
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Si2H radical supported by two N-heterocyclic carbenes†
Marius I.
Arz
a,
Gregor
Schnakenburg
a,
Andreas
Meyer
b,
Olav
Schiemann
b and
Alexander C.
Filippou
*a
aInstitute of Inorganic Chemistry, University of Bonn, Gerhard-Domagk-Str. 1, D-53121, Bonn, Germany. E-mail: filippou@uni-bonn.de
bInstitute of Physical and Theoretical Chemistry, University of Bonn, Wegelerstr. 12, D-53115, Bonn, Germany
First published on 9th May 2016
Abstract
Cyclic voltammetric studies of the hydridodisilicon(0,II) borate [(Idipp)(H)SiII![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si0(Idipp)][B(ArF)4] (1H[B(ArF)4], Idipp = C[N(C6H3-2,6-iPr2)CH]2, ArF = C6H3-3,5-(CF3)2) reveal a reversible one-electron reduction at a low redox potential (E1/2 = −2.15 V vs. Fc+/Fc). Chemical reduction of 1H[B(ArF)4] with KC8 affords selectively the green, room-temperature stable mixed-valent disilicon(0,I) hydride Si2(H)(Idipp)2 (1H), in which the highly reactive Si2H molecule is trapped between two N-heterocyclic carbenes (NHCs). The molecular and electronic structure of 1H was investigated by a combination of experimental and theoretical methods and reveals the presence of a π-type radical featuring a terminal bonded H atom at a flattened trigonal pyramidal coordinated Si center, that is connected via a Si–Si bond to a bent two-coordinated Si center carrying a lone pair of electrons. The unpaired electron occupies the SiSi π* orbital leading to a formal Si–Si bond order of 1.5. Extensive delocalization of the spin density occurs via conjugation with the coplanar arranged NHC rings with the higher spin density lying on the site of the two-coordinated silicon atom.
Si0(Idipp)][B(ArF)4] (1H[B(ArF)4], Idipp = C[N(C6H3-2,6-iPr2)CH]2, ArF = C6H3-3,5-(CF3)2) reveal a reversible one-electron reduction at a low redox potential (E1/2 = −2.15 V vs. Fc+/Fc). Chemical reduction of 1H[B(ArF)4] with KC8 affords selectively the green, room-temperature stable mixed-valent disilicon(0,I) hydride Si2(H)(Idipp)2 (1H), in which the highly reactive Si2H molecule is trapped between two N-heterocyclic carbenes (NHCs). The molecular and electronic structure of 1H was investigated by a combination of experimental and theoretical methods and reveals the presence of a π-type radical featuring a terminal bonded H atom at a flattened trigonal pyramidal coordinated Si center, that is connected via a Si–Si bond to a bent two-coordinated Si center carrying a lone pair of electrons. The unpaired electron occupies the SiSi π* orbital leading to a formal Si–Si bond order of 1.5. Extensive delocalization of the spin density occurs via conjugation with the coplanar arranged NHC rings with the higher spin density lying on the site of the two-coordinated silicon atom.
1. Introduction
Open-shell silicon hydrides are of significant importance as transient intermediates in the chemical vapor deposition (CVD) of silicon or silicon-containing thin films, which are extensively used in the semiconductor industry.1 Fundamental species in the gas phase include the SiHx (x = 1–3) and Si2Hx (x = 1–5) molecules as well as higher aggregated SinHm clusters, which are formed from silane (SiH4) or disilane (Si2H6) in a complex cascade of reactions.1 These species, which are also of interest in astrochemistry,2 are unstable under terrestrial conditions and can only be detected by spectroscopic or mass spectrometric techniques.3 One scarcely studied species in this context is the Si2H molecule, which was so far only detected by vibrationally-resolved photoelectron spectroscopy of Si2H− anions.4 Quantum chemical calculations of Si2H suggest two almost isoenergetic, C2v-symmetric H-bridged structures, in which the unpaired electron occupies either the Si–Si π-bonding orbital (2B1 state) or a σ-type molecular orbital corresponding to the in-phase combination of the Si lone pair orbitals (2A1 state).5Recently, N-heterocyclic carbenes (NHCs) were found to be particularly suitable Lewis bases for the thermodynamic and kinetic stabilization of highly reactive, unsaturated, low-valent Si species, leading to the isolation of a series of novel compounds with intriguing synthetic potential.6,7 Several CAAC-stabilized open-shell silicon compounds (CAAC = cyclic alkyl(amino)carbene) were also reported, in which the unpaired electron is mainly located on the CAAC substituent.8 Trapping of Si2H by NHCs appeared therefore an achievable, albeit very challenging goal, given the fact that isolable molecular hydrides of silicon in an oxidation state <2 are very rare9,10 and open-shell congeners presently unknown. In comparison, three-coordinate SiII hydrides11 and four-coordinate SiII hydrides of the general formula (LB)SiH(X)(LA) (LB = neutral Lewis base; LA = neutral Lewis acid; X = singly bonded substituent)12 are meanwhile well documented.
2. Results and discussion
The hydridodisilicon(0,II) salt [(Idipp)(H)SiIISi0(Idipp)][B(ArF)4] (1H[B(ArF)4], Idipp = C[N(C6H3-2,6-iPr2)CH]2, ArF = C6H3-3,5-(CF3)2), which was isolated recently in our group upon protonation of Si2(Idipp)2 (1),9 appeared to be a suitable starting material to tackle the problem of isolating an NHC-trapped Si2H radical. Quantum chemical studies revealed the same sequence of frontier orbitals in 1H+ and its isolobal phosphorus counterpart [R2PPR]+, according to which the HOMO−1 corresponds to the lone-pair orbital at the two-coordinated E atom (E = Si, P), the HOMO is the EE π-bonding orbital and the LUMO is the EE π* orbital.9 This isolobal interrelationship suggested that 1H+ might be also reversibly reducible as the phosphanylphosphenium cation [Mes*(Me)PPMes*]+ (Mes* = C6H2-2,4,6-tBu3).13 In fact, cyclic voltammetric (CV) studies of 1H[B(ArF)4] in fluorobenzene at room temperature revealed a reversible one-electron reduction at a rather low half-wave potential (E1/2) of −1.63 V as well as an irreversible oxidation at +0.67 V versus the [Fe(η5-C5Me5)2]+1/0 reference electrode (Fig. 1 and ESI†).14 The methyl analogue [(Idipp)(Me)SiIISi0(Idipp)][B(ArF)4] (1Me[B(ArF)4])9 was found also to undergo a reversible one-electron reduction, albeit at a more negative potential (E1/2 = −1.85 V) than 1H[B(ArF)4]. Notably, reduction of 1H+ and 1Me+ occurs at much lower potentials than that of the cation [Mes*(Me)PPMes*]+ (E1/2 = −0.48 V).13 This marked difference in the redox potentials of the Si- and P-based cations can be rationalized with the large increase of the LUMO energy occurring upon replacement of the two PMes* fragments by the much less electronegative isolobal Si(Idipp) fragments as suggested by quantum chemical calculations.9
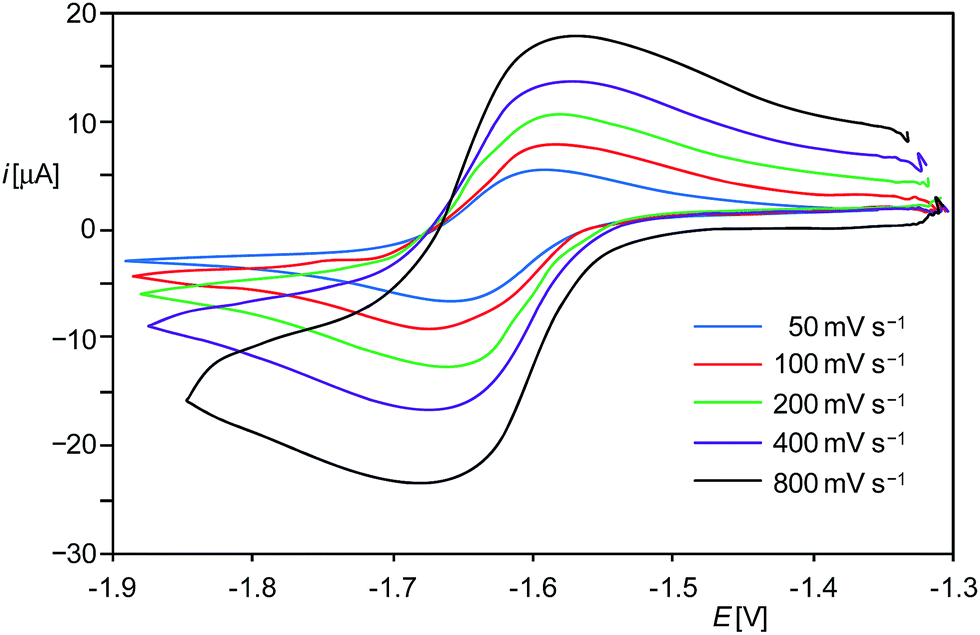 | ||
| Fig. 1 Single-scan cyclic voltammograms of 1H[B(ArF)4] from (−1.9) to (−1.3) V at different scan rates at room temperature in fluorobenzene/0.1 M (nBu4N)PF6 solution; reference electrode: 4 mM [Fe(η5-C5Me5)2]+1/0/0.1 M (nBu4N)PF6 in fluorobenzene. | ||
The CV results prompted us to attempt a chemical one-electron reduction of 1H[B(ArF)4]. Indeed, vacuum transfer of THF to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric mixture of 1H[B(ArF)4] and KC8 at −196 °C followed by warming to −40 °C resulted in a distinct color change of the dark red solution of 1H[B(ArF)4] to give an intensely dark green solution, which after work-up and crystallization from n-hexane at −60 °C afforded Si2(H)(Idipp)2 (1H) as a dark green, almost black crystalline solid in 55% yield (Scheme 1) (see ESI†). Compound 1H is extremely air-sensitive and immediately decolorizes upon contact with air, but can be stored under an atmosphere of argon at −30 °C without any color change or signs of decomposition in its EPR spectrum. Thermal decomposition of 1H in a vacuum-sealed glass capillary was detected upon melting at 147 °C leading to a dark red mass. Analysis of the soluble part of the melting residue in C6D6 by 1H NMR spectroscopy revealed the presence of Idipp (95%) and 1 (5%).
:1 stoichiometric mixture of 1H[B(ArF)4] and KC8 at −196 °C followed by warming to −40 °C resulted in a distinct color change of the dark red solution of 1H[B(ArF)4] to give an intensely dark green solution, which after work-up and crystallization from n-hexane at −60 °C afforded Si2(H)(Idipp)2 (1H) as a dark green, almost black crystalline solid in 55% yield (Scheme 1) (see ESI†). Compound 1H is extremely air-sensitive and immediately decolorizes upon contact with air, but can be stored under an atmosphere of argon at −30 °C without any color change or signs of decomposition in its EPR spectrum. Thermal decomposition of 1H in a vacuum-sealed glass capillary was detected upon melting at 147 °C leading to a dark red mass. Analysis of the soluble part of the melting residue in C6D6 by 1H NMR spectroscopy revealed the presence of Idipp (95%) and 1 (5%).
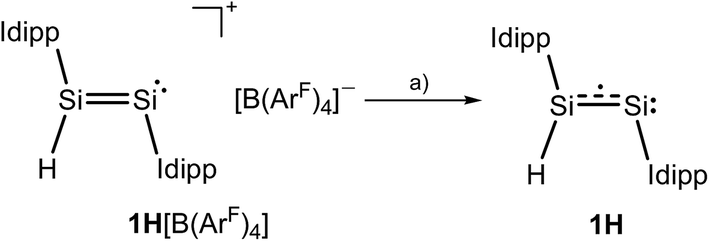 | ||
| Scheme 1 Synthesis of 1H upon one-electron reduction of 1H[B(ArF)4]; (a) +KC8, −K[B(ArF)4], −8C; THF; −196 °C → −40 °C. Two dots indicate a lone pair of electrons and the dotted line indicates the population of the SiSi π* orbital upon reduction; formal charges are omitted for clarity. | ||
Notably, the redox potential of 1H [E1/2 in C6H5F = −2.15 V vs. [Fe(η5-C5H5)2]+1/0 (Fc+/Fc)]15 lies in-between that of the benzophenone radical anion (E1/2 in THF = −2.30 V vs. Fc+/Fc)16 and [Co(η5-C5Me5)2] (E1/2 in MeCN = −1.91 V vs. Fc+/Fc),16 indicating that the radical 1H is a very strong one-electron reducing agent. Consequently, the radical 1H is selectively oxidized back to 1H[B(ArF)4] upon treatment with one equivalent of [Fe(η5-C5Me5)2][B(ArF)4] in THF-d8 (see ESI†). Thereby, the redox pair 1H+/1H provides a very rare example of a chemically reversible Si-based redox system.7c,17
Compound 1H is well soluble in n-hexane, benzene, diethyl ether or THF affording intensely dark-green solutions, even at low concentrations. The origin of this intense color was analyzed by UV-Vis-NIR spectroscopy of 1H in n-hexane (Fig. 2, left and ESI†), which revealed electronic absorptions in the whole spectral range from 220–1100 nm. Six absorption maxima were located at 254 (9970), 305 (8140), 436 (5170), 608 (7110), 704 (6860) and 958 (1440) nm, of which the intense absorptions at 608 and 704 nm are responsible for the green color of 1H (the values of the molar absorption coefficients ελ are given in brackets in L mol−1 cm−1). The UV-Vis-NIR spectrum was also analyzed by time-dependent density functional theory (TdDFT) calculations (see ESI, Fig. S21†).18
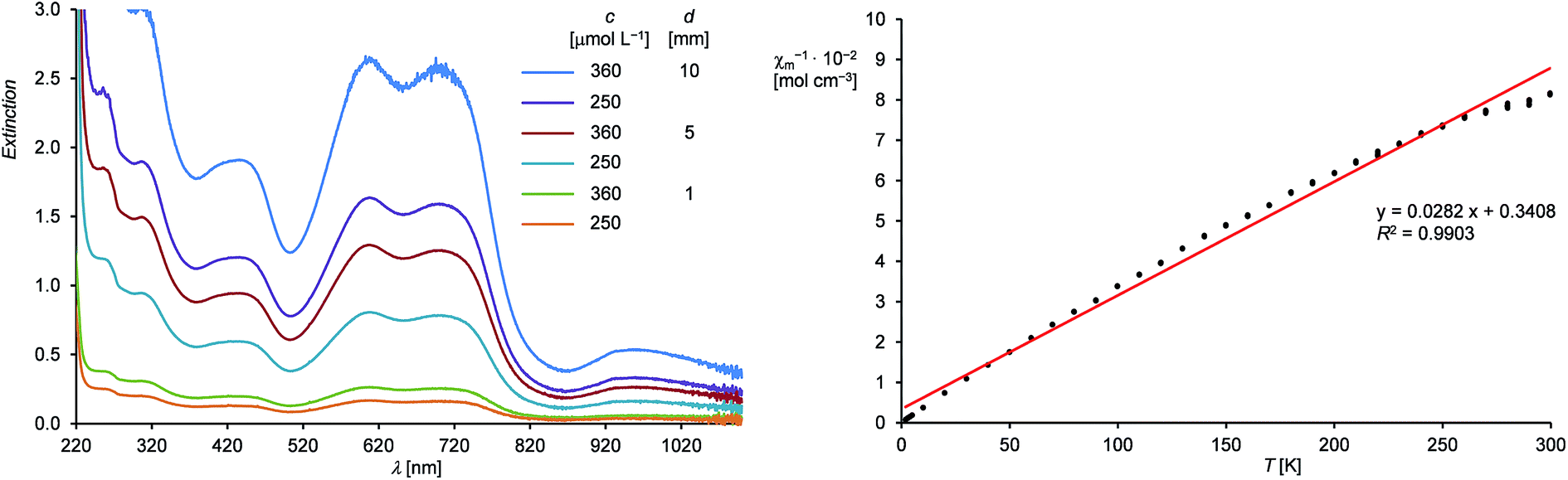 | ||
| Fig. 2 Left: UV-Vis-NIR spectra of 1H in n-hexane from 220–1100 nm at different concentrations (c) and path lengths (d). Right: Plot of the reciprocal molar magnetic susceptibility (χm−1) against the absolute temperature (T) (dotted black line) and the corresponding line (red) and line equation obtained by linear regression. | ||
Magnetic susceptibility measurements of solid 1H from 300.0–1.9 K suggest the presence of a paramagnetic compound with one unpaired electron following Curie's law. A plot of the reciprocal molar magnetic susceptibility (χm−1) against the absolute temperature (T) showed a linear correlation from which the effective magnetic moment μeff was calculated after linear regression and found to be 1.68 μB (Fig. 2, right and ESI†). This value is slightly lower than the value derived from the spin-only formula for one unpaired electron (μeff = 1.73 μB).
The molecular structure of 1H was determined by single crystal X-ray crystallography. The radical features a crystallographically imposed inversion symmetry (space group: P21/c) in marked contrast to the C1-symmetric structure of 1H+ in 1H[B(ArF)4].9 The Si-bonded H atom was located in the difference Fourier map and anisotropically refined with a site occupancy of 1/2 at each Si atom. However, the exact position of this H atom could not be deduced by X-ray crystallography, since structural refinements with either a terminal (Si–H) or a bridging (Si–H–Si) position led to identical wR2 values. 1H features as 1H[B(ArF)4] and 1 a trans-bent planar CNHC–Si–Si–CNHC core (Fig. 3). However, distinct structural differences become apparent upon comparing the three structures. For example, the Si–Si bond of 1H (2.281(3) Å) is considerably longer than that in 1H[B(ArF)4] (2.1873(8) Å)9 or 1 (2.229(1) Å)19 (Table 1), and lies in-between that of a typical SiSi double bond (2.20 Å)20 and a Si–Si single bond (e.g. 2.352 Å in α-Si).21 In comparison, the Si–CNHC bonds in 1H (1.873(4) Å) are shorter than the Si–CNHC bonds of the dicoordinated Si atoms in 1H[B(ArF)4] (1.940(2) Å)9 and 1 (1.927(1) Å)19 (Table 1), and similar to that of the trigonal-planar coordinated Si atom in 1H[B(ArF)4] (1.882(2) Å).9 Reduction of 1H+ results also in a distinct change of the conformation of the NHC substituents. Thus, both N-heterocyclic rings in 1H are arranged coplanar with the trans-bent CNHC–Si–Si–CNHC core as evidenced by the dihedral angle φNHC of 3.3(2)° (Table 1), whereas in 1H+ one of the two N-heterocyclic rings (bonded to the two-coordinated Si atom) adopts an almost orthogonal orientation (Table 1). All these structural changes can be rationalized by quantum theory (vide infra). Thus, reduction of 1H+ leads to a population of the SiSi π* orbital with one electron, reducing thereby the formal Si–Si bond order from 2 in 1H+ to 1.5 in 1H as nicely reflected in the computed Si–Si Wiberg bond indexes (WBI; WBI(Si–Si) of 1H+ = 1.70; WBI(Si–Si) of 1H = 1.17) (see ESI, Tables S11 and S12†). The coplanar arrangement of the N-heterocyclic rings allows for an optimal in-phase interaction (π-conjugation) of the SiSi π* orbital with π*(CN2) orbitals of the NHC substituents in the SOMO of 1H (Fig. 6), providing a rationale for the shortening of the Si–CNHC bonds and the concomitant elongation of the CNHC–NNHC bonds of 1Hversus1H+ (Table 1).
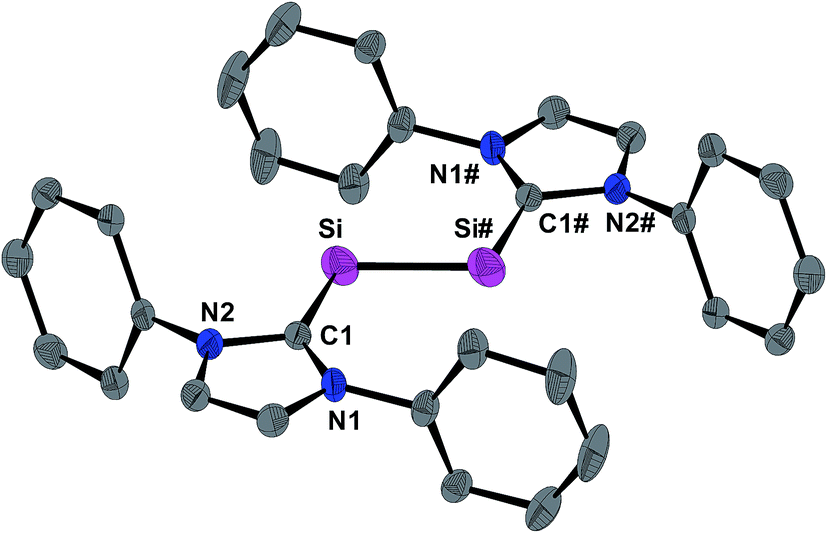 | ||
| Fig. 3 DIAMOND plot of the molecular structure of 1H in the single crystal at 123(2) K. Thermal ellipsoids are set at 30% electronic probability. The hydrogen atoms and the iPr groups are omitted for clarity. The Si-bonded H atom was omitted due to its uncertain position. Selected bond lengths [Å], bond angles [°] and torsion angles [°]: Si–Si# 2.281(3), Si–C1 1.873(4); C1–Si–Si# 109.5(1); C1–Si–Si#–C1# 180.0(3). | ||
| Si–Si [Å] | Si–CNHC [Å] | CNHC–NNHC [Å] | CNHC–Si–Si [°] | φ NHC [°] | |
|---|---|---|---|---|---|
|
a Data taken from ref. 9. Connectivity: [(NHC1)(H)Si1Si2(NHC2)]+.
b Data taken from ref. 19.
c
φ
NHC denotes the dihedral angles between the CNHC–Si–Si–CNHC least-square plane and the respective N-heterocyclic ring least-square planes.
|
|||||
| 1H | 2.281(3) | 1.873(4) | 1.381(4), 1.402(4) | 109.5(1) | 3.3(2) |
| 1H[B(ArF)]4a | 2.1873(8) | 1.882(2) (Si1–CNHC) | 1.356(2), 1.358(2) | 116.73(7) (C1–Si1–Si2) | 8.60(6) (φNHC1) |
| 1.940(2) (Si2–CNHC) | 1.356(2), 1.358(2) | 95.34(6) (C28–Si2–Si1) | 71.06(6) (φNHC2) | ||
| 1 | 2.229(1) | 1.927(2) | 1.368(2), 1.372(2) | 93.37(5) | 87.11(5) |
IR spectroscopy proved to be a very useful method to determine unequivocally the position of the Si-bonded H atom. In fact, the ATR FT-IR spectrum of 1H displayed a ν(Si–H) absorption band at 2089 cm−1, which is characteristic for stretching vibrations of terminal Si–H bonds (see ESI, Fig. S4†). In comparison, the ν(Si–H–Si) band of Si2H is predicted at significantly lower wavenumbers (1592 cm−1 (2A1 state); 1491 cm−1 (2B1 state)),4 and also the ν(Si–H–Si) absorption bands of H-bridged silylium ions are shifted to much lower wavenumbers (ca. 1750–1950 cm−1; e.g. 1900 cm−1 in [Et3Si–H–SiEt3][CHB11Cl11]) compared with the ν(Si–H) bands of the corresponding silanes (ca. 2150 cm−1).22 Notably, the ν(Si–H) absorption band of 1H appears in-between that of 1H[B(ArF)4] containing a trigonal planar coordinated Si atom (ν(Si–H) = 2142 cm−1),9 and the Si(II)-hydride (IMe4)SiH(SitBu3) containing a strongly pyramidal bonded Si atom (IMe4 = C[N(Me)CMe]2: ν(Si–H) in KBr = 1984 cm−1).11d Apparently, the ν(Si–H) frequency decreases with increasing pyramidalization of the Si atom, which according to the quantum chemical calculations can be traced back to the decreasing s-character of the Si hybrid orbital in the Si–H bond (see ESI, Tables S11 and S12†).
Further insight into the structure of 1H was provided by continuous wave (cw) EPR spectroscopy at X-band frequencies. Spectra with a nicely resolved hyperfine coupling pattern could be obtained from samples of 1H in n-hexane solution at 336 K (Fig. 4; see also ESI, Fig. S10† for EPR spectra at different temperatures). Notably, a similar EPR spectrum was obtained in diethyl ether solution at 298 K (see ESI, Fig. S12†), suggesting that solvent coordination effects are negligible. The EPR spectrum of 1H displays a multiplet at a giso value of 2.00562, which could be well simulated assuming coupling of the unpaired electron to one 1H (I = 1/2) nucleus, two different 29Si (I = 1/2) and two pairs of two magnetically equivalent 14N (I = 1) nuclei, respectively (Fig. 4). These observations suggest that 1H has a rigid structure and does not undergo a reversible 1,2-H-migration in solution in contrast to 1H+.9 Remarkably, two quite different a(29Si) hyperfine coupling constants (1.725 and 0.431 mT) were found, indicating an asymmetric spin density distribution over the Si atoms. Both values are smaller than those of other Si-based π type radicals, such as the disilene radical cation [Si2(SitBu2Me)4]+ (2.30 mT)23 or the disilene radical anions [Si2R4]− (2.45–4.83 mT, R = silyl substituent)24 due to extensive delocalization of the spin density into the NHC substituents, and also significantly smaller than that of the σ-type radical cation in 1[B(ArF)4] (5.99 mT),7c indicating a localization of the unpaired electron in a molecular orbital of π-symmetry in agreement with the results of the quantum chemical calculations (vide infra). The two a(14N) hfcc's (0.246 and 0.100 mT) suggest a fast rotation of the magnetically different NHC substituents about the Si–CNHC bonds on the EPR timescale occurring even at low temperature (see ESI, Table S6†).
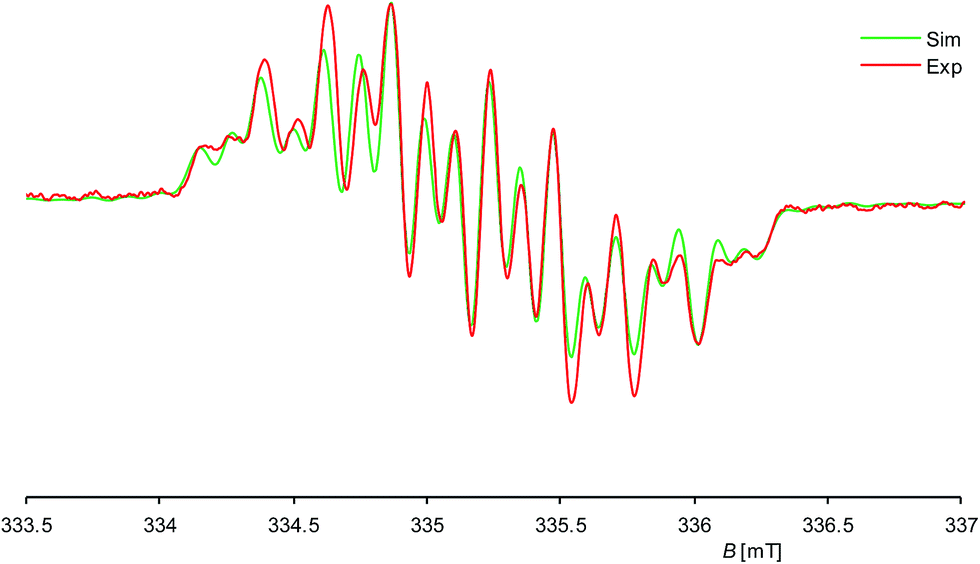 | ||
| Fig. 4 Experimental (red curve) and simulated (green curve) X-band EPR spectra of 1H in n-hexane at 336 K; the ordinate (dA/dB) is omitted for clarity. giso = 2.00562, a(29Si1) = 1.725 mT, a(29Si2) = 0.431 mT, a(14N1) = 0.246 mT, a(14N2) = 0.100 mT, a(1H) = 0.605 mT. | ||
Quantum chemical calculations of 1H were carried out using the B3LYP functional in combination with the 6-311G** basis set for the Si, N, Si-bonded H and NHC ring C atoms and the 6-31G* basis set for the peripheral C and H atoms or the B97-D3 functional in combination with RI-JCOSX approximations and the def2-TZVP basis set for all atoms.25 The levels of theory are abbreviated in the following with B3LYP/I and B97-D3/II. Remarkably, calculations at the B3LYP/I level of theory yielded one minimum structure (1Hcalc), whereas two almost degenerate minimum structures were obtained at the B97-D3/II level of theory (1Hcalc and 1H′calc) (Fig. 5). All calculated minimum structures display a terminally bonded H atom bound to the Si1 atom. No minimum structure with a bridged H atom was found on the potential energy hypersurface of 1H at both levels of theory. The geometrical parameters of the minimum structure calculated at the B3LYP/I level of theory and the global minimum structure at the B97-D3/II level of theory are almost identical (Table 2 and ESI, Table S8†). These structures (1Hcalc) contain a trigonal-pyramidal coordinated Si1 atom with a sum of angles of 335.51° (B3LYP/I) and 342.58° (B97-D3/II), respectively. Remarkably, the calculated structure of the diphosphanyl radical P2(Me)Mes*2, which is isolobal to 1H, displays a trigonal pyramidal geometry at the three-coordinated P atom (sum of angles: 337.5°),13 as found for 1Hcalc. In comparison, the second minimum structure obtained at the B97-D3/II level of theory (1H′calc) is only 5.5 kJ mol−1 higher in energy than 1Hcalc and contains the Si1 atom in a trigonal planar environment (sum of angles: 359.61°). A comparison of the structural parameters of 1Hcalc and 1H′calc with those obtained by single crystal X-ray diffraction reveals a good agreement of the calculated Si–Si, Si–CNHC and CNHC–NNHC bond lengths of both minimum structures (Table 2 and ESI, Table S8†). While the experimental results did not allow to clearly distinguish whether a flattened trigonal-pyramidal or a trigonal-planar geometry of the H-bound Si atom is present in 1H, the theoretical studies suggest a flat energy hypersurface for the planarization of the three-coordinated Si atom.
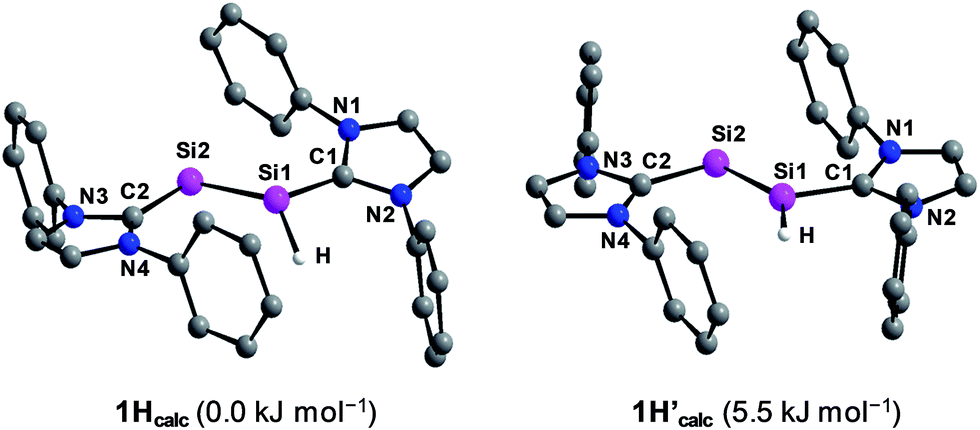 | ||
| Fig. 5 Calculated minimum structures (1Hcalc and 1H′calc) of Si2(H)(Idipp)2 at the B97-D3/RI-JCOSX/def2-TZVP level of theory. The relative energies are given in brackets. The H atoms, except the H atom bonded to Si1, and the iPr substituents are omitted for clarity. | ||
| Si1–Si2 [Å] | Si1–C1 [Å] | Si2–C2 [Å] | ∑Si1c [°] | C1–Si1–Si2–C2 [°] | φ NHC1 [°] | φ NHC2 [°] | |
|---|---|---|---|---|---|---|---|
| a Calculated at the B3LYP/6-311G**/6-31G* level of theory. b Calculated at the B97-D3/RI-JCOSX/def2-TZVP level of theory. c ∑Si1 is the sum of angles around the Si1 atom. d φ NHC1 and φNHC2 denote the dihedral angles between the least-square plane of the atoms C1, Si1, Si2, C2 and the least square plane of the heterocyclic ring atoms of the NHC substituent bonded to Si1 and Si2, respectively. | |||||||
| 1H | 2.281(3) | 1.873(4) | 1.873(4) | — | 180.0(3) | 3.3(2) | 3.3(2) |
| 1Hcalc | 2.339 | 1.885 | 1.907 | 335.51 | 173.69 | 32.71 | 1.26 |
| 1Hcalc | 2.308 | 1.861 | 1.884 | 342.58 | 173.63 | 21.95 | 3.41 |
| 1H′calc | 2.289 | 1.841 | 1.886 | 359.61 | 179.32 | 6.68 | 3.24 |
The calculated quasi-restricted orbitals (QROs) of 1Hcalc at the B3LYP/I level of theory and of 1Hcalc and 1H′calc at the B97-D3/II level of theory are almost identical (Fig. 6 and ESI, Fig. S17–S19†). The SOMO is the SiSi π* orbital, confirming that reduction of 1H+ leads to a population of the empty SiSi π* orbital of 1H+ with one electron (see ESI, Fig. S16†). The SOMO reveals significant contributions of π* NHC orbitals due to π-conjugation. The two lower lying doubly occupied molecular orbitals (DOMOs) are the SiSi π and the n(Si) lone pair orbital, respectively.
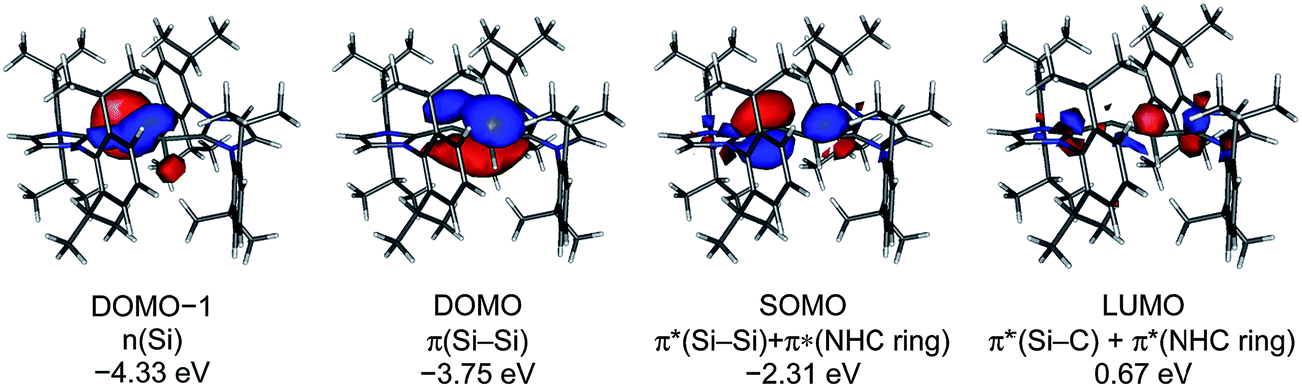 | ||
| Fig. 6 Quasi-restricted orbitals (QROs) of 1Hcalc (B97-D3/RI-JCOSX/def2-TZVP) and their corresponding energy eigenvalues; isosurface value: 0.04 e bohr−3; DOMO = doubly occupied molecular orbital, SOMO = singly occupied molecular orbital, LUMO = lowest unoccupied molecular orbital. | ||
Notably, CASSCF(3,3)/def2-TZVP calculations26 of 1Hcalc revealed that the overall wave function is described by a major ground state configuration of [2-1-0] of the DOMO, SOMO and LUMO with 96% contribution, suggesting that static correlation can be neglected in the electronic description of 1H (see ESI†).
The calculated spin densities of 1Hcalc and 1H′calc at the B97-D3/II level of theory are depicted in Fig. 7. Mulliken analyses27 of the spin densities reveal that the highest spin density is located at the dicoordinated Si2 atom (37% in 1Hcalc, 29% in 1H′calc), whereas the spin density at the Si1 atom is quite small (9% in 1Hcalc, 6% in 1H′calc), which is in full agreement with the observation of one large and one small a(29Si) hfcc in the experimental EPR spectrum of 1H (vide supra) (see ESI, Table S9†).28 Remarkably, a significant amount of the spin density is delocalized into the CNHC and NNHC atoms of the Si1-bonded (17% in 1Hcalc, 27% in 1H′calc) and Si2-bonded (29% in 1Hcalc, 30% in 1H′calc) NHC substituents, which explains the EPR-spectroscopic detection of two a(14N) hfcc's. The calculated giso values of 1Hcalc (2.00483) and 1H′calc (2.00454) agree well with the experimentally obtained giso value (2.00562).
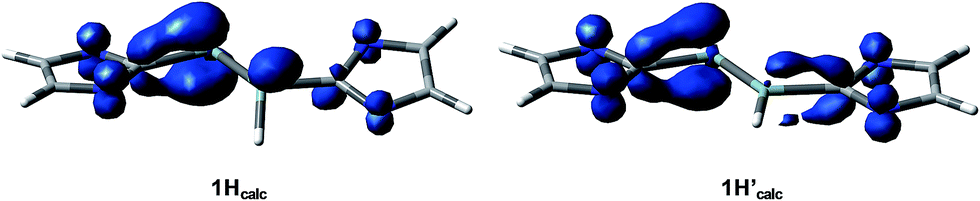 | ||
| Fig. 7 Spin densities of the calculated minimum structures 1Hcalc (left) and 1H′calc (right) at the B97-D3/RI-JCOSX/def2-TZVP level of theory. The N-bonded 2,6-diisopropylphenyl substituents are omitted for clarity. | ||
Further insight into the electronic structure of 1H was provided by a natural bond orbital (NBO) analysis at the B3LYP/I level of theory (see ESI, Table S12†).25k The Si–Si bond is composed of a Si–Si σ bond and a SiSi π bond with an occupancy of 1.95 and 0.82 electrons, respectively, which indicates indirectly a population of the SiSi π* orbital with one electron leading thereby to a decrease of the formal Si–Si bond order from 2 in 1H+ to 1.5 in 1H (vide supra). The Si2 atom in 1Hcalc bears a lone pair of high s-character (72%) as similarly found for 1H+calc (75%). Remarkably, both Si–CNHC bonds in 1Hcalc are composed of one doubly occupied Si–CNHC σ NBO and one singly occupied SiCNHC π NBO, of which the latter is absent in 1H+calc. These additional Si–CNHC π contributions rationalize the shortening and strengthening of the Si–CNHC bonds in 1H, which is also reflected in the higher Si–CNHC WBI indexes (1H: WBI(Si–CNHC) = 1.01 and 0.95; 1H+: WBI(Si–CNHC) = 0.86 and 0.74).
Comparative analyses of the charge by natural population analyses (NPA) of 1Hcalc and 1H+calc at the B3LYP/I level of theory reveal that the positive partial charges at the Si atoms of 1H+calc (q(Si1) = 0.27e, q(Si2) = 0.21e) are decreased by the reduction (1H: q(Si1) = 0.14e, q(Si2) = 0.03e) (see ESI, Table S13†). Furthermore, the one-electron reduction leads to a significant decrease of the overall charges of the NHC substituents (1H+calc: q(NHC1) = 0.36e, q(NHC2) = 0.30e; 1H: q(NHC1) = 0.05e, q(NHC2) = −0.04e), whereas the hydridic character of the Si1-bonded H atom is retained (1H+calc: q(H) = −0.14e; 1H: q(H) = −0.18e).
3. Conclusions
The isolation and full characterization of NHC-trapped Si2H (1H) can be considered as a major advance in low-valent silicon hydride chemistry, given the intermediacy of Si2H in the chemical vapor deposition of amorphous hydrogenated silicon that is widely used in solar cell and thin film transistors technology. Whereas Si2H features a C2v-symmetric H-bridged ground state structure and is a σ-type radical with a symmetric distribution of the spin density over the two silicon atoms, its NHC-trapped counterpart Si2(H)(Idipp)2 (1H) features a terminal Si–H bond and is a π-type radical, in which the unpaired electron occupies the SiSi π* orbital (SOMO), leading to a formal Si–Si bond order of 1.5. Significant delocalization of the spin density into the NHC substituents occurs via π-conjugation of the SiSi π* orbital with the π* orbitals of the coplanar arranged N-heterocyclic rings leading to a stabilization of the radical, in which the spin density is higher at the two-coordinated Si site. The mixed valent disilicon(0,I) hydride 1H can be alternatively regarded as a H atom trapped in the closed shell compound Si2(Idipp)2. Implications of this view in hydrogen atom transfer chemistry29 are currently investigated.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (SFB813, “Chemistry at Spin Centers”) for financial support of this study. We also thank Dr. Burhanshah Lewall for cyclic voltammetric studies, Martin Straβmann for recording the UV-Vis-NIR spectra and Norbert Wagner for the magnetic susceptibility measurements.Notes and references
- (a) J. M. Jasinski and S. M. Gates, Acc. Chem. Res., 1991, 24, 9 CrossRef CAS; (b) M. Moravej, S. E. Babayan, G. R. Nowling, X. Yang and R. F. Hicks, Plasma Sources Sci. Technol., 2004, 13, 8 CrossRef CAS.
- M. C. McCarthy, C. A. Gottlieb and P. Thaddeus, Mol. Phys., 2003, 101, 697 CrossRef CAS and refs. therein.
- (a) J. M. Jasinski, R. Becerra and R. Walsh, Chem. Rev., 1995, 95, 1203 CrossRef CAS; (b) H. Stafast, G. Andrä, F. Falk and E. Witkowicz, in Silicon Chemistry. From the Atom to Extended Systems, ed. P. Jutzi and U. Schubert, Wiley-VCH, Weinheim, 2003, ch. 3, pp. 33–43 Search PubMed.
- C. Xu, T. R. Taylor, G. R. Burton and D. M. Neumark, J. Chem. Phys., 1998, 108, 7645 CrossRef CAS.
- (a) J. Kalcher and A. F. Sax, Chem. Phys. Lett., 1993, 215, 601 CrossRef CAS; (b) B. Ma, N. L. Allinger and H. F. Schaefer III, J. Chem. Phys., 1996, 105, 5731 CrossRef CAS; (c) C. Pak, S. S. Wesolowski, J. C. Rienstra-Kiracofe, Y. Yamaguchi and H. F. Schaefer III, J. Chem. Phys., 2001, 115, 2157 CrossRef CAS; (d) Z. T. Owens, J. D. Larkin and H. F. Schaefer III, J. Chem. Phys., 2006, 125, 164322 CrossRef PubMed.
- Selected recent reviews: (a) R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444 CrossRef CAS PubMed; (b) E. Rivard, Struct. Bonding, 2014, 156, 203 CrossRef CAS; (c) Y. Wang and G. H. Robinson, Inorg. Chem., 2014, 53, 11815 CrossRef CAS PubMed.
- (a) D. Geiß, M. I. Arz, M. Straßmann, G. Schnakenburg and A. C. Filippou, Angew. Chem., Int. Ed., 2015, 54, 2739 ( Angew. Chem. , 2015 , 127 , 2777 ) CrossRef PubMed; (b) P. Ghana, M. I. Arz, U. Das, G. Schnakenburg and A. C. Filippou, Angew. Chem., Int. Ed., 2015, 54, 9980 ( Angew. Chem. , 2015 , 127 , 10118 ) CrossRef CAS PubMed; (c) M. I. Arz, M. Straßmann, A. Meyer, G. Schnakenburg, O. Schiemann and A. C. Filippou, Chem.–Eur. J., 2015, 21, 12509 CrossRef CAS PubMed and refs. therein; (d) M. I. Arz, D. Geiß, M. Straßmann, G. Schnakenburg and A. C. Filippou, Chem. Sci., 2015, 6, 6515 RSC and refs. therein.
- (a) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020 RSC; (b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed; (c) K. C. Mondal, S. Roy and H. W. Roesky, Chem. Soc. Rev., 2016, 45, 1080 RSC.
- M. I. Arz, M. Straßmann, D. Geiß, G. Schnakenburg and A. C. Filippou, J. Am. Chem. Soc., 2016, 138, 4589 CrossRef CAS PubMed.
- R. Kinjo, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2007, 129, 26 CrossRef CAS PubMed.
- (a) N. Wiberg, W. Niedermayer, H. Nöth and M. Warchhold, Z. Anorg. Allg. Chem., 2001, 627, 1717 CrossRef CAS; (b) R. Rodriguez, D. Gau, Y. Contie, T. Kato, N. Saffon-Merceron and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 11492 ( Angew. Chem. , 2011 , 123 , 11694 ) CrossRef CAS PubMed; (c) T. Agou, Y. Sugiyama, T. Sasamori, H. Sakai, Y. Furukawa, N. Takagi, J.-D. Guo, S. Nagase, D. Hashizume and N. Tokitoh, J. Am. Chem. Soc., 2012, 134, 4120 CrossRef CAS PubMed; (d) S. Inoue and C. Eisenhut, J. Am. Chem. Soc., 2013, 135, 18315 CrossRef CAS PubMed.
- (a) A. Jana, D. Leusser, I. Objartel, H. W. Roesky and D. Stalke, Dalton Trans., 2011, 40, 5458 RSC; (b) M. Y. Abraham, Y. Wang, Y. Xie, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2011, 133, 8874 CrossRef CAS PubMed; (c) S. M. I. Al-Rafia, A. C. Malcolm, R. McDonald, M. J. Ferguson and E. Rivard, Angew. Chem., Int. Ed., 2011, 50, 8354 ( Angew. Chem. , 2011 , 123 , 8504 ) CrossRef CAS PubMed; (d) M. Stoelzel, C. Präsang, S. Inoue, S. Enthaler and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 399 ( Angew. Chem. , 2012 , 124 , 411 ) CrossRef CAS PubMed; (e) S. M. I. Al-Rafia, A. C. Malcolm, R. McDonald, M. J. Ferguson and E. Rivard, Chem. Commun., 2012, 48, 1308 RSC; (f) S. M. I. Al-Rafia, R. McDonald, M. J. Ferguson and E. Rivard, Chem.–Eur. J., 2012, 18, 13810 CrossRef CAS PubMed; (g) B. Blom, S. Enthaler, S. Inoue, E. Irran and M. Driess, J. Am. Chem. Soc., 2013, 135, 6703 CrossRef CAS PubMed; (h) E. Rivard, Chem. Soc. Rev., 2016, 45, 989 RSC.
- S. Loss, A. Magistrato, L. Cataldo, S. Hoffmann, M. Geoffroy, U. Röthlisberger and H. Grützmacher, Angew. Chem., Int. Ed., 2001, 40, 723 (
Angew. Chem.
, 2001
, 113
, 749
) CrossRef CAS . The redox potential of [Mes*(Me)PPMes*]+versus the saturated calomel electrode (SCE) was deduced from this work (E1/2 in MeCN = −0.57 V) and converted to the [Fe(η5-C5Me5)2]+1/0 redox scale using the half-wave potential of the redox couple [Fe(η5-C5Me5)2]+1/0versus SCE (E1/2 in MeCN = −0.09 V) determined in our laboratory (see ESI†).
- The [Fe(η5-C5Me5)2]+1/0 was chosen as the reference standard for the CV experiments of 1H[B(ArF)4] owing to its favorable properties versus the [Fe(η5-C5H5)2]+1/0 redox couple: (a) I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, J. Phys. Chem. B, 1999, 103, 6713 CrossRef CAS; (b) J. R. Aranzaes, M.-C. Daniel and D. Astruc, Can. J. Chem., 2006, 84, 288 CrossRef.
- For comparison reasons, the half-wave potential of the [Fe(η5-C5H5)2]+1/0 (Fc+/Fc) redox couple was determined in C6H5F under the same conditions and found to be +0.520 V versus the redox couple [Fe(η5-C5Me5)2]+1/0.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- (a) T. Matsuno, M. Ichinohe and A. Sekiguchi, Angew. Chem., Int. Ed., 2002, 41, 1575 ( Angew. Chem. , 2002 , 114 , 1645 ) CrossRef CAS; (b) H. Maruyama, H. Nakano, M. Nakamoto and A. Sekiguchi, Angew. Chem., Int. Ed., 2014, 53, 1324 ( Angew. Chem. , 2014 , 126 , 1348 ) CrossRef CAS PubMed.
- The TdDFT calculations
suggest that the absorption bands of 1H centered at 608, 704 and 958 nm originate from several electronic transitions including those from the SiSi π* (HOMO(α)) and SiSi π (HOMO−1(α)) orbitals into antibonding π* orbitals of the N-bonded 2,6-diisopropylphenyl substituents (for details see ESI†). The SOMO → LUMO transition is predicted to give rise to a band at 1295 nm.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069 CrossRef CAS PubMed.
- SiSi bond lengths range between 2.118(1)–2.2700(5) Å: T. Iwamoto and S. Ishida, Struct. Bonding, 2014, 156, 125 CrossRef CAS.
- A. F. Holleman, E. Wiberg and N. Wiberg, Inorganic Chemistry, Academic Press, San Diego/London, 2001 Search PubMed; A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, 102. Auflage, deGruyter, Berlin, 2007 Search PubMed.
- (a) S. P. Hoffmann, T. Kato, F. S. Tham and C. A. Reed, Chem. Commun., 2006, 767 RSC; (b) A. Y. Khalimon, Z. H. Lin, R. Simionescu, S. F. Vyboishchikov and G. I. Nikonov, Angew. Chem., Int. Ed., 2007, 46, 4530 ( Angew. Chem. , 2007 , 119 , 4614 ) CrossRef CAS PubMed; (c) N. Kordts, C. Borner, R. Panisch, W. Saak and T. Müller, Organometallics, 2014, 33, 1492 CrossRef CAS.
- S. Inoue, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2008, 130, 6078 CrossRef CAS PubMed.
- (a) M. Kira and T. Iwamoto, J. Organomet. Chem., 2000, 611, 236 CrossRef CAS; (b) A. Sekiguchi, S. Inoue, M. Ichinohe and Y. Arai, J. Am. Chem. Soc., 2004, 126, 9626 CrossRef CAS PubMed; (c) A. Tsurusaki and S. Kyushin, Chem.–Eur. J., 2016, 22, 134 CrossRef CAS PubMed.
- ORCA 3.0.0: (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS; B3LYP functional: (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; 6-311G**/6-31G* basis sets: (d) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS; B97-D3 functional: (e) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed; (f) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; RI-JCOSX approximation: (g) F. Neese, J. Comput. Chem., 2003, 24, 1740 CrossRef CAS PubMed; (h) F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98 CrossRef CAS; def2-TZVP basis set: (i) A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef; (j) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; NBO 3.1 program: (k) E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833 CrossRef CAS.
- The higher spin density at the Si2 atom in 1H can be rationalized considering the polarization of the SiSi π-orbital in 1H+ towards the Si1 atom due to the hydride substituent. This leads to a reversed polarization of the SiSi π*-orbital in 1H+ with a higher contribution
at the Si2 atom, which upon population with one electron gives rise to a higher spin density at Si2 in 1H.
- (a) A. Gansäuer, L. Shi, M. Otte, I. Huth, A. Rosales, I. Sancho-Sanz, N. M. Padial and J. E. Oltra, Top. Curr. Chem., 2012, 320, 93 CrossRef; (b) A. Simonneau and M. Oestreich, Angew. Chem., Int. Ed., 2015, 54, 3556 ( Angew. Chem. , 2015 , 127 , 3626 ) CrossRef CAS PubMed and refs. therein.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammetric studies of 1H[B(ArF)4]; synthesis, analytical data and illustrations of the IR and UV-Vis spectra of 1H; details of the magnetic susceptibility measurements and single crystal X-ray diffraction analysis of 1H; details of the EPR spectroscopic measurements and illustrations of the EPR spectra of 1H; details of the quantum chemical calculations. CCDC 1471165. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01569g |
| This journal is © The Royal Society of Chemistry 2016 |