPh]: [SiOH].
We conclude that dry aniline reacts with strained siloxane bridges to generate vicinal N-phenylsilanamine and silanol groups. These results are consistent with those obtained with FT-IR spectroscopy.
In the 13C CP-MAS NMR spectrum (Fig. S4A†), the signals of the aromatic ring appear clearly at 118, 120, 129 and 143 ppm corresponding to the CoH, CpH, CmH and to the quaternary carbon linked to the nitrogen surface Si–NH–CIV, respectively. These assignments are in accordance with those of the silsesquioxane model (see ESI, Fig. S2†). The 1H–13C HETCOR spectrum demonstrates a strong correlation between these four carbon resonances and the proton resonances at 6.6 and 7 ppm attributed to the proton of aromatic group (see ESI, Fig. S4B†). These results confirm that the integrity of the organic fragment is maintained under the reaction conditions.
To identify the formation of a covalent bond between the silica surface and the organic fragment, SiNHPh, 29Si and 15N solid state NMR spectra are required, but are not practical using conventional methods due to their low sensitivity at natural isotopic abundance.
Dynamic nuclear polarization surface enhanced NMR (DNP SENS)44,45 has recently been introduced and demonstrated to overcome these difficulties for the characterization of hybrid materials, and was used here.46–49 In this work, DNP yielded εH ∼ 256 (defined as the ratio of signal intensities of spectra acquired with and without microwave irradiation, Fig. S5, ESI†). The natural abundance 29Si DNP SENS spectrum of 1 (Fig. 3a) displays a signal centered at −100 ppm (intense) and a signal at −18 ppm (weak). According to the literature the former is attributed to both [(SiO)3SiOE], with E = Si or H (commonly dubbed Q4 and Q3)50,51 and the latter is assigned to the [SiNHPh].52
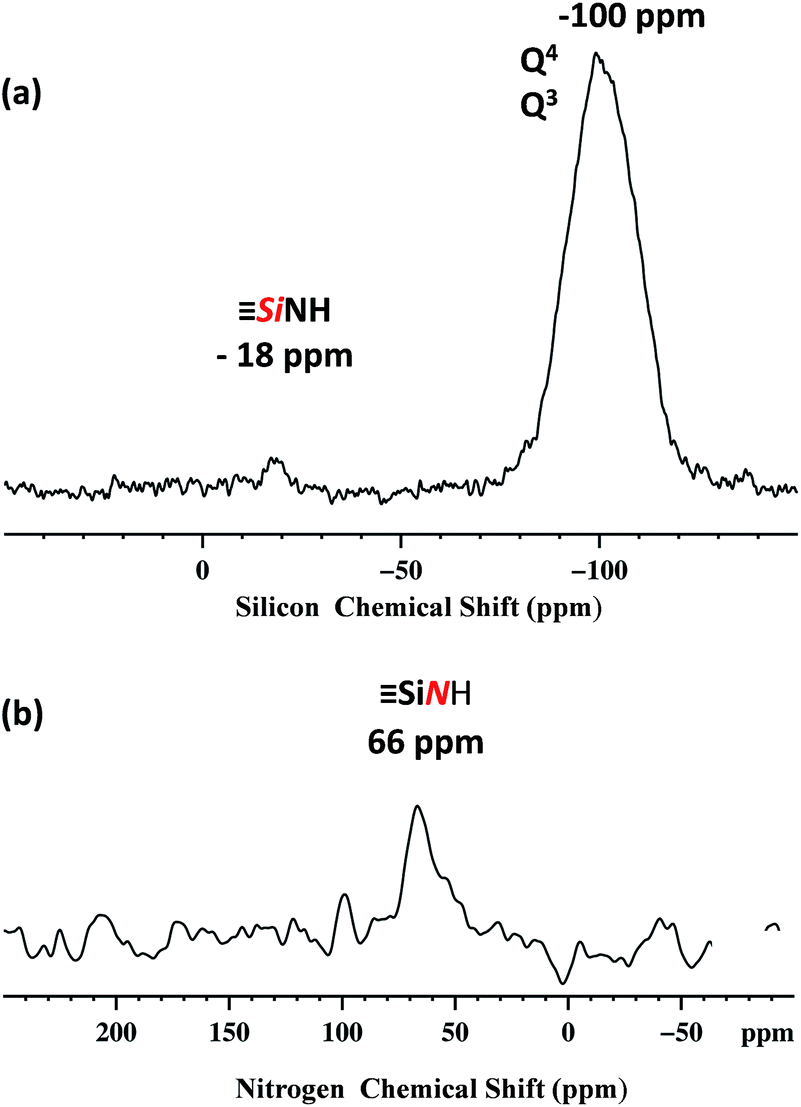 | ||
Fig. 3 400 MHz DNP SENS spectra of 1 (20 mg) impregnated with a 16 mM solution of TEKPOL53 in 1,1,2,2-tetrachloroethane at 8 kHz MAS frequency with a sample temperature of 100 K. (a) 29Si DNP enhanced CP/MAS with a CP contact time of 5 ms, a 3 s polarization delay and 1024 scans. Exponential line broadening of 60 Hz was applied prior to Fourier transformation. (b) 15N DNP enhanced CP/MAS with 5 ms CP contact time, a 3 polarization delay and 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans. Exponential line broadening of 150 Hz was applied prior to Fourier transformation. In both (a) and (b) for comparison, spectra are shown with both for both μwave on and off. 000 scans. Exponential line broadening of 150 Hz was applied prior to Fourier transformation. In both (a) and (b) for comparison, spectra are shown with both for both μwave on and off. | ||
The natural abundance 15N DNP SENS spectrum shows a single peak at 66 ppm (Fig. 3b) and it is in agreement with the 15N liquid-state NMR spectrum of the model molecular silsesquioxane, for which the 15N chemical shift appears at 62 ppm (Fig. S3, ESI†).
Textural characterization is used to evaluate the preservation of the mesoporous materials, here by nitrogen sorption porosimetry, small angle X-ray diffraction (XRD), and Transmission Electronic Microscopy (TEM). The small angle X-ray diffraction patterns of 1 (Fig. S6, ESI†) exhibit three clear peaks (d100, d110 and d200) in the 2θ range of 0.7–4°. They confirm the presence of a well-ordered hexagonal mesophase with a d100 spacing of 86.28 Å (see ESI, Table S1†). The structure of the mesoporous materials is thus maintained throughout the chemisorption of dry aniline.
Analyses of the nitrogen adsorption/desorption isotherms yielded BET surface areas of 1 of approximately 512 m2 g−1 (versus 679 m2 g−1 for SBA1100) and pore volumes of 0.65 cm3 g−1 (versus 0.9 cm3 g−1 for SBA1100). Also, 1 showed type IV isotherms (Fig. S7, ESI†), with clear H1-type hysteresis loops associated with capillary condensation in the mesopores and with regular pore sizes of 50 Å. The textural parameters of sample 1 are summarized in Table S1† and are characteristic of mesoporous materials. The surface coverage α of the organic moieties based on the carbon content was calculated as described by Jaroniec et al.54 Using eqn (S1) (see ESI†), a carbon content of 1.42 wt% was determined for 1, which translates into a surface coverage of 0.3 μmol m−2. The low surface coverage supports the results of N2 sorption experiments and indicates the functionalization of the SBA151100.
Further evidence for a well-ordered hexagonal mesostructure is provided by the TEM images (Fig. 4), which are representative of mesoporous SBA15. After the high thermal treatment (1100 °C, 10−5 mbar) and the dissociative chemisorption of aniline (80 °C, toluene, 20 h), the mesoporous structure is still regular over the whole particle of 1.
 | ||
| Fig. 4 Transmission electron micrographs of 1 at different tilt angles: in the direction perpendicular to the pore axis (B), and in the direction of the mesopores axis (B). | ||
A materials with atomic organization of acid–base pairs should exhibit cooperative catalytic behavior for the Knoevenagel condensation of benzaldehyde with diethyl malonate (pKa = 13) (Scheme 3).55–57 The Knoevenagel condensation between a carbonyl group and activated methylene compounds is one of the most useful CC bond forming reactions. It produces several important key intermediates such as α, β unsaturated products widely used for the synthesis of therapeutic drugs, functional polymers and fine chemicals.58,59 The Knoevenagel reaction is the right catalytic reaction as it is considered as the model reaction to evaluate the basic strength of bifunctional acid/base materials.55–57
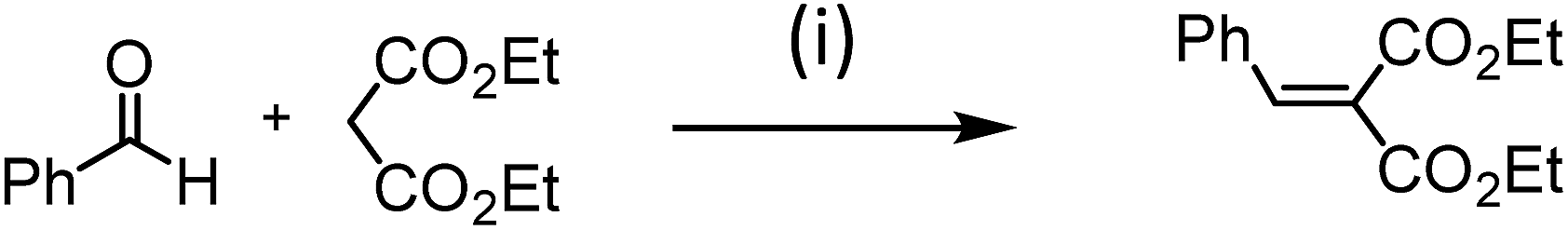 | ||
| Scheme 3 Knoevenagel condensation between benzaldehyde and diethylmalonate. (i) The reaction was performed in sealed flask in which each reactant (2 mmol) and the catalyst (20 mg) were added to dry ethanol (5 mL) and the reaction mixture was refluxed at 80 °C for 24 h. | ||
In the literature, several studies have revealed an efficient catalysis by cooperative acid–base pairs well organized on mesoporous silica.16,60 Those previous studies have been supported by recent work from Jones et al.61 where the catalytic activity of amino-propyl functionalized MCM-41 decreases drastically when the Si–OH are capped with trimethylsilyl group (Si–OSiMe3). So, weakly acidic silanols play a vital role in the cooperative catalytic cycle as well as the spatial organisation of the acid–base functionalities.60 In the mechanism, activation of the carbonyl group occurs on weak Brönsted acid sites and the basic sites extract the proton from methylene (Scheme S1, ESI†). In this case, the control of the distance is a key parameter to enhance the reactivity of the Knoevenagel condensation.12,14
For comparison purposes, a series of bifunctional mesoporous materials with different electronic properties were successfully synthesized through the same approach (Scheme 4). All the materials were characterized by FT-IR and 1H-MAS solid state NMR spectroscopy (Fig. S8 and S9†).
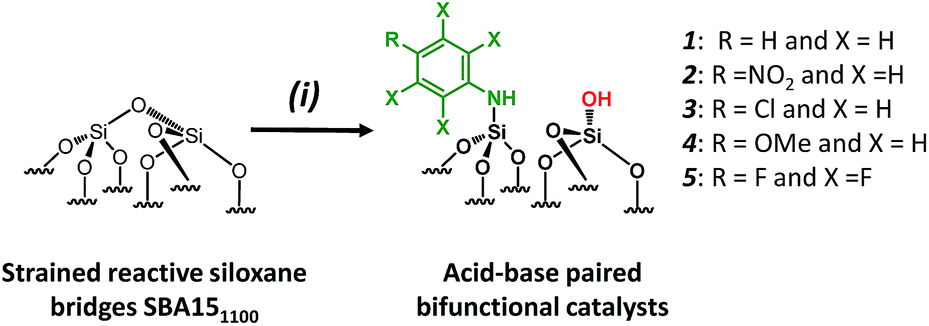 | ||
| Scheme 4 Synthesis of acid–base paired catalysts via the chemisorption of dry aniline derivatives on SBA151100 in toluene at 80 °C for 20 h. | ||
All the FT-IR spectra of catalysts 2–5 display the characteristic vibration bands of ν(OH) (3745 cm−1), ν(NH) and δ(NH), 3435 and 1500 cm−1 respectively. Vibrational bands of the aromatic group are still present at 3089–3023 cm−1 [ν(CH)], at 1606 and 1500 cm−1 [δ(CC)] (overlapping a NH band). The shoulder characteristic of the electronic interactions of the π system of the aromatic group with the newly formed adjacent silanol (π–OH interactions) in the range of 3690–3585 cm−1 is again observed for all catalysts (weak in the case of catalyst 5).
All the 1H NMR spectra feature the characteristic signal of SiOH and SiNH at around 2 ppm and 3.5–3.9 ppm, respectively. As expected, the protons in ortho and meta position to electron donating (OMe) and electron withdrawing (Cl, NO2) substituents show distinct upfield and downfield shifts (Fig. S9†).
Their catalytic performance were tested (Table 1, entry 1–5) and all the samples showed good catalytic ability. Entry 1 showed higher activity than entry 2. The nitro group is a strongly electron-withdrawing group (EWG) and thus, catalyst (2) is a weaker base than catalyst (1). A chloro group in the para position is a slightly EWG, so catalyst (3) exhibits better activity than (2) and is slightly less active than (1). Introducing an electron-donating group (EDG) as a p-methoxy group in the catalyst (4) enhances the catalytic performance in the Knoevenagel reaction. Among all these catalysts, (4) exhibits the best performance whereas (5) exhibits the lowest due to the base weakening effect.
| Entry | Catalyst | N loadinga (mmol g−1) | Yieldb (%) | TONc |
|---|---|---|---|---|
| a Determined by elemental analysis. b Determined by GC analysis after 24 h. c Turnover number (TON) = number of moles of product per number of moles of active amine site. | ||||
| 1 |
|
0.23 | 59 | 257 |
| 2 |
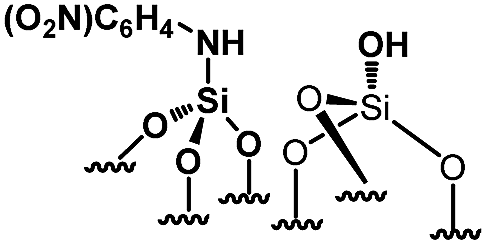
|
0.22 | 35 | 159 |
| 3 |
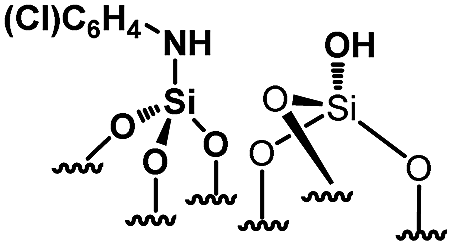
|
0.18 | 43 | 238 |
| 4 |
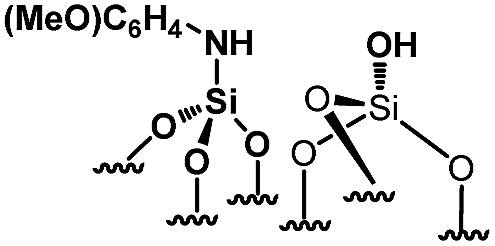
|
0.21 | 64 | 304 |
| 5 |
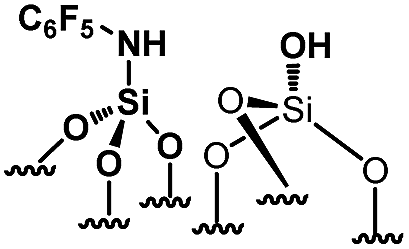
|
0.18 | 24 | 133 |
| 6 |
|
0 | 0 | 0 |
| 7 |
|
0.35 | 18 | 51 |
Besides this, the catalytic results of this series of acid–base paired catalysts (Table 1, entry 1–5) were compared to two other materials (Table 1, entry 6 and 7): an unmodified SBA15 displaying different silanols (vicinal, geminal) (6), [N,O]SBA15 where primary amine and silanol groups are proximal (7).27 As expected (6) shows no activity as no basic sites are present. (7) contains primary amines which are supposed to be the strongest base; yet it gives only 18% conversion after 24 h. (1) and (4) yield better conversion although their basicity is lower than that of (7). These results are explained by the higher stability of these catalysts under the experimental conditions (ethanol is the solvent and water is produced during the Knoevenagel reaction). Indeed, the SiNH2 group is well-known to be easily hydrolyzed.32,34
In parallel, the stability of (7) and (1) towards ethanol was monitored by FT-IR spectroscopy. After 5 min in contact with ethanol, the FT-IR spectrum (Fig. S10, ESI†) of (7) shows complete disappearance of the characteristic bands of the SiNH2 group [νs(NH2) = 3535, νs(NH2) = 3445 and δ(NH2) = 1550 cm−1]. However the FT-IR spectrum of (1) shows the characteristic bands of Si–NHPh, [ν(NH) = 3435 cm−1] even after 1 h in contact with dry ethanol. In addition, during the catalytic test with catalyst 1, no leaching of aniline was detected by both GC-FID and GC-MD (Fig. S11, ESI†).
Conclusions
The opening siloxane bridges approach was successfully established to create an atomic organization of well-defined bi-functional acid–base pairs on mesoporous SBA15. This approach is based on an analogy between organic epoxides and strained siloxanes (Si–O–Si) of mesoporous SBA151100. The generation of well-defined adjacent N-phenylsilanamine–silanol pairs was unambiguously determined through FTIR, 2D solid state NMR, XRD, nitrogen sorption and TEM. This way to design bi-functionalized mesoporous surface offers new opportunities to modify the electronic and steric properties of mesoporous silica useful for heterogeneous catalysis.
Acknowledgements
This work received support from the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. CRG_R2_13_BASS_KAUST_1. We thank Dr Olivier Ouari and Paul Tordo for providing the TeKPol radical.References
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- A. Kuschel, M. Drescher, T. Kuschel and S. Polarz, Chem. Mater., 2010, 22, 1472–1482 CrossRef CAS.
- D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS PubMed.
- A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589–3614 CrossRef CAS PubMed.
- I. Sierra and D. Perez-Quintanilla, Chem. Soc. Rev., 2013, 42, 3792–3807 RSC.
- M. Ogawa, K. Saito and M. Sohmiya, Eur. J. Inorg. Chem., 2015, 2015, 1126–1136 CrossRef CAS.
- J. D. A. Pelletier and J.-M. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS PubMed.
- J. M. Basset, R. Psaro, D. Roberto and R. Ugo, Modern surface organometallic chemistry (hardback), 2009 Search PubMed.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- F. Lefebvre, in Atomically-Precise Methods for Synthesis of Solid Catalysts, The Royal Society of Chemistry, 2015, pp. 1–26, 10.1039/9781782628439-00001.
- P. Sautet and F. Delbecq, Chem. Rev., 2010, 110, 1788–1806 CrossRef CAS PubMed.
- E. L. Margelefsky, A. Bendjériou, R. K. Zeidan, V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2008, 130, 13442–13449 CrossRef CAS PubMed.
- V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2003, 125, 9403–9413 CrossRef CAS PubMed.
- E. L. Margelefsky, R. K. Zeidan, V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2007, 129, 13691–13697 CrossRef CAS PubMed.
- R. K. Zeidan, S.-J. Hwang and M. E. Davis, Angew. Chem., 2006, 118, 6480–6483 CrossRef.
- N. A. Brunelli, K. Venkatasubbaiah and C. W. Jones, Chem. Mater., 2012, 24, 2433–2442 CrossRef CAS.
- A. T. Dickschat, F. Behrends, M. Bühner, J. Ren, M. Weiß, H. Eckert and A. Studer, Chem.–Eur. J., 2012, 18, 16689–16697 CrossRef CAS PubMed.
- A. T. Dickschat, F. Behrends, S. Surmiak, M. Weiß, H. Eckert and A. Studer, Chem. Commun., 2013, 49, 2195–2197 RSC.
- Y. Huang, S. Xu and V. S. Y. Lin, Angew. Chem., Int. Ed., 2011, 50, 661–664 CrossRef CAS PubMed.
- K. K. Sharma and T. Asefa, Angew. Chem., 2007, 119, 2937–2940 CrossRef.
- N. R. Shiju, A. H. Alberts, S. Khalid, D. R. Brown and G. Rothenberg, Angew. Chem., Int. Ed., 2011, 50, 9615–9619 CrossRef CAS PubMed.
- V. Vidal, A. Théolier, J. Thivolle-Cazat and J.-M. Basset, Science, 1997, 276, 99–102 CrossRef CAS PubMed.
- V. Dufaud and J.-M. Basset, Angew. Chem., Int. Ed., 1998, 37, 806–810 CrossRef CAS.
- J. A. A. Sales, G. C. Petrucelli, F. J. V. E. Oliveira and C. Airoldi, J. Colloid Interface Sci., 2007, 315, 426–433 CrossRef CAS PubMed.
- M. Pastó, B. Rodríguez, A. Riera and M. A. Pericàs, Tetrahedron Lett., 2003, 44, 8369–8372 CrossRef.
- M. Chini, P. Crotti, L. A. Flippin and F. Macchia, J. Org. Chem., 1991, 56, 7043–7048 CrossRef CAS.
- A. Bendjeriou-Sedjerari, J. M. Azzi, E. Abou-Hamad, D. H. Anjum, F. A. Pasha, K.-W. Huang, L. Emsley and J.-M. Basset, J. Am. Chem. Soc., 2013, 135, 17943–17951 CrossRef CAS PubMed.
- F. A. Pasha, A. Bendjeriou-Sedjerari, K.-W. Huang and J.-M. Basset, Organometallics, 2014, 33, 3320–3327 CrossRef CAS.
- F. A. Pasha, A. Bendjeriou-Sedjerari, E. Abou-Hamad, K.-W. Huang and J.-M. Basset, Chem. Commun., 2016, 52, 2577–2580 RSC.
- S. Rana, S. Maddila, R. Pagadala and S. Jonnalagadda, J. Porous Mater., 2015, 22, 353–360 CrossRef CAS.
- E. Gianotti, U. Diaz, A. Velty and A. Corma, Catal. Sci. Technol., 2013, 3, 2677–2688 CAS.
- K. Sugino, N. Oya, N. Yoshie and M. Ogura, J. Am. Chem. Soc., 2011, 133, 20030–20032 CrossRef CAS PubMed.
- L.-B. Sun, X.-Q. Liu and H.-C. Zhou, Chem. Soc. Rev., 2015, 44, 5092–5147 RSC.
- Y. Furukawa and M. Ogura, J. Am. Chem. Soc., 2014, 136, 119–121 CrossRef CAS PubMed.
- Y. Xia and R. Mokaya, Angew. Chem., Int. Ed., 2003, 42, 2639–2644 CrossRef CAS PubMed.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- B. C. Bunker, D. M. Haaland, T. A. Michalske and W. L. Smith, Surf. Sci., 1989, 222, 95–118 CrossRef CAS.
- B. A. Morrow, I. A. Cody and L. S. M. Lee, J. Phys. Chem., 1976, 80, 2761–2767 CrossRef CAS.
- L. T. Zhuravlev, Colloids Surf., A, 2000, 173, 1–38 CrossRef CAS.
- D. L. Pavia, G. M. Lampman, G. S. Kriz and J. R. Vyvyan, Introduction to Spectroscopy, Cengage Learning, 4th edn , 2009 Search PubMed.
- M. J. D. Low and V. V. S. Rao, Can. J. Chem., 1969, 47, 1281–1287 CrossRef CAS.
- M. J. D. Low and M. Hasegawa, J. Colloid Interface Sci., 1968, 26, 95–101 CrossRef CAS.
- M. J. Fuchter, C. J. Smith, M. W. S. Tsang, A. Boyer, S. Saubern, J. H. Ryan and A. B. Holmes, Chem. Commun., 2008, 2152–2154, 10.1039/b801537f.
- A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Miéville, J. Alauzun, A. Roussey, C. Thieuleux, A. Mehdi, G. Bodenhausen, C. Coperet and L. Emsley, J. Am. Chem. Soc., 2010, 132, 15459–15461 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS PubMed.
- T. Gutmann, J. Liu, N. Rothermel, Y. Xu, E. Jaumann, M. Werner, H. Breitzke, S. T. Sigurdsson and G. Buntkowsky, Chem.–Eur. J., 2015, 21, 3798–3805 CrossRef CAS PubMed.
- F. A. Perras, T. Kobayashi and M. Pruski, J. Am. Chem. Soc., 2015, 137, 8336–8339 CrossRef CAS PubMed.
- T. Kobayashi, F. A. Perras, I. I. Slowing, A. D. Sadow and M. Pruski, ACS Catal., 2015, 5, 7055–7062 CrossRef CAS.
- A. J. Rossini, A. Zagdoun, M. Lelli, J. Canivet, S. Aguado, O. Ouari, P. Tordo, M. Rosay, W. E. Maas, C. Copéret, D. Farrusseng, L. Emsley and A. Lesage, Angew. Chem., Int. Ed., 2012, 51, 123–127 CrossRef CAS PubMed.
- S. Leonardelli, L. Facchini, C. Fretigny, P. Tougne and A. P. Legrand, J. Am. Chem. Soc., 1992, 114, 6412–6418 CrossRef CAS.
- M. Lelli, D. Gajan, A. Lesage, M. A. Caporini, V. Vitzthum, P. Miéville, F. Héroguel, F. Rascón, A. Roussey, C. Thieuleux, M. Boualleg, L. Veyre, G. Bodenhausen, C. Coperet and L. Emsley, J. Am. Chem. Soc., 2011, 133, 2104–2107 CrossRef CAS PubMed.
- A. Y. Nikonov, I. V. Sterkhova and N. F. Lazareva, Russ. J. Gen. Chem., 2015, 85, 1866–1869 CrossRef CAS.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Copéret, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
- B. Buszewski, M. Jaroniec and R. K. Gilpin, J. Chromatogr. A, 1994, 673, 11–19 CrossRef CAS.
- S. L. Hruby and B. H. Shanks, J. Catal., 2009, 263, 181–188 CrossRef CAS.
- A. Katz and M. E. Davis, Nature, 2000, 403, 286–289 CrossRef CAS PubMed.
- J. D. Bass, S. L. Anderson and A. Katz, Angew. Chem., Int. Ed., 2003, 42, 5219–5222 CrossRef CAS PubMed.
- L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed.
- F. Liang, Y.-J. Pu, T. Kurata, J. Kido and H. Nishide, Polymer, 2005, 46, 3767–3775 CrossRef CAS.
- E. L. Margelefsky, R. K. Zeidan and M. E. Davis, Chem. Soc. Rev., 2008, 37, 1118–1126 RSC.
- V. E. Collier, N. C. Ellebracht, G. I. Lindy, E. G. Moschetta and C. W. Jones, ACS Catal., 2016, 6, 460–468 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc01229a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |