
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A metal–organic framework immobilised iridium pincer complex†
Martino
Rimoldi
a,
Akitake
Nakamura
a,
Nicolaas A.
Vermeulen
a,
James J.
Henkelis
a,
Anthea K.
Blackburn
a,
Joseph T.
Hupp
a,
J. Fraser
Stoddart
*a and
Omar K.
Farha
*ab
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: jfstoddart@gmail.com; omarkfarha@gmail.com
bDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 10th May 2016
Abstract
An iridium pincer complex has been immobilised in the metal–organic framework NU-1000 using a technique called solvent assisted ligand-incorporation (SALI). The framework proved to be stable under the conditions required to activate the iridium complex and spectroscopic investigations showed formation of the catalytically active iridium dihydride. The Ir-pincer modified NU-1000 is an active catalyst for the condensed phase hydrogenation of a liquid alkene (1-decene and styrene) and shows enhanced activity with respect to a homogeneous analogue. Additionally, the Ir-pincer immobilised inside NU-1000 operated as an efficient heterogenous catalyst under flow conditions.
Introduction
Pincer complexes have been widely used and investigated for application in homogeneous catalytic transformations.1 Iridium pincer complexes are of special interest and were first prepared two decades ago.2 Their chemistry is still the object of numerous investigations3 and this class of complexes are extremely effective in applications involving hydrocarbon conversions.4 Heterogenisation of organometallic complexes has been under extensive study, especially employing amorphous metal oxides as supports.5Generally, supporting an organometallic species requires the use of organic ligands designed to enable anchoring on a heterogeneous substrate.6 The ligands are often complex and require a significant investment in synthesis. This approach aims to yield single sites or usually well-defined systems, whose characterisation is not always straightforward.
Recently this approach has also been applied to the post-synthetic metalation of MOFs to afford, amongst others, Co(II)- and Fe(II)-based single-site catalysts that were found active in various transformations. In particular, high turnover numbers were obtained in alkene hydrogenations. Catalytic tests where typically conducted at 40 bar, room temperature and using THF as solvent.7
To date, only a few heterogeneous materials containing pincer complexes have been prepared and used in catalytic processes.8
Herein, we describe the incorporation of a catalytically active iridium pincer complex into a metal–organic framework (MOF). We selected NU-10009 – a mesoporous MOF (Fig. 1) featuring 30 Å channels with Zr6 oxide metal nodes and 1,3,6,8,-tetrakis(p-benzoic acid)pyrene as the tetratopic linkers—as the heterogenization platform. Detailed physical and structural characterisations, as well as the elucidation of the proton topology around the node, have been described9,10 recently and a post-synthetic modification, called solvent assisted ligand-incorporation11 (SALI) has been used to decorate the metal nodes of NU-1000 with carboxylic acid containing molecules. SALI makes use of carboxyl groups to displace labile –OH and H2O ligands on the Zr6 node and form strong chelating metal–ligand bond. In this report, an Ir(III) complex featuring a bis-phosphinite pincer ligand, similar to those introduced and developed by Brookhart and co-workers,12 has been incorporated into NU-1000 using SALI and its catalytic activity tested in a hydrogenation reaction.
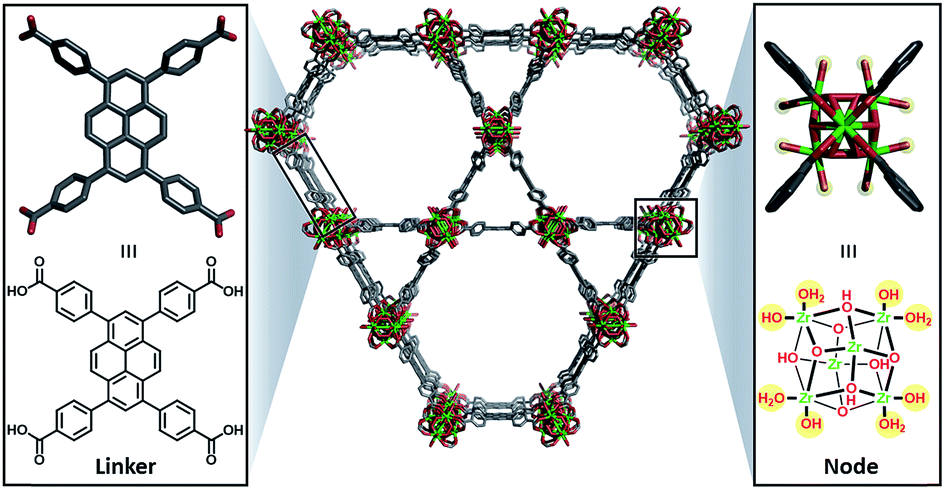 | ||
| Fig. 1 Unfunctionalised NU-1000 showing the linker (left) and node (right) with the displaceable –OH and H2O ligands highlighted in yellow. | ||
Results and discussion
The synthetic route outlined in Scheme 1 was used to prepare the carboxylic acid functionalised pincer complex 3. Firstly, the TBDMS-protected bis-phosphinite ligand (see ESI†) was reacted with [IrCl(COD)]2 to afford the complex 1. Deprotection of the phenol silyl ether using Cu(II) chloride led to the formation of the phenolic derivative 2 as the major product. At this stage we observed that one of the tert-butyl phosphinite substituents had reacted with the metal center to form a cyclometallated four-membered ring compound 2. Similar complexes have been reported13 and their distinctive characterisation data described. The 31P NMR spectrum of 2 (and of the complexes obtained in the subsequent steps) shows a set of resonances (two doublets) at δ 160.4 and 119.3, confirming the asymmetry of the complex and the existence of two heterotopic phosphorous atoms. Notably, the hydride signal (δ −41.9) that characterises the complex 1 is no longer observed. Single crystal X-ray diffraction analysis confirms (Fig. 2) the structural assignment of 2.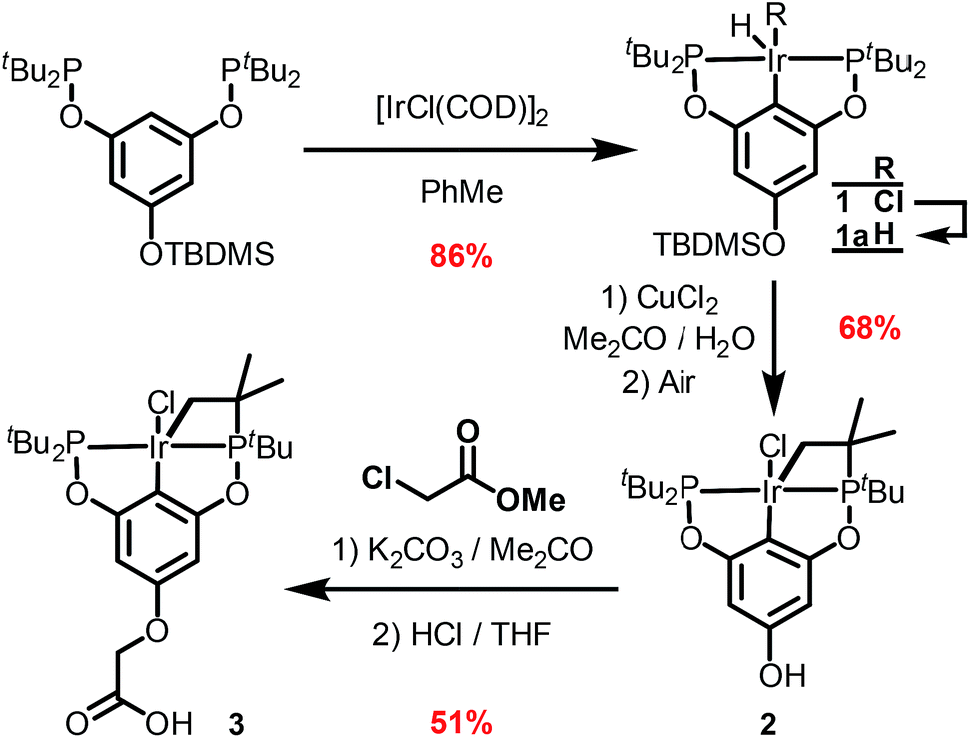 | ||
| Scheme 1 Synthesis of the complex 3. | ||
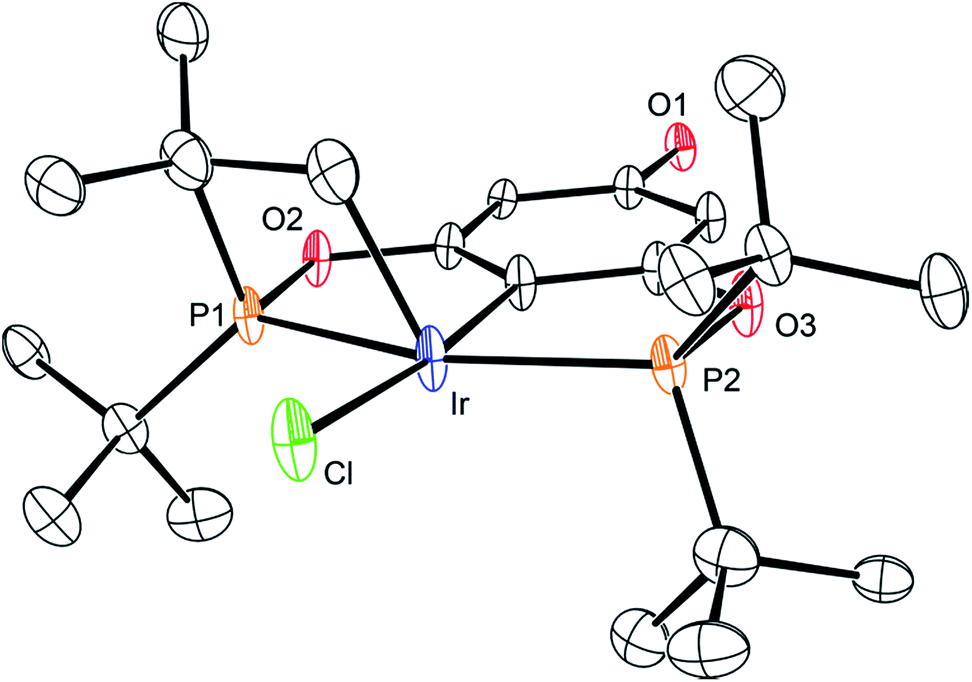 | ||
| Fig. 2 ORTEP drawing of 2. Hydrogen atoms are omitted for clarity, and thermal ellipsoids are set to 20% probability. Only one set of disordered tert-butyl groups and chlorides are shown for clarity. | ||
Etherification, using methyl chloroacetate and subsequent hydrolysis completes the synthesis of the desired carboxylic acid functionalised pincer complex 3. Hydride iridium pincer complexes are known14 to activate a variety of unreactive bonds and to prevent the pincer complex from reacting in an uncontrolled fashion with the zirconium nodes or with the organic linkers in the MOF, we decided to perform SALI with the iridium–chloride complex 3 and to proceed to its activation in a subsequent step.15
NU-1000 was suspended in a PhMe solution of 3 and allowed to react at room temperature (see ESI†). After 24 h, the MOF changed colour from yellow to orange, indicating (Scheme 2) the incorporation of the iridium complex and the formation of 4. The solid was thoroughly washed16 with PhMe. Inductively coupled plasma – optical emission spectrometry (ICP-OES) of 4 showed an Ir![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P:Zr ratio of 0.8:1.5:6 (in theory a 1:2:6 ratio is expected for incorporation of one iridium pincer complex per node) and confirmed the incorporation of the iridium complex into the framework with a loading of 0.8 iridium complexes per Zr6 node. Scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDS) confirms that following SALI, crystallites of NU-1000 retain their morphology while the iridium complex is evenly distributed (see ESI†) within the crystal. The identity of the heterogenised complex was probed by solid-state 31P MAS NMR spectroscopy. A set of two signals at δ 158 and 116 was observed, in good agreement with those found (δ 160.9 and 119.5 see Fig. 3) for the homogenous starting material 3. This spectroscopic evidence proves that the pincer complexes remain intact following SALI. Employing procedures previously reported12 for the activation of homogenous iridium pincer complexes, 4 was exposed to tBuOK in THF under an atmosphere of H2 at room temperature (see ESI†). Adoption of this procedure (Scheme 2) allowed us to (i) activate the supported iridium chloride complex17,18 and (ii) form the corresponding metal hydride 5.
:P:Zr ratio of 0.8:1.5:6 (in theory a 1:2:6 ratio is expected for incorporation of one iridium pincer complex per node) and confirmed the incorporation of the iridium complex into the framework with a loading of 0.8 iridium complexes per Zr6 node. Scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDS) confirms that following SALI, crystallites of NU-1000 retain their morphology while the iridium complex is evenly distributed (see ESI†) within the crystal. The identity of the heterogenised complex was probed by solid-state 31P MAS NMR spectroscopy. A set of two signals at δ 158 and 116 was observed, in good agreement with those found (δ 160.9 and 119.5 see Fig. 3) for the homogenous starting material 3. This spectroscopic evidence proves that the pincer complexes remain intact following SALI. Employing procedures previously reported12 for the activation of homogenous iridium pincer complexes, 4 was exposed to tBuOK in THF under an atmosphere of H2 at room temperature (see ESI†). Adoption of this procedure (Scheme 2) allowed us to (i) activate the supported iridium chloride complex17,18 and (ii) form the corresponding metal hydride 5.
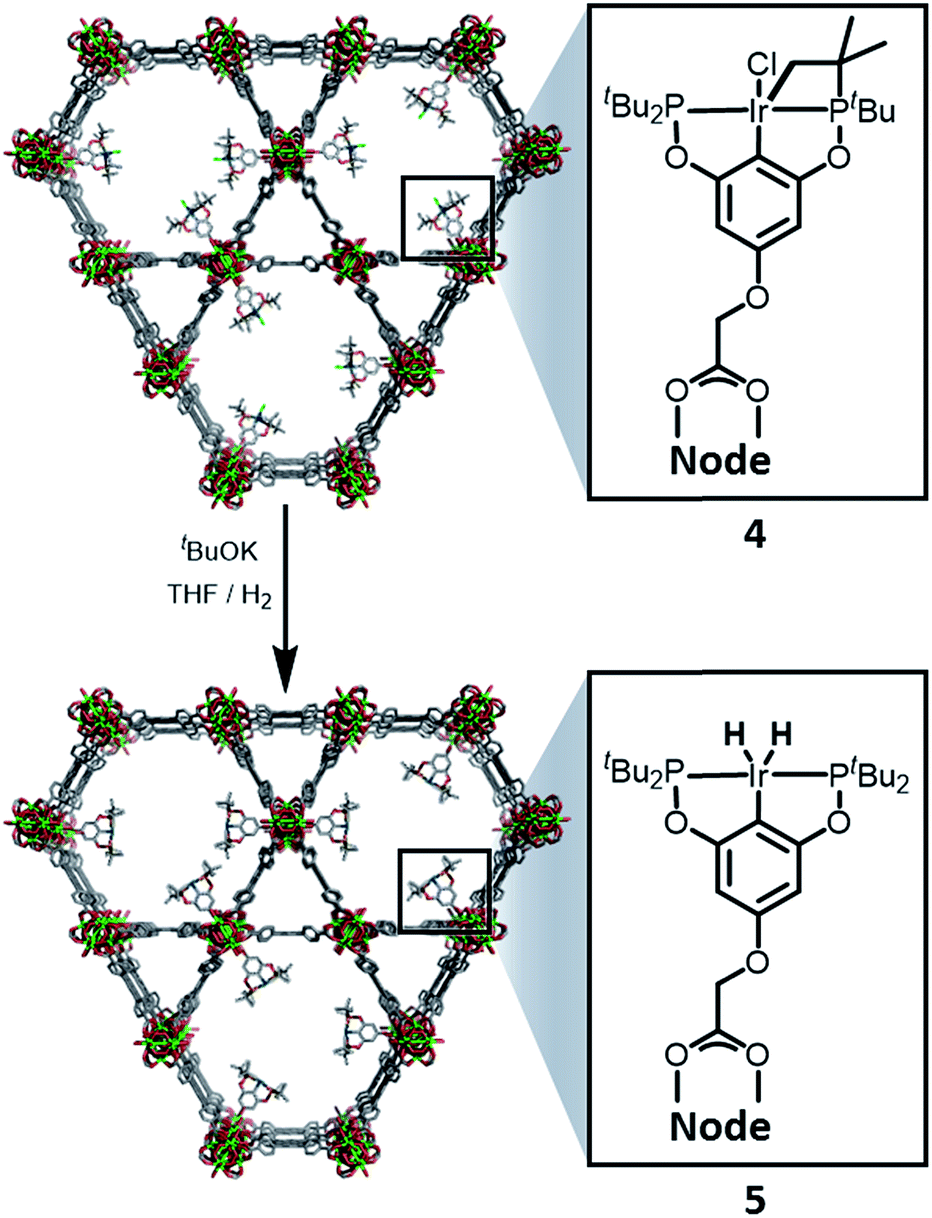 | ||
| Scheme 2 Reactivity of 4 with tBuOK and H2 to give the NU-1000 supported dihydride iridium pincer complex, 5. | ||
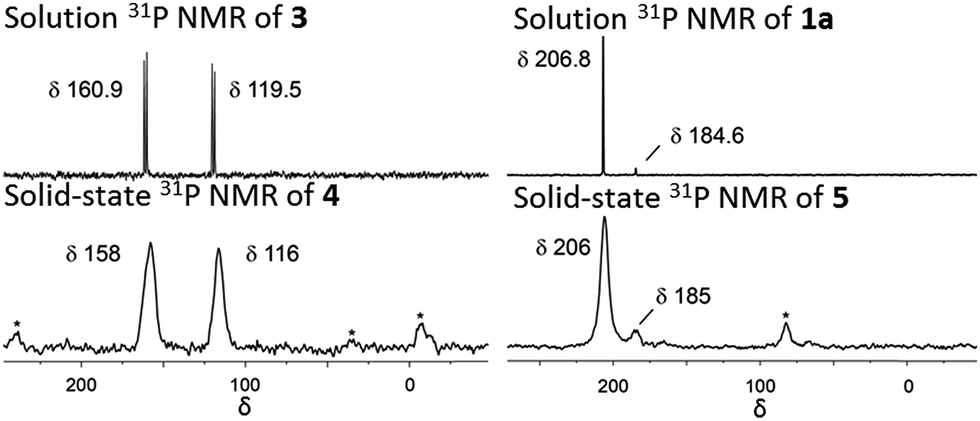 | ||
| Fig. 3 Solution phase 31P NMR of 3 and 1a in C6D6 at 243 MHz. Solid-state 31P MAS NMR of 4 and 5 at 162 MHz. Asterisks denote spinning side bands. | ||
Solid-state 31P MAS NMR spectroscopy (Fig. 3) revealed that treatment of 4 with tBuOK and H2 produces a material with resonances at δ 206 and 185. These signals correspond18 to the dihydride complex (δ 206, major) and the tetrahydride complex (δ 185, minor). Additionally, infrared spectroscopy revealed (see ESI†) the appearance of a band at 2006 cm−1 in good agreement with the formation of a metal hydride. In addition, powder X-ray diffraction of 4 confirmed the retention of crystallinity following SALI and, more importantly, demonstrated that the framework's extended structure is maintained even after the activation step with tBuOK to give 5 (see ESI†). Moreover, the porosity of 5 was retained as shown in Fig. S2–3.†
For comparison, we prepared (Scheme 1) the homogeneous dihydride 1a (see ESI†) from 1. Solution and solid-state 31P MAS NMR spectra showed (Fig. 3) similar resonances for 1a and 5, confirming the presence of analogous Ir(III) complexes in both a homogeneous solution and a heterogeneous material.
Condensed-phase hydrogenation of 1-decene and styrene, catalysed by 1a and 5 at 1 bar (abs.) and 23 °C, were used to examine the relative catalytic activities of the homogenous and heterogeneous Ir(III) catalysts (see ESI†). With 1-decene as the substrate, turnover frequencies (TOFs) of 0.3 and 2.3 h−1 for 1a and 5, respectively, were calculated. Catalysis with styrene reveals TOFs of 0.1 and 7.2 h−1 in the case of 1a and 5, respectively. In both examples, 5 proved (Fig. 4) to be more active than the homogeneous catalyst 1a.
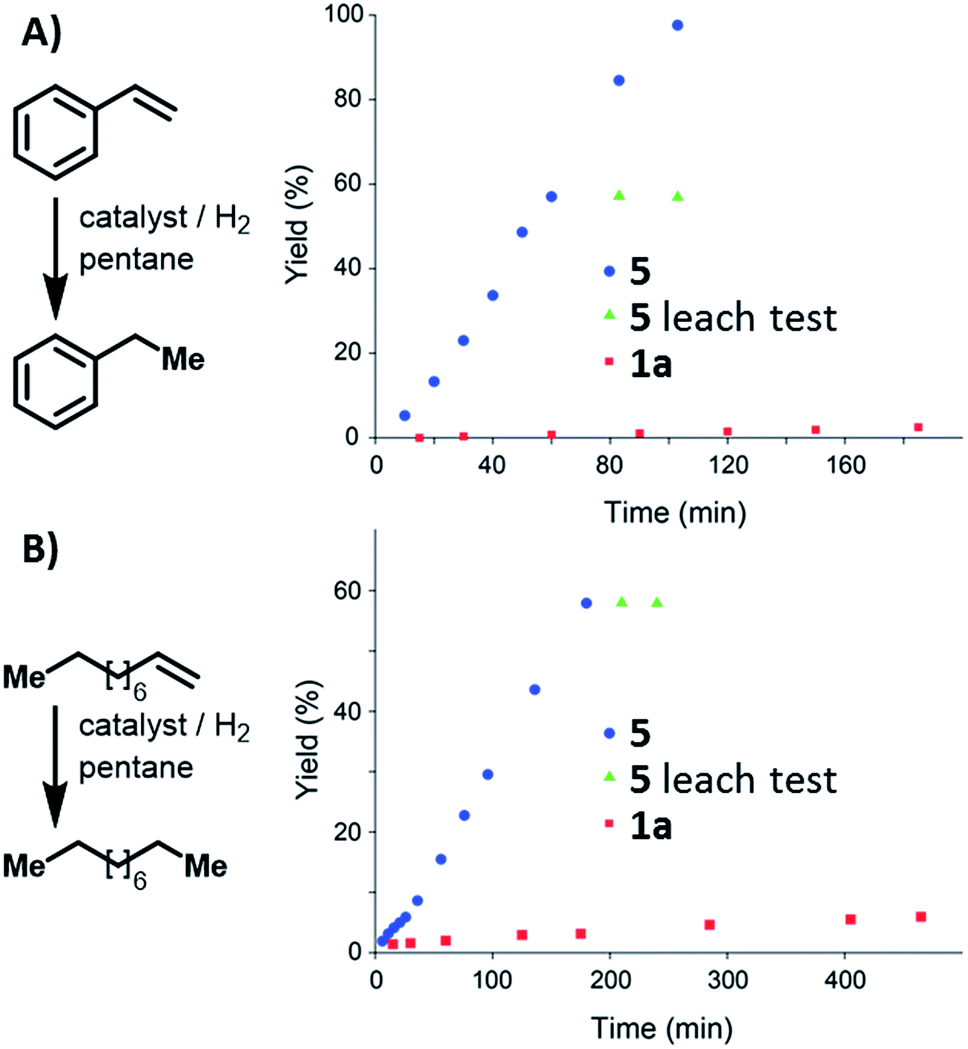 | ||
| Fig. 4 Reaction profile of styrene (A) and 1-decene (B) hydrogenation catalysed by 1a, and 5. Leaching tests performed using 5 indicate that the solution did not contain active catalyst. | ||
Leaching tests on the heterogeneously catalysed reactions showed that the liquid phase is not catalytically active, confirming that the Ir(III) pincer complex is not released into the solution.
Although a thorough explanation of such enhanced activity is not trivial, we speculate that site isolation caused by the heterogenization of the molecular complex reduces intermolecular interactions that could deactivate the catalyst or interfere in the catalytic cycle. Similar behaviour has recently been reported.8f,19
In order to probe the activity of catalyst 5, we tested gas-phase ethene hydrogenation in a tubular flow reactor. The TOF was determined under differential conditions at conversions below 5% (see ESI†). Catalyst 5 showed a TOF value of 0.32 s−1 at 50 °C, 1 bar (rel.), and with an ethene:hydrogen ratio of 1:1.20 Little deactivation was observed over time and stability tests (Fig. 5) showed only minor losses (4%) of activity over the course of 24 h.
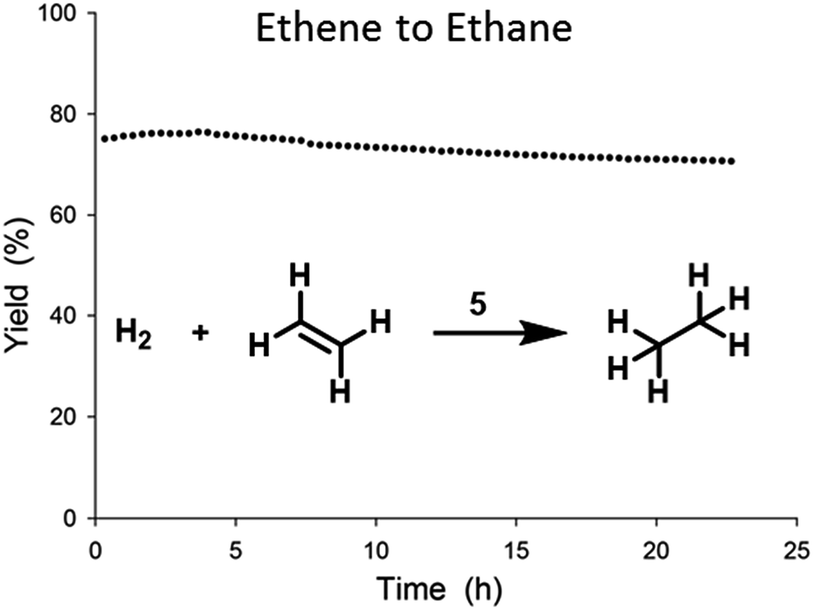 | ||
| Fig. 5 Stability test of ethene hydrogenation catalysed by 5 in flow reactor at 0.5 bar (rel.) and at 23 °C, with an ethene:hydrogen ratio of 1:1 and a total flow of 80 mL min−1. | ||
Conclusions
We report attaching an iridium pincer complex inside a MOF without having to synthesise the extended structure de novo. The complex can be activated after its incorporation to yield the active dihydrido-iridium catalyst. Despite the basic conditions required to carry out the chloride-abstraction, the crystallinity and porosity of the framework are retained and spectroscopic investigations corroborate the similarity between the solid and the related homogeneous catalyst. This observation establishes that the catalytically active site characteristic of the molecular compound has been preserved upon its heterogenization. In order to demonstrate the accessibility to the active sites, we tested this material in the hydrogenation of liquid substrates and with ethene (gas) under flow conditions – the latter being possible only with the catalyst in heterogeneous form. It is noteworthy that the heterogeneous catalyst shows enhanced activity with respect to its homogeneous counterpart and reveals stability upon prolonged use. This work serves as a proof-of-concept that well studied organometallic complexes can be installed and activated within a crystalline metal–organic framework.Acknowledgements
This work was supported as part of the Inorganometallic Catalyst Design Center, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under award DE-SC0012702. This research is part of the Joint Center of Excellence in Integrated Nano-Systems (JCIN) at King Abdulaziz City for Science and Technology (KACST) at Northwestern University (NU). The authors would like to thank both KACST and NU for their continued support of this research which made use of the IMSERC facility supported by the National Science Foundation (NSF, DMR-0521267); the J.B.Cohen X-Ray Diffraction Facility at the Materials Research Center of NU supported by the MRSEC (NSF, DMR-1121262); the EPIC facility of the NUANCE Center at NU, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF, NNCI-1542205); the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. Metal analysis was performed at the NU Quantitative Bio-element Imaging Center. M.R. was supported by the Swiss National Science Foundation with an “Early Postdoc Mobility Fellowship”. A.K.B. thanks Fulbright New Zealand for a Fulbright Graduate Award and the New Zealand Federation of Graduate Women for a Postgraduate Fellowship Award.Notes and references
- (a) R. Langer, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 9948–9952 CrossRef CAS PubMed; (b) N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed; (c) R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed; (d) P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 CrossRef CAS PubMed; (e) C.-I. Lee, J. Zhou and O. V. Ozerov, J. Am. Chem. Soc., 2013, 135, 3560–3566 CrossRef CAS PubMed.
- (a) M. Gupta, C. Hagen, W. C. Kaska, R. E. Cramer and C. M. Jensen, J. Am. Chem. Soc., 1997, 119, 840–841 CrossRef CAS; (b) C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- M. C. Haibach, S. Kundu, M. Brookhart and A. S. Goldman, Acc. Chem. Res., 2012, 45, 947–958 CrossRef CAS PubMed.
- (a) A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257–261 CrossRef CAS PubMed; (b) R. Ahuja, B. Punji, M. Findlater, C. Supplee, W. Schinski, M. Brookhart and A. S. Goldman, Nat. Chem., 2011, 3, 167–171 CrossRef CAS PubMed.
- J. M. Thomas, Design and Application of Single-Site Heterogeneous Catalysts, Imperial College Press, London, 2012 Search PubMed.
- G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) K. Manna, T. Zhang, M. Carboni, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2014, 136, 13182–13185 CrossRef CAS PubMed; (b) T. Zhang, K. Manna and W. Lin, J. Am. Chem. Soc., 2016, 138, 3241–3249 CrossRef CAS PubMed; (c) N. C. Thacker, Z. Lin, T. Zhang, J. C. Gilhula, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2016, 138, 3501–3509 CrossRef CAS PubMed.
- (a) Z. Huang, M. Brookhart, A. S. Goldman, S. Kundu, A. Ray, S. L. Scott and B. C. Vicente, Adv. Synth. Catal., 2009, 351, 188–206 CrossRef CAS; (b) J. Ternel, L. Delevoye, F. Agbossou-Niedercorn, T. Roisnel, R. M. Gauvin and C. M. Thomas, Dalton Trans., 2010, 39, 3802–3804 RSC; (c) E. K. Huang, W. M. Cheung, K. W. Chan, F. L. Y. Lam, X. J. Hu, Q. F. Zhang, I. D. Williams and W. H. Leung, Eur. J. Inorg. Chem., 2013, 2013, 2893–2899 CrossRef CAS; (d) M. Rimoldi, D. Fodor, J. A. van Bokhoven and A. Mezzetti, Chem. Commun., 2013, 49, 11314–11316 RSC; (e) M. Rimoldi and A. Mezzetti, Inorg. Chem., 2014, 53, 11974–11984 CrossRef CAS PubMed; (f) S. A. Burgess, A. Kassie, S. A. Baranowski, K. J. Fritzsching, K. Schmidt-Rohr, C. M. Brown and C. R. Wade, J. Am. Chem. Soc., 2016, 138, 1780–1783 CAS.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed.
- N. Planas, J. E. Mondloch, S. Tussupbayev, J. Borycz, L. Gagliardi, J. T. Hupp, O. K. Farha and C. J. Cramer, J. Phys. Chem. Lett., 2014, 5, 3716–3723 CrossRef CAS PubMed.
- (a) P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS PubMed; (b) P. Deria, Y. G. Chung, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Sci., 2015, 6, 5172–5176 RSC; (c) S. T. Madrahimov, J. R. Gallagher, G. Zhang, Z. Meinhart, S. J. Garibay, M. Delferro, J. T. Miller, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2015, 5, 6713–6718 CrossRef CAS.
- I. Gottker-Schnetmann, P. White and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 1804–1811 CrossRef PubMed.
- (a) A. V. Polukeev, S. A. Kuklin, P. V. Petrovskii, A. S. Peregudov, F. M. Dolgushin, M. G. Ezernitskaya and A. A. Koridze, Russ. Chem. Bull., 2010, 59, 745–749 CrossRef CAS; (b) H. A. Y. Mohammad, J. C. Grimm, K. Eichele, H.-G. Mack, B. Speiser, F. Novak, M. G. Quintanilla, W. C. Kaska and H. A. Mayer, Organometallics, 2002, 21, 5775–5784 CrossRef CAS.
- (a) A. C. Sykes, P. White and M. Brookhart, Organometallics, 2006, 25, 1664–1675 CrossRef PubMed; (b) I. Gottker-Schnetmann and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 9330–9338 CrossRef PubMed.
- To obtain an active species, iridium-chloride pincer complexes are commonly activated in solution by halogen abstraction under hydrogen atmosphere. This procedure usually affords the corresponding highly reactive dihydride species.
- The remaining reaction solution was shown by 31P NMR to contain exclusively unreacted starting material.
- Phosphinite iridium hydride pincer complexes are known to be stable in the form of dihydride and to loosely coordinate an additional dihydrogen molecule when exposed to hydrogen.
- I. Gottker-Schnetmann, P. S. White and M. Brookhart, Organometallics, 2004, 23, 1766–1776 CrossRef.
- M. Rimoldi, D. Fodor, J. A. van Bokhoven and A. Mezzetti, Catal. Sci. Technol., 2015, 5, 4574–4586 Search PubMed.
- A TOF of 0.14 s−1 was obtained at 23 °C and 0.5 bar (rel.).
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures. CCDC 1465323. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01376g |
| This journal is © The Royal Society of Chemistry 2016 |