
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guest and solvent modulated photo-driven charge separation and triplet generation in a perylene bisimide cyclophane†
Peter
Spenst
a,
Ryan M.
Young
b,
Michael R.
Wasielewski
*b and
Frank
Würthner
*a
aInstitut für Organische Chemie and Center for Nanosystems Chemistry, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: wuerthner@chemie.uni-wuerzburg.de
bDepartment of Chemistry and Argonne-Northwestern Solar Energy Research (ANSER) Center, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208-3113, USA. E-mail: m-wasielewski@northwestern.edu
First published on 18th May 2016
Abstract
Cofacial positioning of two perylene bisimide (PBI) chromophores at a distance of 6.5 Å in a cyclophane structure prohibits the otherwise common excimer formation and directs photoexcited singlet state relaxation towards intramolecular symmetry-breaking charge separation (τCS = 161 ± 4 ps) in polar CH2Cl2, which is thermodynamically favored with a Gibbs free energy of ΔGCS = −0.32 eV. The charges then recombine slowly in τCR = 8.90 ± 0.06 ns to form the PBI triplet excited state, which can be used subsequently to generate singlet oxygen in 27% quantum yield. This sequence of events is eliminated by dissolving the PBI cyclophane in non-polar toluene, where only excited singlet state decay occurs. In contrast, complexation of electron-rich aromatic hydrocarbons by the host PBI cyclophane followed by photoexcitation of PBI results in ultrafast electron transfer (<10 ps) from the guest to the PBI in CH2Cl2. The rate constants for charge separation and recombination increase as the guest molecules become easier to oxidize, demonstrating that charge separation occurs close to the peak of the Marcus curve and the recombination lies far into the Marcus inverted region.
Introduction
The precise positioning of the light absorbing chlorophylls and their associated redox cofactors in photosynthetic reaction center proteins using weak metal–ligand and hydrogen bonds results in optimized electronic interactions between them that result in efficient charge separation to form radical ion pairs (RPs).1–4 For example, photoexcitation of the chlorophyll special pair dimer in some reaction center proteins leads to symmetry-breaking charge separation.5 There is a long and rich history involving the design, synthesis, and characterization of covalent electron donor–acceptor systems that model the electron transfer (ET) processes within reaction center proteins.6–12 In general, however, efforts to employ non-covalent molecular interactions and self-assembly strategies to understand photo-driven charge separation have had less emphasis.3,13–16One factor thus far limiting studies of non-covalent supramolecular ensembles has been the lack of suitable dye-based hosts bearing sufficiently large cavities to complex redox-active guest molecules. In our recent work we approached this challenge with a cyclic perylene bisimide (PBI) trimer for which biomimetic intramolecular symmetry-breaking charge separation (SB-CS)12,17 could be observed in τCS = 12 ps, although the free energy of charge separation, ΔGCS, is barely negative.17 The charge recombination (CR) back to the PBI ground state (GS) is much slower (τCR = 1.12 ns), despite the larger ΔGCR of this process. According to Marcus theory,18–20 ET can occur in the normal region (−ΔGET < λ) or in the inverted region (−ΔGET > λ) depending on the relative magnitudes of the Gibbs free energy (−ΔGET) and the reorganization energy (λ).6,21,22 The PBI trimer data imply that its charge recombination reaction is in the Marcus inverted region.18–20,23,24 Unfortunately, no guest encapsulation could be achieved within the PBI trimer, in contrast to the PBI cyclophane, Cy-PBI, whose fluorescence is quenched by the encapsulation of electron rich aromatic guests.25 Accordingly, in the present study we elucidate the electronic interactions of Cy-PBI and its corresponding host–guest complexes by steady-state absorption, fluorescence and transient absorption (TA) spectroscopy to identify the ET processes and the individual lifetimes of the states formed. While Cy-PBI itself fluoresces strongly in low-polarity toluene, it undergoes intramolecular SB-CS followed by CR to the PBI triplet state upon excitation in polar solvents like CH2Cl2 (Fig. 1). Binding of electron-rich guests within Cy-PBI leads to intermolecular CS followed by CR back to the GS. Thus, the lowest excited singlet state of Cy-PBI decays in a complex sequence of events that can be modulated by solvent polarity or the presence of electron-rich guest molecules.
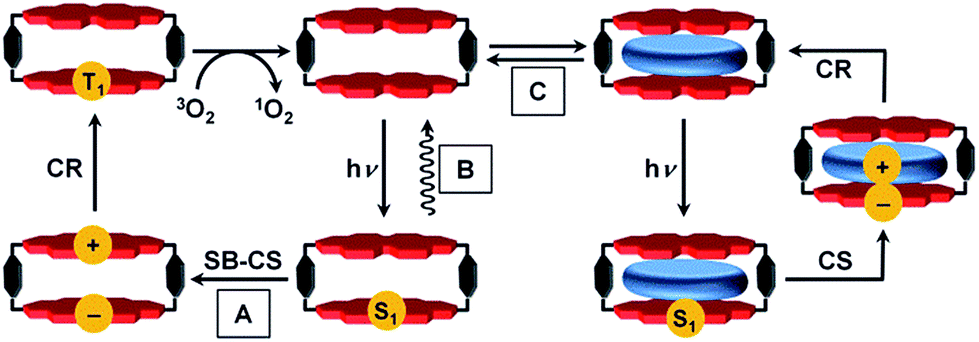 | ||
| Fig. 1 Schematic of the different excited state photophysics upon excitation; (A) symmetry-breaking charge separation and recombination to the PBI triplet within the PBI cyclophane Cy-PBI, in CH2Cl2 that can be used for singlet oxygen generation; (B) emission in toluene; (C) encapsulation of aromatic guests and photo-driven charge separation between guest and Cy-PBI and charge recombination to the ground state. | ||
Results and discussion
Steady state spectroscopy and electrochemistry
The steady-state UV-vis absorption spectrum of Cy-PBI has its maximum at 582 nm in CH2Cl2, which is comparable to the tetraphenoxy-substituted monomeric PBI (Ref-PBI). Similar to other multi-chromophoric PBI systems, the 0–1 vibronic band of Cy-PBI at 543 nm is significantly enhanced with respect to the 0–0 transition that can be related to excitonic coupling of the two cofacial PBI units (Fig. 2a).17,26,27 The corresponding fluorescence spectrum has its maximum at 627 nm, which is bathochromically shifted by 12 nm and has a significantly quenched 7% fluorescence quantum yield compared to Ref-PBI (ϕFl(Ref-PBI) = 75% in CH2Cl2), indicative of an efficient nonradiative decay process. No long-lived and red-shifted excimer-like emission26,27 was observed, which is attributed to the stiff linkage and the relatively long, 6.5 Å interplanar distance between the two PBI chromophores.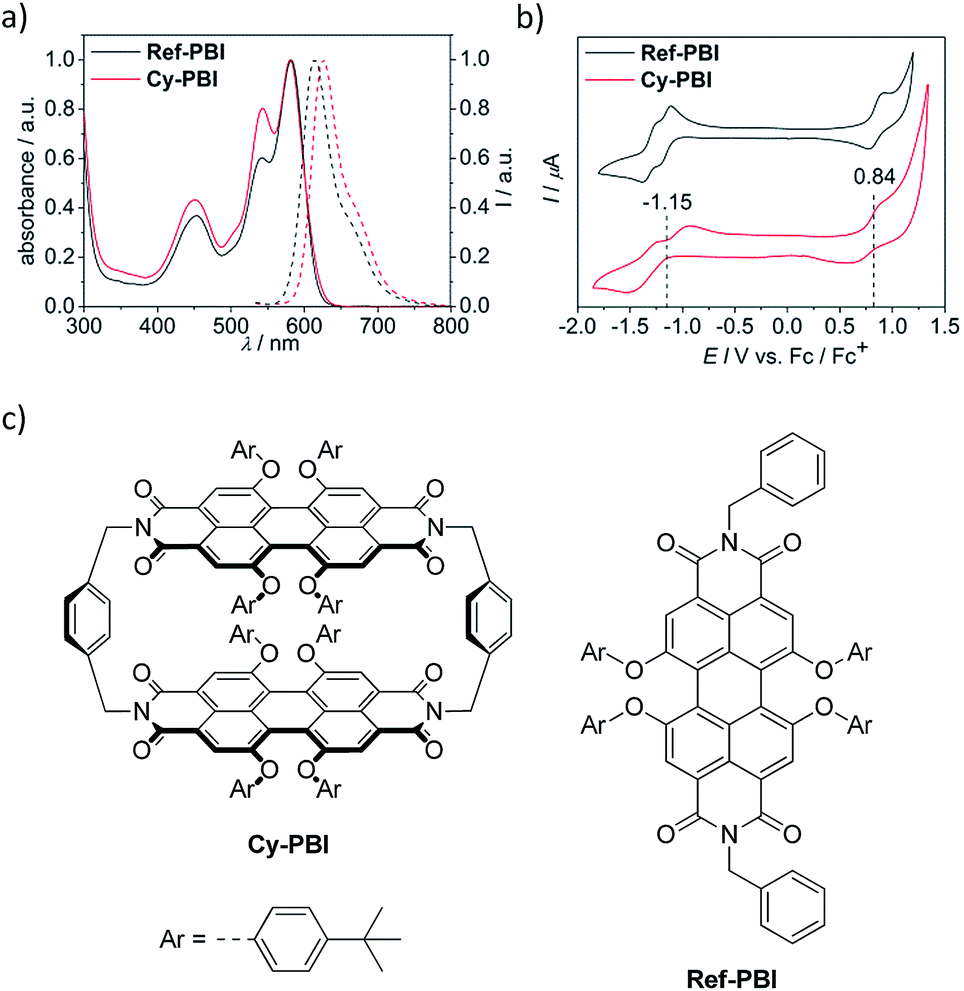 | ||
| Fig. 2 Normalized UV-vis absorption (solid line) and fluorescence (dashed line) spectra (a) of Cy-PBI and Ref-PBI (CH2Cl2, RT, c = 5 × 10−5 M); cyclic voltammograms (b) in CH2Cl2 (reference electrode: Ag/AgCl, working and auxiliary electrode: Pt; 0.1 M TBAHFP, Fc+/Fc, RT, c ∼ 2 × 10−4 M) and chemical structures (c) of Cy-PBI and Ref-PBI. | ||
To calculate the free energy ΔGCS we performed cyclic voltammetry on Cy-PBI and Ref-PBI in dry CH2Cl2 under argon (Fig. 2b). Cy-PBI shows similar behaviour to the monomer with two reversible reduction and one reversible oxidation waves, which are slightly cathodically shifted by 20 mV compared to Ref-PBI. The broadening of the Cy-PBI voltammogram can be related to multi-electron processes.26 To quantify ΔGCS and ΔGCR for the cyclophane we applied the Weller equation (eqn (1) and (2)):28
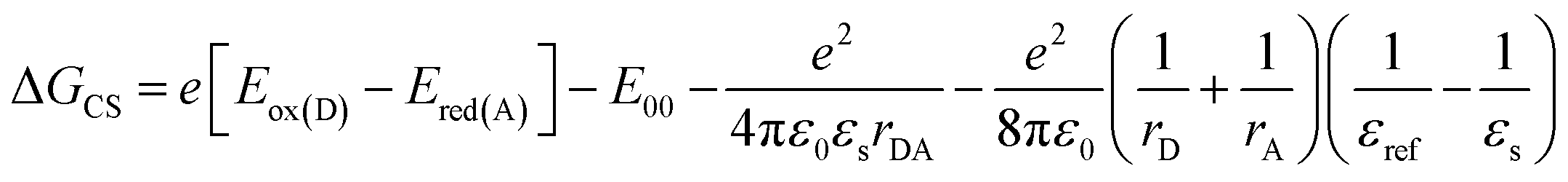 | (1) |
| ΔGCR = −(ΔGCS + E00) | (2) |
Transient absorption spectroscopy of the free host Cy-PBI
To elucidate the excited state dynamics in Cy-PBI, we performed femtosecond (fs) and nanosecond (ns) TA studies (Fig. 3, S3 and S4† in the ESI†). The fs TA spectra (Fig. 3a) show the ground state bleach (GB) at 461, 543 and 583 nm and the stimulated emission (SE) at 611 and 664 nm as shoulders in the spectra. The excited singlet state 1*PBI absorption (ESA) has a positive signal at 710 nm and two characteristic strong maxima in the NIR region at 959 and 1035 nm. While the TA spectra and high fluorescence quantum yield for 1*Ref-PBI indicate that it decays back to the ground state primarily by emission (Fig. S2†), Cy-PBI shows very different excited state dynamics with a fast decay of the 1*PBI state in 161 ± 4 ps to a new transient species. Here the SE and ESA signals fully disappear, while new bands arise in the visible region at 486 and 628 nm, in the NIR region at 797, 993 and 1100 nm with a broad feature at ∼1220 nm.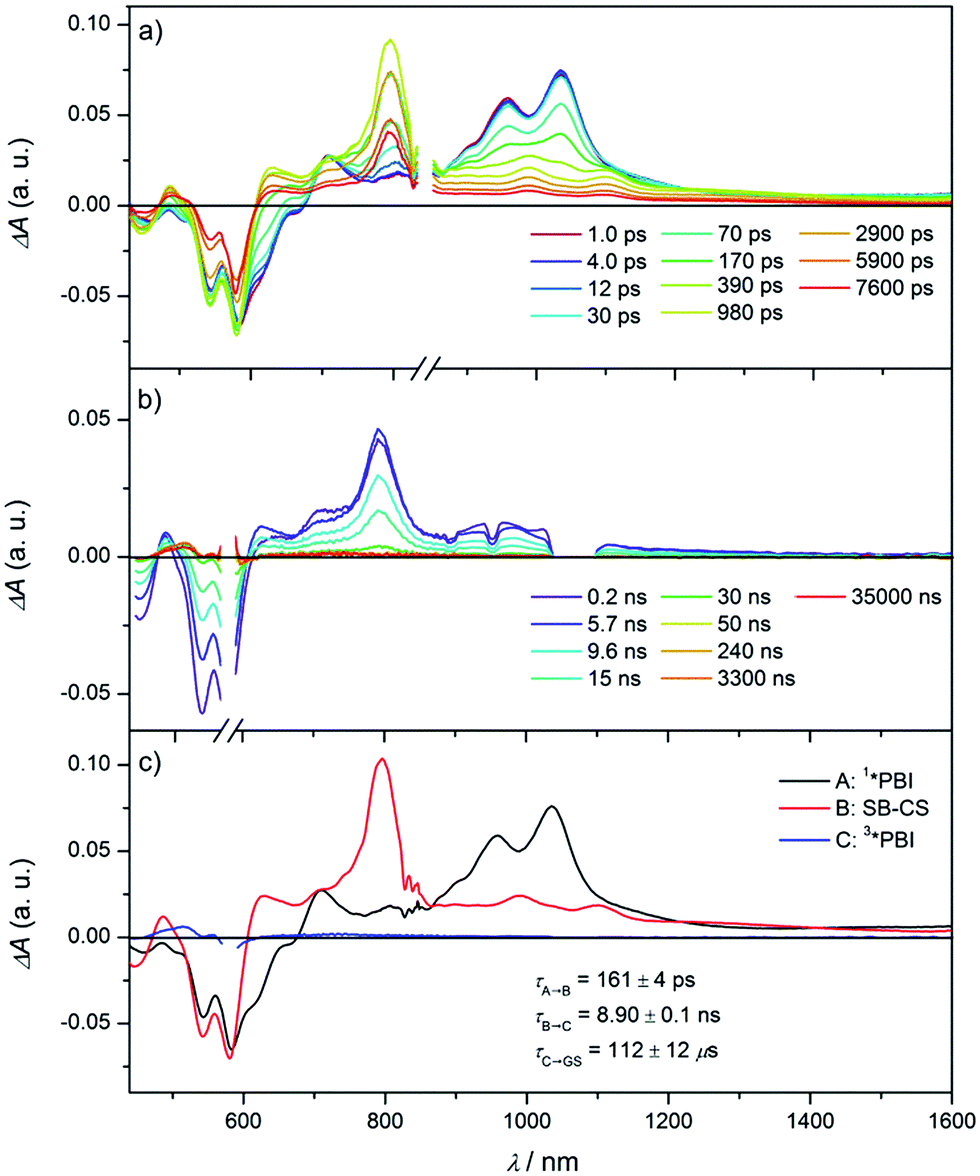 | ||
| Fig. 3 Femtosecond (a) and nanosecond (b) transient absorption spectra of Cy-PBI in CH2Cl2 showing the excited state dynamics after photoexcitation. Species-associated spectra (c) reconstructed from global fits to the sequential A → B → C → ground state model, where A is 1*PBI, B is SB-CS state and C is 3*PBI (λex = 580 nm, 1.0 μJ per pulse, CH2Cl2, 298 K, degassed for ns TA). The C → ground state time was determined from ns TA. | ||
By comparison to the PBI radical cation and anion absorption spectra these bands can clearly be attributed to PBI˙+ (486, 628, 1220 nm) and PBI˙− (797, 993, 1100 nm).17,29 From these ultrafast transient dynamics data we conclude that photo-driven intramolecular SB-CS occurs in Cy-PBI with a high quantum yield.
Most interestingly and presumably caused by the long distance between the PBI moieties, CR between PBI˙+ and PBI˙− in Cy-PBI occurs only slowly with 8.90 ± 0.06 ns to produce a significant yield of the PBI triplet state (3*PBI) characterized by positive absorptions at 515 and 556 nm, a bleach at 579 nm and a weak positive and broad absorption at 734 nm (Fig. 3b).30
Triplet formation mechanism
Spin–orbit induced intersystem crossing (SO-ISC) is slow in PBIs, leading to very low triplet quantum yields for common PBIs (<1%).31–33 Since formation of PBI+˙–PBI−˙ is a prerequisite for populating 3*PBI, either spin–orbit charge transfer intersystem crossing (SOCT-ISC) or radical pair intersystem crossing (RP-ISC) are responsible for 3*PBI formation.30 The SOCT-ISC mechanism requires large changes in orbital angular momentum upon formation of PBI+˙–PBI−˙, which would require the π systems of the two PBI molecules to be nearly orthogonal,34–36 which is not the case in Cy-PBI. In contrast, the RP-ISC mechanism requires a relatively weak magnetic interaction between the two PBI radicals within PBI+˙–PBI−˙.37,38 Photoexcitation of Cy-PBI produces 1(PBI+˙–PBI−˙), whose spin dynamics depend strongly on the isotropic spin–spin exchange interaction, 2J = ES − ET, where ES and ET are the energies of 1(PBI+˙–PBI−˙) and 3(PBI+˙–PBI−˙), respectively.39 Due to the long through-space and through-bond distances between the PBI subunits, 2J for PBI+˙–PBI−˙ should be small enough to enable RP-ISC of 1(PBI+˙–PBI−˙) to 3(PBI+˙–PBI−˙).37,38 Moreover, since 2J ∝ V2,39 and kET ∝ V2 (eqn (3)), the relatively long 8.90 ns CR time is also consistent with a small value of 2J. The subsequent CR process is spin selective in that 1(PBI+˙–PBI−˙) recombines back to the singlet ground state, whereas 3(PBI+˙–PBI−˙) recombines to 3*PBI within Cy-PBI.40 Our experimental findings, thus, indicate that RP-ISC is the most likely mechanism producing 3*PBI within Cy-PBI. Unfortunately, the 8.90 ns PBI+˙–PBI−˙ lifetime is too short to observe this RP directly by time-resolved EPR spectroscopy.The lifetime of 3*PBI within Cy-PBI is very long (τ ≥ 112 μs) in a degassed solution at room temperature. Isolating Cy-PBI in a glassy solvent matrix is necessary to prohibit quenching by diffusion; however, this also prohibits the SB-CS process and thus, the intrinsic triplet lifetime could not be investigated. To estimate the quantum yield for the triplet formation, singlet oxygen emission was measured and compared to that of a methylene blue standard (MB).41 From this experiment, the singlet oxygen quantum yield, ϕΔ = 0.27, which also serves as the lower limit of the 3*PBI yield, and is consistent with the weak triplet signal in the TA spectra (Fig. 3c). This result and the low fluorescence quantum yield indicate that the main pathway back to the ground state is by singlet RP recombination. Furthermore, no photobleaching of Cy-PBI with singlet oxygen was observed, verifying the great photostability of PBIs against oxidation. The SB-CS process is disfavoured in non-polar solvents such as toluene as evidenced by the increase in Cy-PBI fluorescence quantum yield to 64%.42 Consistent with the increased emission, the transient absorption spectra of Cy-PBI in toluene show only singlet excited state decay directly back to the GS in τ = 4.5 ± 0.6 ns without the population of other transient species (Fig. S5†).
Transient absorption spectroscopy of the host–guest complexes
By adding electron-rich guests, such as carbazole, pyrene, anthracene, and perylene to Cy-PBI, host–guest complexes are formed, leading to a slight bathochromic shift of the Cy-PBI absorption maximum and the appearance of a new band at longer wavelength that can be attributed to a charge transfer transition. Furthermore, the PBI fluorescence is almost fully quenched in the presence of these guests (Fig. S1†).25 The oxidation potentials of carbazole, pyrene, anthracene, and perylene are 0.64,43 0.91,44 0.88,45 and 0.59 V46vs. Fc+/Fc, respectively. Using these data, eqn (1), and the calculated 0.35 nm PBI–guest distance in the complex, we calculate ΔGCS = −0.73, −0.46, −0.49 and −0.78 eV, respectively, and ΔGCR = −1.33, −1.60, −1.57 and −1.28 eV for the host–guest complexes (guest@Cy-PBI), which clearly show that the electron transfer processes in the complexes are highly favored thermodynamically.In the fsTA spectra, an ultrafast CS components of τCS = 6.7 ± 0.2, 3.6 ± 0.3, 1.1 ± 0.2 and 0.9 ± 0.1 ps were observed for Cy-PBI complexed with carbazole, pyrene, anthracene and perylene, respectively (Fig. 4 and S6–S9†). The fsTA spectra of the perylene@Cy-PBI complex in Fig. 4 show the PBI−˙ absorptions along with a strong positive absorption at 542 nm that can be attributed to perylene+˙.47 The radical cation features of the other hydrocarbons are much weaker in the observed spectral window and strongly overlap with the GSB and PBI˙− absorption changes, and were thus not observed. However, no PBI+˙ bands were detected, confirming that the CS now takes place between Cy-PBI and the guest molecule alone.
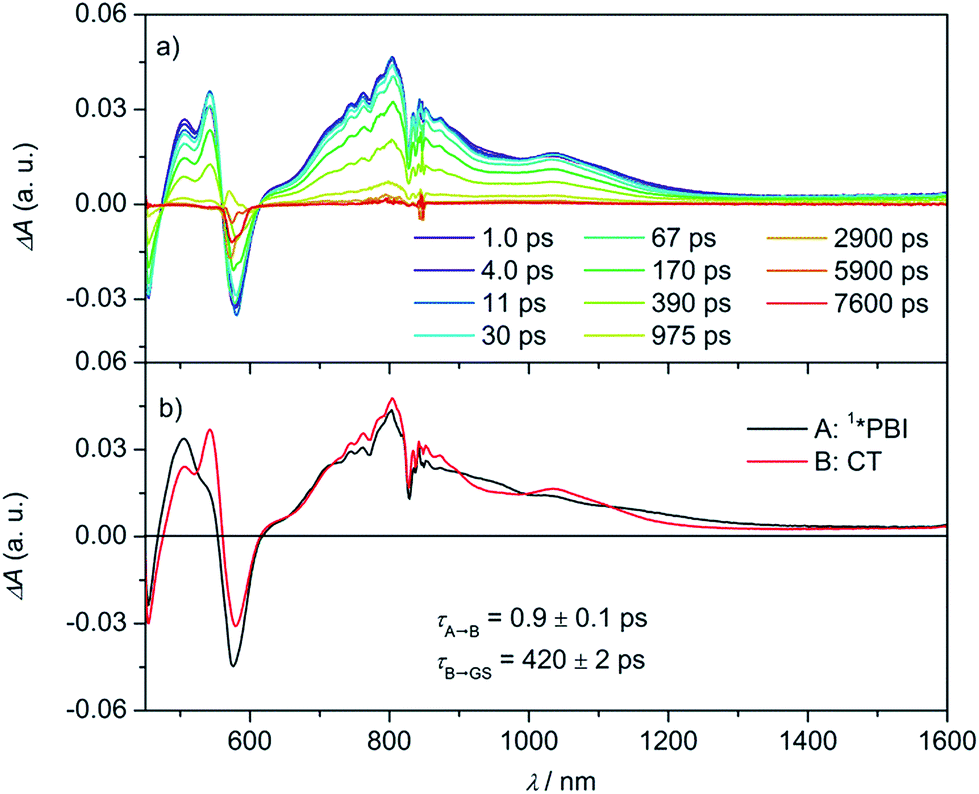 | ||
| Fig. 4 Femtosecond (a) transient absorption spectra of the perylene@Cy-PBI complex showing the excited state dynamics after photoexcitation. Species-associated spectrum (b) reconstructed from global fits to the sequential A → B → ground state (GS) model, where A is 1*PBI and B is CT state (λex = 580 nm, 1.0 μJ per pulse, CH2Cl2, 298 K, air equilibrated). | ||
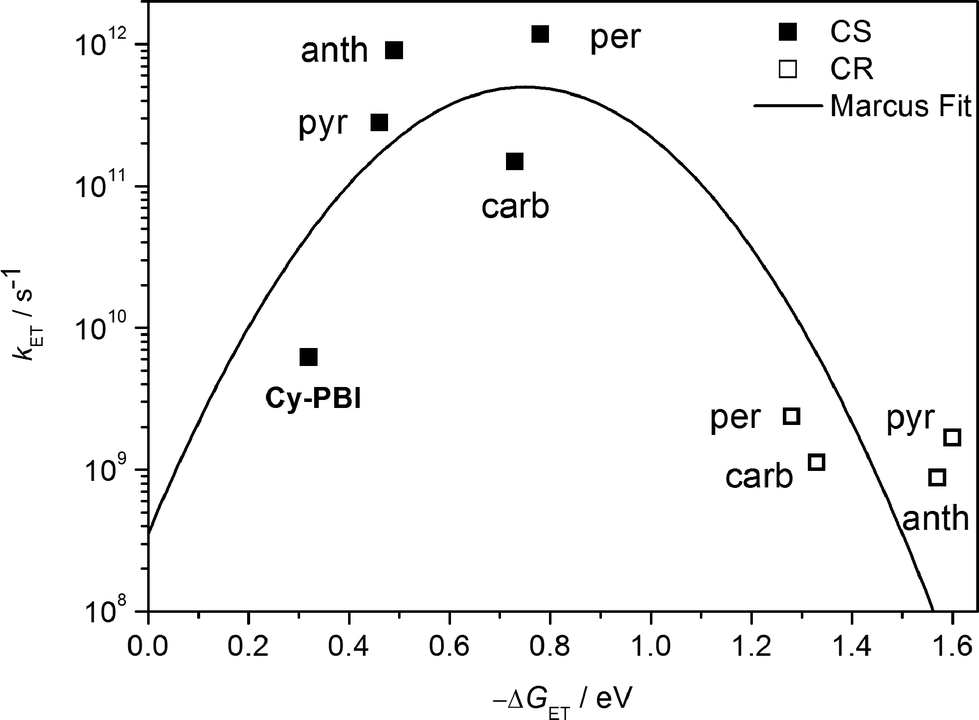 | ||
| Fig. 5 Plot of electron transfer rate constants in the PBI cyclophane and the corresponding host–guest complexes vs. the thermodynamic driving force (−ΔGET) for charge separation (solid squares) and charge recombination (open squares); the line represents the fit according to eqn (3) with λ = 0.75 eV and V = 15 cm−1. | ||
The ultrafast charge separation processes in the complexes are close to our detection limit (∼200 fs); thus the spectra of 1*PBI are difficult to observe for the perylene and anthracene complexes, but are clearer for pyrene and carbazole. The second time constant gives the CR lifetime directly back to the GS of PBI without any indication of triplet formation. This is consistent with a large 2J in the guest+˙–PBI˙− RP, which precludes RP-ISC. The corresponding CR times using the carbazole, pyrene, anthracene and perylene guests are τCR = 892 ± 46, 593 ± 42, 1140 ± 120 to 420 ± 2 ps, respectively.
The data show that the CS rate increases with increasing ΔGCS for ET from the HOMO of the respective electron-donating guest to the photo-excited electron-accepting PBI (Fig. 5). Using τCS and τCR obtained from the transient absorption kinetics and the corresponding values of ΔGCS and ΔGCR, the total reorganization energy λ = λS + λI, where λS and λI are the solvent and internal reorganization energies, respectively, and the electronic coupling matrix element V can be calculated according to Marcus theory by applying eqn (3):19
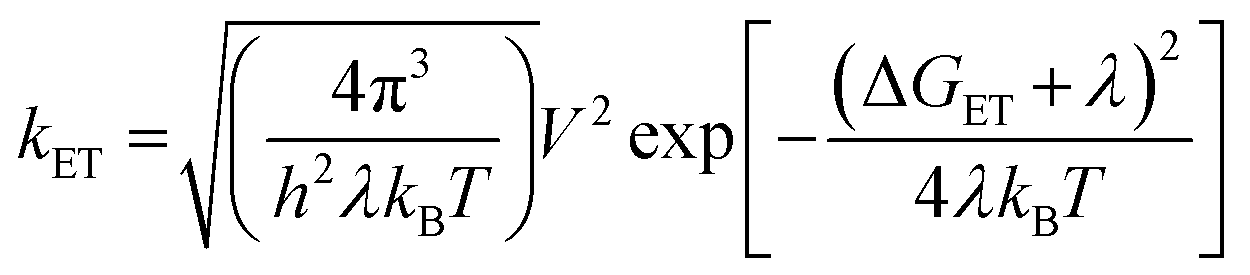 | (3) |
Conclusions
In summary, we have shown that the PBI cyclophane Cy-PBI undergoes intramolecular symmetry-breaking charge separation and slow charge recombination, which is accompanied by RP-ISC leading to 3*PBI that can be used to generate singlet oxygen with a 27% quantum yield.Since 3*PBI is not accessible by conventional SO-ISC,48 the RP-ISC pathway to 3*PBI offers the possibility of developing an entirely new set of PBI applications, as demonstrated here with singlet oxygen generation. In contrast, the CS reaction within Cy-PBI is endergonic in a non-polar solvent like toluene, resulting in a high Cy-PBI fluorescence quantum yield. Binding electron-rich guest molecules within the Cy-PBI host affords a complete change of the photoexcited state relaxation pathway leading to intermolecular charge separation within a few picoseconds with formation of the radical cation of the guest and the PBI radical anion. Our findings show that the PBI cyclophane is indeed a special dye pair whose excited state properties are effectively modulated by its solvent environment as well as host–guest complex formation with electron donors.
Experimental methods
Synthesis
The tetraphenoxy-substituted perylene bisimide cyclophane (Cy-PBI) and the monomeric reference compound (Ref-PBI) were prepared according to literature.25Steady-state spectroscopy
UV-vis absorption spectroscopy was performed on a Perkin-Elmer Lambda 35 or Lambda 950 spectrometer. Solvents for spectroscopic studies were of spectroscopic grade and used without further purification. Fluorescence spectroscopy was performed on a PTI QM-4/2003 spectrofluorimeter. The fluorescence quantum yields were determined by optical dilution method49 (ODmax < 0.05) as the average value of four different excitation wavelengths using N,N′-(2,6-di-iso-propylphenyl)-1,6,7,12-tetraphenoxyperylen-3,4:9,10-tetracarboxylic acid bisimide (ϕfl = 0.96 in chloroform) as reference. Singlet oxygen emission was recorded on a PTI spectrofluorimeter. The quantum yield of singlet oxygen was determined in an air-equilibrated solution of Cy-PBI in dichloromethane (ODmax ∼ 0.5) as the average value of four different excitation wavelength using methylene blue as reference (ϕΔ = 0.57 in dichloromethane).41Electrochemistry
Cyclic voltammetry was carried out with a standard commercial electrochemical analyzer (EC epsilon; BAS Instruments, UK) in a three electrode single-compartment cell. The supporting electrolyte tetrabutylammonium hexafluorophosphate (TBAHFP) was recrystallized from ethanol/water. As an internal standard for the calibration of the potential ferrocenium/ferrocene (Fc+/Fc) was used. As reference electrode Ag/AgCl and as working and auxiliary electrodes a Pt disc and a Pt wire were used.Transient absorption spectroscopy
Femtosecond and nanosecond transient absorption experiments were performed using an instrument as previously described50 with an approximately 120 fs output of a commercial Ti:sapphire oscillator/amplifier (Tsunami/Spitfire, Spectra-Physics) that was split to seed and pump a laboratory-constructed optical parametric amplifier used to generate the 569 nm excitation (“pump”) beam and a femtosecond continuum probe, by using a 3 mm sapphire plate for the visible range or a proprietary crystal for the near-infrared (NIR) spectral region (Ultrafast Systems, LLC). Transient spectra were collected by using customized commercial detectors (Helios, Ultrafast Systems, LLC). Experiments were performed with a depolarized pump to eliminate contributions from orientational dynamics. The kinetic analysis is based on a global fit to selected single-wavelength kinetics. Several kinetic traces at different wavelengths were chosen and fitted globally to a kinetic model. The differential equations were solved and then convoluted with the instrument response function, before employing a least-square fitting to find the parameters which result in matches to the same functions for all selected wavelengths (MATLAB). These parameters are then fed directly into the differential equations, which were solved for the populations of the states in model. Finally, the raw data matrix (with all the raw data) is deconvoluted with the populations as functions of time to produce the species-associated spectra.Molecular modelling
DFT calculations were performed by using the Gaussian 09 program package51 with B3-LYP52–54 as functional and 6-31+G* as basis set. The structures were geometry optimized, followed by frequency calculations on the optimized structures which confirmed the existence of a minimum (one very small imaginary frequency of 4i cm−1 was obtained for Cy-PBI. Small imaginary frequencies (<100i cm−1) are considered most likely to be an artefact of the calculation instead of an indication of a transition state55).Acknowledgements
This work has been supported by the DFG in the framework of the research training group FOR 1809 at the Universität Würzburg. Work at Northwestern University was supported by the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, US DOE under grant no. DE-FG02-99ER14999 (M. R. W.).Notes and references
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2–28 CrossRef PubMed.
- R. Bhosale, J. Misek, N. Sakai and S. Matile, Chem. Soc. Rev., 2010, 39, 138–149 RSC.
- P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- D. Noy, C. C. Moser and P. L. Dutton, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 90–105 CrossRef CAS PubMed.
- M. R. Wasielewski, M. P. Niemczyk, W. A. Svec and E. B. Pewitt, J. Am. Chem. Soc., 1985, 107, 1080–1082 CrossRef CAS.
- T. Kircher and H. G. Löhmannsröben, Phys. Chem. Chem. Phys., 1999, 1, 3987–3992 RSC.
- J. M. Giaimo, A. V. Gusev and M. R. Wasielewski, J. Am. Chem. Soc., 2002, 124, 8530–8531 CrossRef CAS PubMed.
- D. I. Schuster, P. Cheng, P. D. Jarowski, D. M. Guldi, C. Luo, L. Echegoyen, S. Pyo, A. R. Holzwarth, S. E. Braslavsky, R. M. Williams and G. Klihm, J. Am. Chem. Soc., 2004, 126, 7257–7270 CrossRef CAS PubMed.
- J. M. Baumes, J. J. Gassensmith, J. Giblin, J.-J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025–1030 CrossRef CAS PubMed.
- C. C. Hofmann, S. M. Lindner, M. Ruppert, A. Hirsch, S. A. Haque, M. Thelakkat and J. Köhler, J. Phys. Chem. B, 2010, 114, 9148–9156 CrossRef CAS PubMed.
- M. T. Whited, N. M. Patel, S. T. Roberts, K. Allen, P. I. Djurovich, S. E. Bradforth and M. E. Thompson, Chem. Commun., 2012, 48, 284–286 RSC.
- M. H. Schwartz, J. Inclusion Phenom. Mol. Recognit. Chem., 1990, 9, 1–35 CrossRef CAS.
- M. Brouwer Albert, M. Fazio Sandro, C. Frochot, G. Gatti Francesco, A. Leigh David, K. Y. Wong Jenny and W. H. Wurpel George, Pure Appl. Chem., 2003, 75, 1055–1060 Search PubMed.
- D. M. Bassani, L. Jonusauskaite, A. Lavie-Cambot, N. D. McClenaghan, J.-L. Pozzo, D. Ray and G. Vives, Coord. Chem. Rev., 2010, 254, 2429–2445 CrossRef CAS.
- M. Kumar, K. V. Rao and S. J. George, Phys. Chem. Chem. Phys., 2014, 16, 1300–1313 RSC.
- Y. Wu, R. M. Young, M. Frasconi, S. T. Schneebeli, P. Spenst, D. M. Gardner, K. E. Brown, F. Würthner, J. F. Stoddart and M. R. Wasielewski, J. Am. Chem. Soc., 2015, 137, 13236–13239 CrossRef CAS PubMed.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1965, 43, 679–701 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef CAS.
- J. R. Miller, L. T. Calcaterra and G. L. Closs, J. Am. Chem. Soc., 1984, 106, 3047–3049 CrossRef CAS.
- E. H. A. Beckers, S. C. J. Meskers, A. P. H. J. Schenning, Z. Chen, F. Würthner and R. A. J. Janssen, J. Phys. Chem. A, 2004, 108, 6933–6937 CrossRef CAS.
- T. Häberle, J. Hirsch, F. Pöllinger, H. Heitele, M. E. Michel-Beyerle, C. Anders, A. Döhling, C. Krieger, A. Rückermann and H. A. Staab, J. Phys. Chem., 1996, 100, 18269–18274 CrossRef.
- A. Osuka, G. Noya, S. Taniguchi, T. Okada, Y. Nishimura, I. Yamazaki and N. Mataga, Chem.–Eur. J., 2000, 6, 33–46 CrossRef CAS.
- P. Spenst and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 10165–10168 CrossRef CAS PubMed.
- F. Schlosser, M. Moos, C. Lambert and F. Würthner, Adv. Mater., 2013, 25, 410–414 CrossRef CAS PubMed.
- C. M. Pochas, K. A. Kistler, H. Yamagata, S. Matsika and F. C. Spano, J. Am. Chem. Soc., 2013, 135, 3056–3066 CrossRef CAS PubMed.
- A. Weller, Z. Phys. Chem., 1982, 133, 93–98 CrossRef CAS.
- F. Würthner, A. Sautter, D. Schmid and P. J. A. Weber, Chem.–Eur. J., 2001, 7, 894–902 CrossRef.
- D. Veldman, S. M. A. Chopin, S. C. J. Meskers, M. M. Groeneveld, R. M. Williams and R. A. J. Janssen, J. Phys. Chem. A, 2008, 112, 5846–5857 CrossRef CAS PubMed.
- W. E. Ford and P. V. Kamat, J. Phys. Chem., 1987, 91, 6373–6380 CrossRef CAS.
- A. A. Rachford, S. Goeb and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 2766–2767 CrossRef CAS PubMed.
- Y. Wu, Y. Zhen, Y. Ma, R. Zheng, Z. Wang and H. Fu, J. Phys. Chem. Lett., 2010, 1, 2499–2502 CrossRef CAS.
- M. A. El-Sayed, J. Chem. Phys., 1974, 60, 4502–4507 CrossRef CAS.
- T. Okada, I. Karaki, E. Matsuzawa, N. Mataga, Y. Sakata and S. Misumi, J. Phys. Chem., 1981, 85, 3957–3960 CrossRef CAS.
- K. M. Lefler, K. E. Brown, W. A. Salamant, S. M. Dyar, K. E. Knowles and M. R. Wasielewski, J. Phys. Chem. A, 2013, 117, 10333–10345 CrossRef CAS PubMed.
- C. D. Buckley, D. A. Hunter, P. J. Hore and K. A. McLauchlan, Chem. Phys. Lett., 1987, 135, 307–312 CrossRef CAS.
- M. T. Colvin, A. B. Ricks, A. M. Scott, A. L. Smeigh, R. Carmieli, T. Miura and M. R. Wasielewski, J. Am. Chem. Soc., 2011, 133, 1240–1243 CrossRef CAS PubMed.
- E. A. Weiss, M. J. Ahrens, L. E. Sinks, A. V. Gusev, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 5577–5584 CrossRef CAS PubMed.
- E. A. Weiss, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2003, 107, 3639–3647 CrossRef CAS.
- Y. Usui, Chem. Lett., 1973, 2, 743–744 CrossRef.
- N. Banerji, G. Angulo, I. Barabanov and E. Vauthey, J. Phys. Chem. A, 2008, 112, 9665–9674 CrossRef CAS PubMed.
- K. Karon and M. Lapkowski, J. Solid State Electrochem., 2015, 19, 2601–2610 CrossRef CAS.
- J. Daub, R. Engl, J. Kurzawa, S. E. Miller, S. Schneider, A. Stockmann and M. R. Wasielewski, J. Phys. Chem. A, 2001, 105, 5655–5665 CrossRef CAS.
- V. D. Parker and O. Hammerich, Acta Chem. Scand., Ser. B, 1977, 31, 883–889 CrossRef.
- B. S. Veldkamp, W.-S. Han, S. M. Dyar, S. W. Eaton, M. A. Ratner and M. R. Wasielewski, Energy Environ. Sci., 2013, 6, 1917–1928 CAS.
- V. V. Danilov, A. S. Panfutova, A. I. Khrebtov, S. Ambrosini and D. A. Videnichev, Opt. Lett., 2012, 37, 3948–3950 CrossRef CAS PubMed.
- F. N. Castellano, Dalton Trans., 2012, 41, 8493–8501 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, New York, 2nd edn, 1999 Search PubMed.
- R. M. Young, S. M. Dyar, J. C. Barnes, M. Juríček, J. F. Stoddart, D. T. Co and M. R. Wasielewski, J. Phys. Chem. A, 2013, 117, 12438–12448 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery JrJ. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- E. A. Cobar, R. Z. Khaliullin, R. G. Bergman and M. Head-Gordon, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6963–6968 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including steady-state and transient absorption spectera as described in the text. See DOI: 10.1039/c6sc01574c |
| This journal is © The Royal Society of Chemistry 2016 |