
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of chiral β-alkynyl carbonyl and sulfonyl derivatives via sequential palladium and copper catalysis†
Barry M.
Trost
*,
James T.
Masters
,
Benjamin R.
Taft
and
Jean-Philip
Lumb
Department of Chemistry, Stanford University, Stanford, CA 94305-5080, USA. E-mail: bmtrost@stanford.edu
First published on 10th June 2016
Abstract
We present a full account detailing the development of a sequential catalysis strategy for the synthesis of chiral β-alkynyl carbonyl and sulfonyl derivatives. A palladium-catalyzed cross coupling of terminal alkyne donors with acetylenic ester, ketone, and sulfone acceptors generates stereodefined enynes in high yield. These compounds are engaged in an unprecedented, regio- and enantioselective copper-catalyzed conjugate reduction. The process exhibits a high functional group tolerance, and this enables the synthesis of a broad range of chiral products from simple, readily available alkyne precursors. The utility of the method is demonstrated through the elaboration of the chiral β-alkynyl products into a variety of different molecular scaffolds. Its value in complex molecule synthesis is further validated through a concise, enantioselective synthesis of AMG 837, a potent GPR40 receptor agonist.
Introduction
Conjugate addition reactions are fundamental bond forming processes that are widely employed in the synthesis of functionalized molecules.1 As a result, the development of efficient methods for asymmetric conjugate addition remains of paramount importance in modern organic synthesis. Transition metal catalysis is a well-established means of effecting the conjugate addition of a carbon nucleophile,2 and such catalytic processes often enable the rapid assembly of molecular complexity in a highly atom economic3 fashion.The asymmetric addition of a terminal alkyne to an activated olefin—for example, to an α,β-unsaturated ester, ketone, sulfone, aldehyde, or a synthetic equivalent thereof—is a conjugate addition reaction that has attracted significant attention. Novel methods involving the activation of a terminal alkyne as a more nucleophilic boron,4 aluminum,5 or zinc6 acetylide have been developed toward this end (Scheme 1A). However, strategies based on transition metal catalysis have the potential to improve functional group tolerance while avoiding the generation of stoichiometric metal waste.
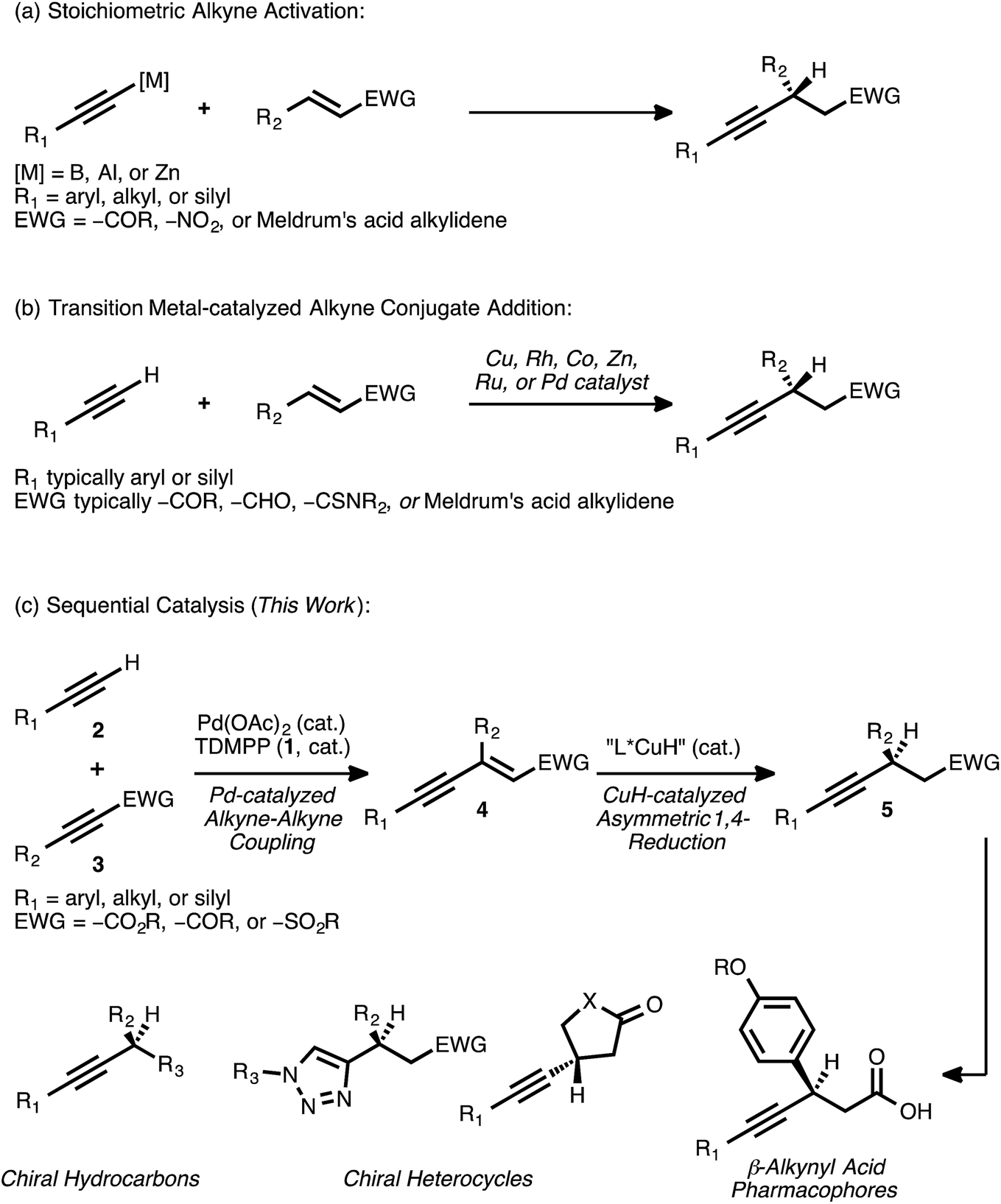 | ||
| Scheme 1 Strategies for the asymmetric conjugate addition of alkynes to activated olefins. | ||
Over the past several years, numerous methods for the transition metal-catalyzed asymmetric conjugate addition of a terminal alkyne to an activated olefin have been described (Scheme 1B). Carreira and co-workers reported the Cu-catalyzed addition of aryl acetylenes to Meldrum's acid alkylidenes with high yields and enantioselectivities,7 with an example of an alkyl acetylene addition that proceeded with more modest yield (29%) and enantioselectivity (68% ee).7a Shibasaki and co-workers disclosed the Cu-catalyzed addition of various terminal alkynes to α,β-unsaturated thioamides.8 Hayashi and Fillion have reported the asymmetric conjugate addition of silyl acetylenes to enones, enals, and Meldrum's acid alkylidenes under Rh9 and Co10 catalysis. Very recently, the asymmetric addition of triisopropylsilylacetylene to select α,β-unsaturated lactone and lactam substrates was accomplished using Rh catalysis, with diphenyl[(triisopropylsilyl)ethynyl]methanol serving as the alkynylating reagent.9e Blay and Pedro have reported asymmetric additions of primarily aryl acetylenes to arylidenediketones,11 coumarins,12 β-trifluoromethyl-α,β-unsaturated enones,13 and other electron-deficient enones14 under Zn or Cu catalysis. Mascareñas and co-workers recently described the Pd-catalyzed addition of various terminal alkynes to enones, enals, and acrylates in a racemic fashion, but investigations toward an asymmetric variant of the process were met with modest success (38–39% ee).15 In a similar vein, Ito and Nishiyama have reported racemic additions of terminal alkynes to different activated olefins using Ru pincer catalysts.16 These conditions were extended to one example of an asymmetric addition, the reaction of phenylacetylene with 3-penten-2-one (49% yield, 82% ee).
These reports illustrate how transition metal catalysis can be effective for the asymmetric conjugate addition of select types of alkynes to certain activated olefins. However, they also demonstrate how such direct approaches can be limited in terms of substrate scope. As these examples illustrate, the highest levels of reactivity are often observed when aryl or silyl acetylenes are employed as donors. Major restrictions regarding the scope of the acceptor olefin are also apparent. Significantly, no general strategy currently exists for the catalytic asymmetric addition of a terminal alkyne to an acyclic α,β-unsaturated ester. The catalytic asymmetric conjugate alkynylation of an α,β-unsaturated sulfone is, similarly, unknown.
A procedure to effect the formal asymmetric conjugate addition of a broad set of terminal alkynes to various acceptor olefins would complement these strategies. Importantly, the products obtained—chiral β-alkynyl carbonyl or sulfonyl derivatives—are valuable both on their own and as precursors to more functionalized molecules. For example, enantioenriched β-aryl, β-alkynyl carboxylic acids have been identified as potent pharmacophores and have been investigated for the treatment of type II diabetes.17 In addition, chemoselective18 functional group manipulations that engage either the alkyne group or the carbonyl group are possible. In an early illustration of this concept, Hayashi and co-workers reported the conversion of a β-alkynyl ketone to a 1,2,3-triazole through a Cu-catalyzed azide-alkyne cycloaddition; this transformation proceeded in high yield and without interference by the carbonyl moiety.9b The prospect of converting β-alkynyl carbonyl derivatives into different heterocyclic scaffolds via such catalytic processes is attractive, and it further motivates interest in a general method for the synthesis of these compounds.
Recognizing the need for a broadly applicable method for the synthesis of chiral β-alkynyl carbonyl and sulfonyl derivatives—and cognizant of the rich synthetic utility associated with these products—we considered a new approach. We envisioned that the subject compounds might be accessible through a sequential catalysis process involving: (a) a Pd-catalyzed cross coupling of alkynes to generate stereodefined enynes, and (b) a regio- and enantioselective CuH-catalyzed asymmetric conjugate reduction of these intermediates (Scheme 1C).
Our research group has established that combinations of palladium acetate and tris(2,6-dimethoxyphenylphosphine) (TDMPP, 1) can efficiently promote the cross coupling of a diverse set of terminal alkynes 2 with various acceptor alkynes 3 (EWG = –CO2R, –COR, –SO2Ph).19 In this completely atom-economic addition reaction, the enyne products 4 are typically formed in very high yields. Except in certain rare cases,20 this reaction proceeds with complete regio- and stereoselectivity, yielding solely the enyne isomer shown. The ability to generate stereodefined olefins in this manner is crucial to the proposed second stage of transition metal-catalyzed conjugate reduction, as a reduction reaction performed using a mixture of olefin isomers would be expected to result in poor enantioselectivity.21
Although CuH-catalyzed asymmetric conjugate reductions of α,β-unsaturated esters, ketones, and sulfones are known,22 the extended π-system borne by an activated enyne of the type 4 presents a critical issue of regioselectivity. Such a substrate has the potential to undergo an undesired 1,6-reduction of the alkyne group in competition with the desired 1,4-reduction event. Indeed, previous research has revealed that Cu-mediated conjugate addition and reduction reactions of activated enynes typically proceed with high selectivity for the 1,6-pathway.23 A 1,4-selective asymmetric conjugate reduction of an activated enyne would thus constitute a reversal of typical reaction trends, and it would provide direct access to the chiral β-alkynyl product of interest. Combining such a selective reduction event with the Pd-catalyzed coupling of alkynes would thus provide efficient access to a broad range of chiral β-alkynyl carbonyl and sulfonyl derivatives starting from simple alkyne precursors.
We previously reported preliminary results toward this end, highlighting the utility of dual Pd and CuH catalysis in the synthesis of selected examples of chiral β-alkynyl esters.24 Herein, we provide a comprehensive account describing not only the development of that process but also further progress in this area. These subsequent studies have enabled significant expansions in both the scope and the synthetic utility of this sequential catalysis strategy. Specifically, the detailed reaction optimization efforts that revealed the critical influence of ligand selection on the regioselectivity of the reduction event are presented. The application of the optimized reaction conditions to the synthesis of many new chiral β-alkynyl esters bearing valuable functionality to truly highlight the selectivity now demonstrates the very broad breadth of the methodology. We show, for the first time, the introduction of further structural diversity at the enyne stage via Pd0-catalyzed allylic alkylation followed by the asymmetric reduction. Moreover, efficient syntheses of chiral β-alkynyl ketones and sulfones via the sequential catalysis approach are reported here for the first time. The chemoselective elaboration of these latter products provides access to new chemical scaffolds that are not readily accessible from the corresponding β-alkynyl esters. In addition, the value of the method in a more complex molecular setting is demonstrated through a concise, asymmetric synthesis of a β-alkynyl, β-aryl carboxylic acid derivative of pharmaceutical relevance.
Results and discussion
Reaction optimization
Our reaction discovery efforts began with the preparation of a model enyne via the Pd-catalyzed alkyne–alkyne coupling reaction. Given the lack of known methods for the direct asymmetric addition of alkynes to acyclic α,β-unsaturated esters, we were attracted to propiolates as acceptor alkynes. The Pd-catalyzed alkyne–alkyne coupling was thus performed using 4-phenyl-1-butyne (6a) as the donor alkyne and methyl 2-nonynoate (7a) as the acceptor alkyne (Scheme 2).19b Ynenoate 8aa was obtained as a single regio- and stereoisomer in excellent yield (96%).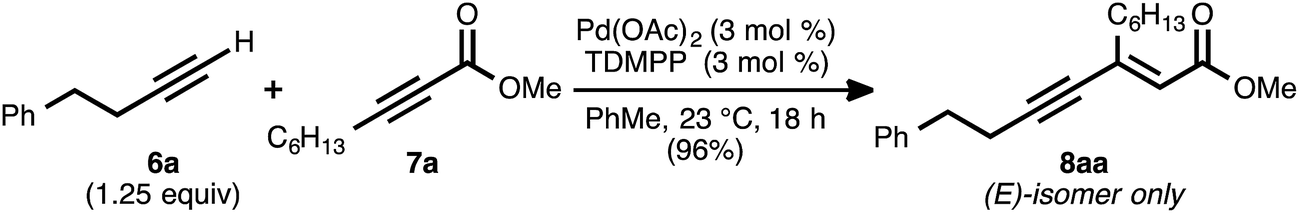 | ||
| Scheme 2 Pd-catalyzed alkyne–alkyne coupling to generate a model substrate. | ||
The asymmetric conjugate reduction stage was then explored, using 8aa as the substrate of interest. Drawing on prior reports of CuH-catalyzed reductions, Cu(OAc)2·H2O was selected as the Cu pre-catalyst, diethoxymethylsilane was used as the stoichiometric reductant, and the reactions were conducted in toluene at 4 °C.22 In addition, tert-butanol was included in the reaction mixture, as it had been reported that this additive could improve the rate of conjugate reduction reactions.22c,d We anticipated that the steric and electronic properties of the bisphosphine ligand might significantly impact the regioselectivity of the process. To this end, the nature of the ligand was systematically varied in these optimization experiments, which were assayed in terms of reaction conversion and regioselectivity (Table 1 and Fig. 1).
|
|
|||
|---|---|---|---|
| Entry | Ligand | Conversiona (%) | 1,4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Σ1,6 selectivityb :Σ1,6 selectivityb |
| a Conversion was determined by 1H NMR analysis of the crude reaction mixture relative to mesitylene as an internal standard. b Determined by 1H NMR analysis of the crude reaction mixture. N. D. = not determined. | |||
| 1 | 10 | 91 | 1.3:1 |
| 2 | 11 | 58 | 1:8.6 |
| 3 | 12 | 71 | 1:5.1 |
| 4 | 13 | 92 | 1:1.3 |
| 5 | 14 | 93 | 1.1:1 |
| 6 | 15 | 60 | 1:3.7 |
| 7 | 16 | 80 | 1:4.6 |
| 8 | 17 | >95 |
>19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 (99% ee) :1 (99% ee)
|
| 9 | 18 | 10 | N. D. |
| 10 | 19 | 11 | N. D. |
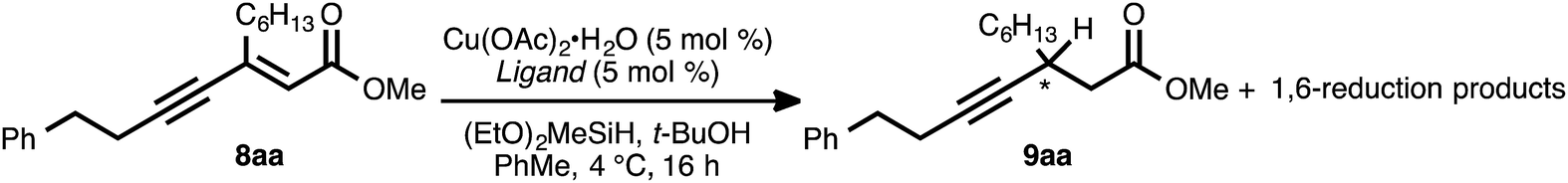
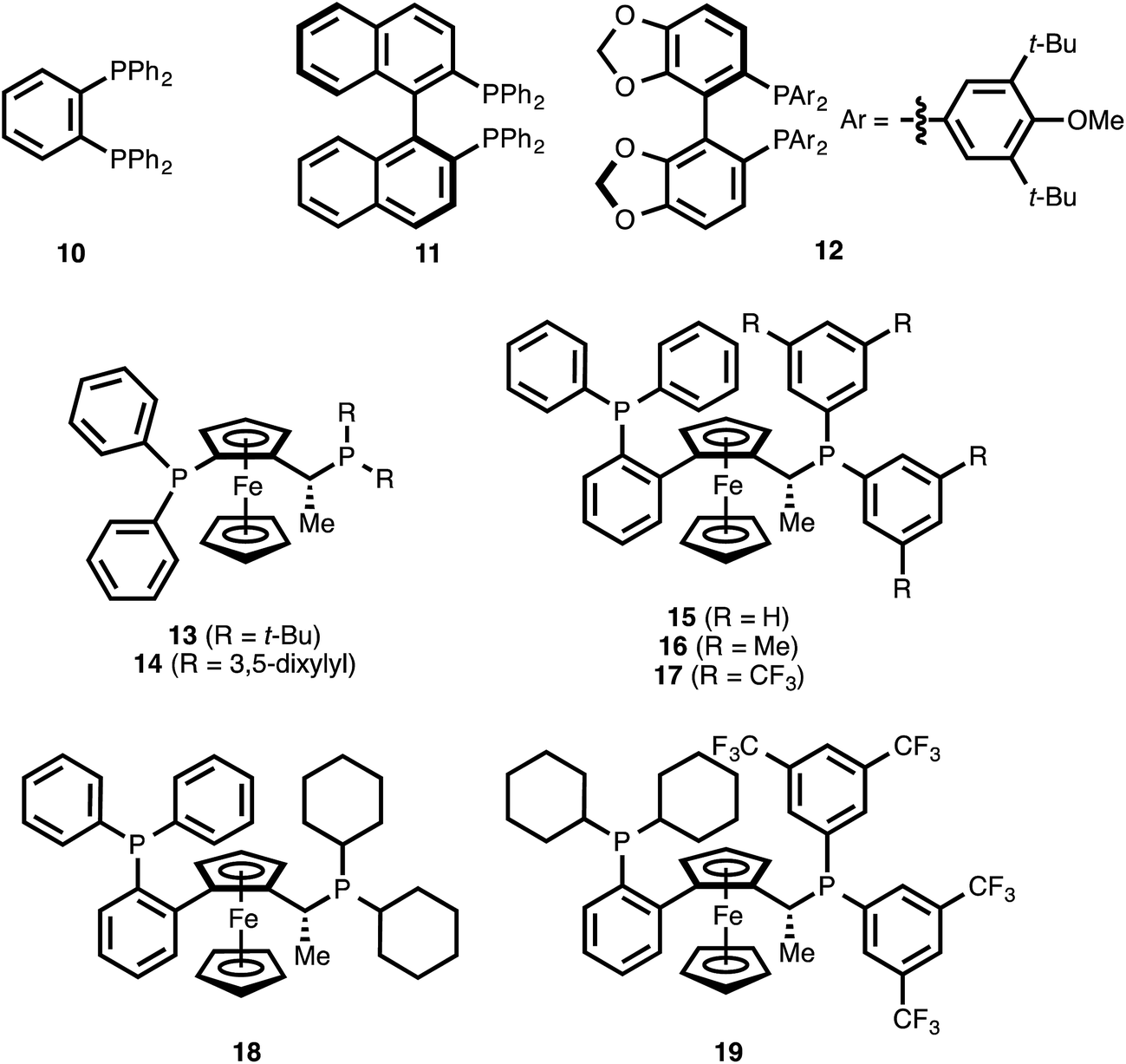 | ||
| Fig. 1 Ligands examined in the CuH-catalyzed conjugate reduction of ynenoate 8aa. | ||
The use of the achiral ligand 1,2-bis(diphenylphosphino)benzene (10)25 led to high conversion of ynenoate 8aa, but the 1,4-reduction product—β-alkynyl ester 9aa—was obtained with only moderate selectivity (entry 1). The mass balance comprised an intractable mixture of compounds, the 1H NMR spectra of which were consistent with 1,6-reduction byproducts. When axially chiral, C2-symmetric ligands such as BINAP22b (11) or DTBM-SEGPHOS (12)22d were employed, significant decreases in both conversion and 1,4-selectivity were observed (entries 2 and 3). The complexity of these product mixtures, together with the fact that the various products were inseparable via chromatography, precluded a determination of the enantioselectivity of the 1,4-reduction event. The use of ferrocene-derived JOSIPHOS ligands26 led to more promising results. Specifically, both tert-butyl- and xylyl-derived ligands 13 and 14 delivered high conversion, and they provided the highest levels of regioselectivity observed among the chiral ligands examined up this point (entries 4 and 5).
The fact that xylyl-substituted ligand 14 delivered a greater level of 1,4-selectivity than did tert-butyl-substituted ligand 13 suggested that aromatic substitution on the eastern portion of the bisphosphine might be beneficial for 1,4-selectivity. It was therefore postulated that modulation of this specific aromatic subunit might yield improved 1,4-selectivity. To this end, various commercially available ligands of the related WALPHOS ligand family (15–19) were examined. Relative to the JOSIPHOS ligands, the WALPHOS series offers greater variation in terms of aromatic substituents on the easternmost phosphine. However, these ligands differ from those in the JOSIPHOS family in that the western portion of the ferrocene unit is not bonded directly to the phosphorous atom but is, instead, annealed onto the 2-position of a triphenylphosphino unit. This gross structural change on its own was sufficient to direct the reduction event toward the 1,6-pathway (entry 6 vs. entry 5). That effect was magnified upon replacement of this eastern phenyl substituent with a xylyl unit (entry 7 vs. entries 5 and 6). However, a significant electronic influence was observed within the WALPHOS series: the use of the 3,5-bis(trifluoromethyl) WALPHOS ligand 17 delivered quantitative reaction conversion, essentially complete 1,4-selectivity, and excellent enantioselectivity (99% ee, entry 8).
One possible explanation for this result is that the electron-withdrawing nature of ligand 17 generates a more Lewis acidic Cu catalyst, one that is better able to coordinate to the carbonyl oxygen atom of ynenoate 8aa. Such secondary binding could help to promote the 1,4-reduction event by increasing the electrophilicity of the β-carbon or by directing hydride delivery to this position through a template effect.
It is noteworthy that both the western triphenylphosphino substituent and the eastern bis(3,5-bis(trifluoromethyl)phenyl) groups of ligand 17 were necessary for reactivity: the use of the related bis(dicyclohexyl)phosphino ligands 18 and 19 resulted in low levels of reaction conversion (entries 9 and 10).
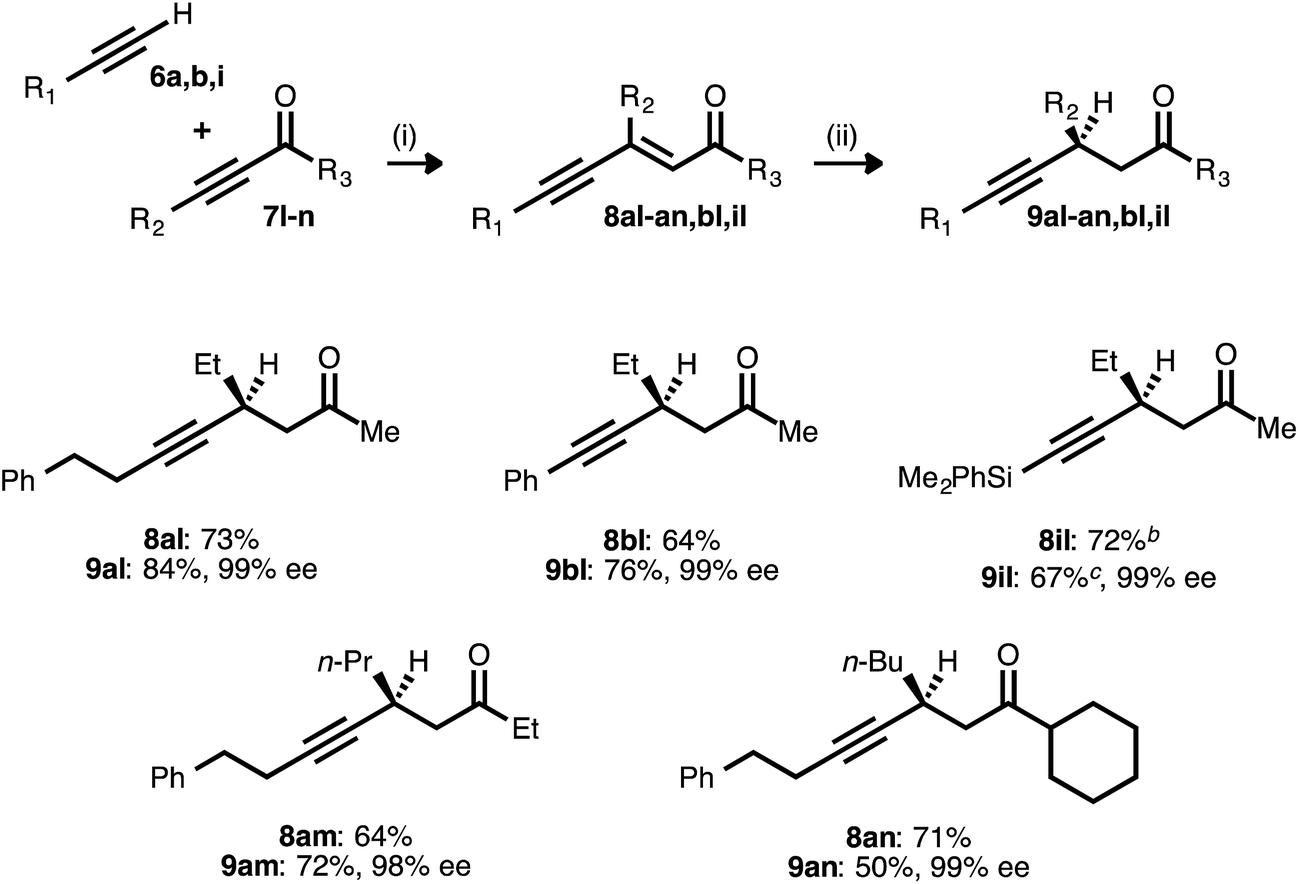
Synthesis of chiral β-alkynyl esters
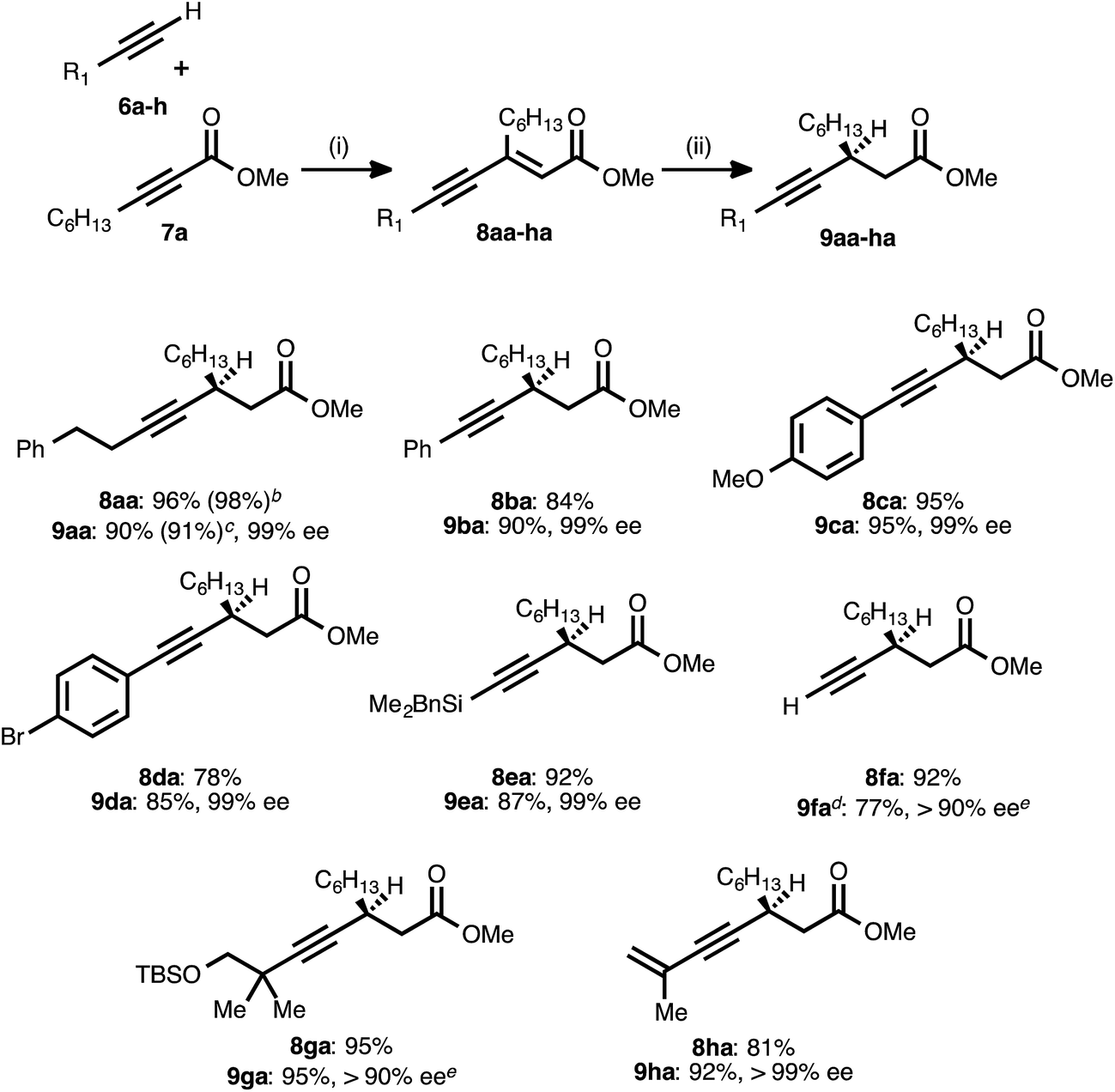
With the optimized conditions for the asymmetric conjugate reduction stage established, the scope of this new synthesis of chiral β-alkynyl esters was investigated. A range of donor alkynes 6 was examined, using methyl 2-nonynoate (7a) as the acceptor alkyne (Table 2). In addition to alkyl alkyne 6a, various aryl alkyne donors reacted successfully, including electron-neutral, electron-rich, and electron-deficient examples. Of particular note is the fact that p-bromophenylacetylene (6d) underwent both the PdII-catalyzed alkyne–alkyne coupling and the CuI-catalyzed reduction without competitive oxidative addition. Silyl substituted donor alkynes were also compatible, with both benzyldimethylsilylacetylene (6e) and trimethylsilylacetylene (6f) reacting smoothly. In the latter case, the corresponding trimethylsilylacetylene-derived β-alkynyl ester (9fa) was identified after the reduction stage (1H NMR analysis of an aliquot), but the crude product was treated with TBAF to liberate the terminal alkyne. The latter was isolated in very good yield after column chromatography. The more sterically encumbered donor alkyne 6g was also employed without complication.|
a Reaction conditions: (i) 6a–h:7a ratio = 1.25–1.75:1.0, 3 mol% Pd(OAc)2/TDMPP, 1.0 M in PhMe at 23 °C for 1–18 h. (ii) 5 mol% Cu(OAc)2·H2O/17, (EtO)2MeSiH (2 equiv.), t-BuOH (2 equiv.), 0.20 M in PhMe at 4 °C for 14–20 h. Yields are of isolated products after chromatography. Enatiomeric excesses were determined by chiral HPLC analysis.
b Result obtained from a 15 mmol-scale reaction using 1.5 mol% Pd(OAc)2/TDMPP.
c Result obtained from a 10 mmol-scale reaction using 2.5 mol% Cu(OAc)2 mol% Cu(OAc)2·H2O/17.
d The crude reduction product was treated with TBAF (1 M in THF), and the corresponding terminal alkyne (R1 = H) was isolated after chromatography.
e The ee was determined after conversion of the product into a diastereomeric mixture of amides using (S)-α-methylbenzylamine (>95:5 dr observed).
|
|---|
|
To further probe the regioselectivity of the reduction stage, an ynenoate bearing an extended (1,7-) π-system was prepared using enynyl donor alkyne 6h. The CuH-catalyzed reduction of this substrate maintained excellent regioselectivity, delivering 1,4-reduction product 9ha in high yield and with excellent enantioselectivity.
To further assess reaction robustness and scalability, each stage was examined on larger scale using a reduced catalyst loading. A 15 mmol-scale coupling between donor 6a and acceptor 7a was performed using 1.5 mol% Pd(OAc)2/TDMPP, and this delivered ynenoate 8aa in 98% yield. A 10 mmol-scale conjugate reduction of the latter compound was performed using 2.5 mol% Cu(OAc)2·H2O/17, and this furnished ester 9aa in 91% yield and 99% ee. Importantly, these results meet or exceed those obtained on smaller scale (1.0 mmol and 0.30 mmol, respectively) and with higher catalyst loadings.
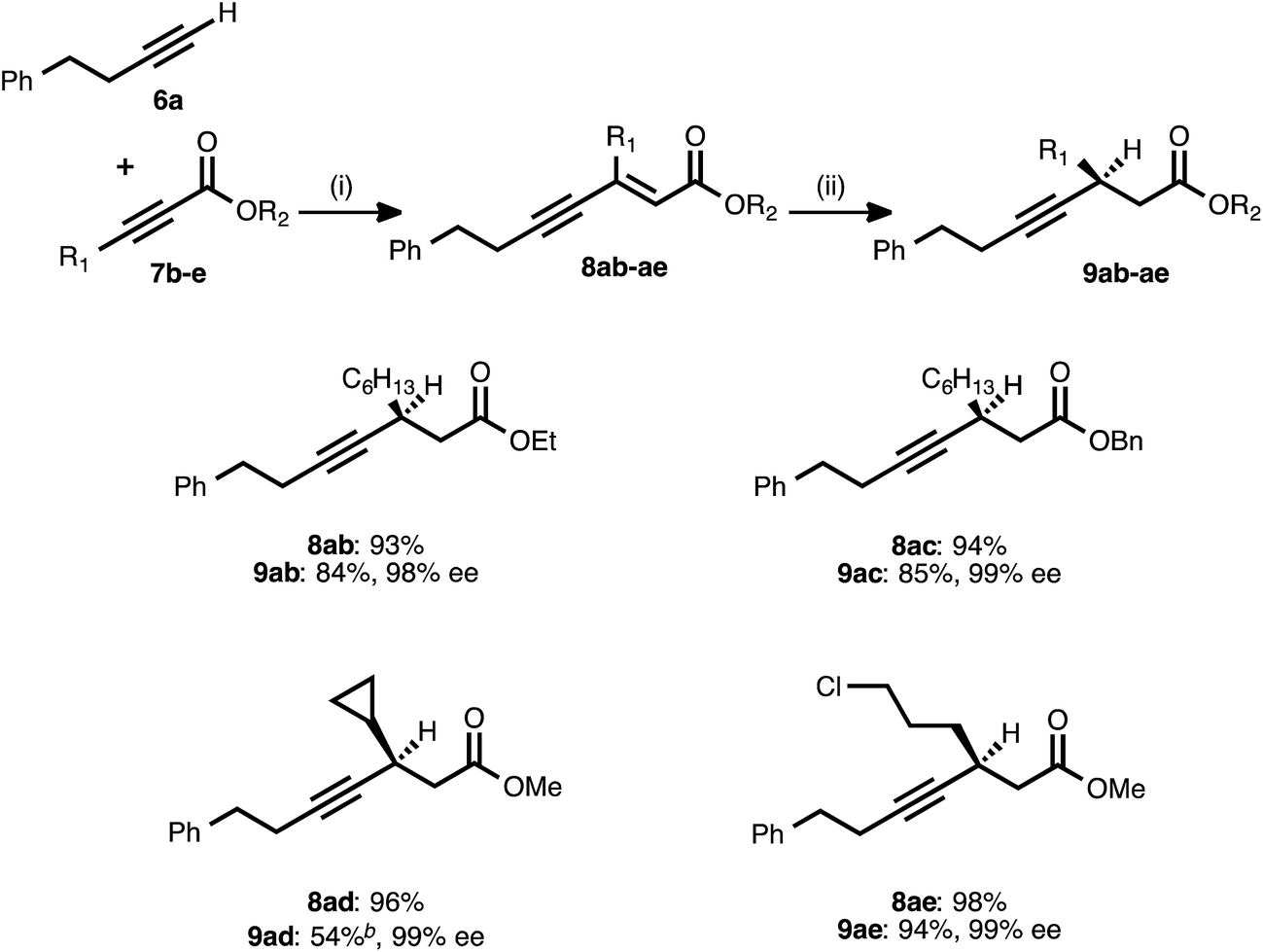
Attention next turned to the scope of the process with regard to other propiolate acceptors, using 4-phenyl-1-butyne (6a) as the donor alkyne (Table 3). The steric bulk around the ester unit could be increased without ill effect: in addition to the methyl ester examples shown previously, ethyl and benzyl esters reacted smoothly in both stages and delivered excellent enantioselectivity. The sterically demanding, cyclopropyl-derived acceptor 7d also underwent the alkyne–alkyne coupling in high yield. When the resulting ynenoate (8ad) was subjected to the conjugate reduction stage, the reaction reached 80% conversion and delivered a 4:1 mixture of 1,4- to 1,6-reduction products from which pure 9ad was isolated in 54% yield. Importantly, the enantioselectivity of the event remained high (99% ee). In addition to these hydrocarbon-substituted acceptor alkynes, a propiolate bearing a primary alkyl chloride was prepared and was found to react smoothly in both stages.
|
a Reaction conditions: (i) 6a:7b–e ratio = 1.25:1.0, 3 mol% Pd(OAc)2/TDMPP, 1.0 M in PhMe at 23 °C for 1–18 h. (ii) 5 mol% Cu(OAc)2·H2O/17, (EtO)2MeSiH (2 equiv.), t-BuOH (2 equiv.), 0.20 M in PhMe at 4 °C for 14–20 h. Yields are of isolated products after chromatography. Enantiomeric excesses were determined by chiral HPLC analysis.
b The reaction reached 80% conversion (1H NMR) and produced a mixture of 1,4- and 1,6-reduction products (4:1 1,4:Σ1,6, 1H NMR analysis of the crude reaction mixture), from which pure 9ad was isolated in 54% isolated yield.
|
|---|
|
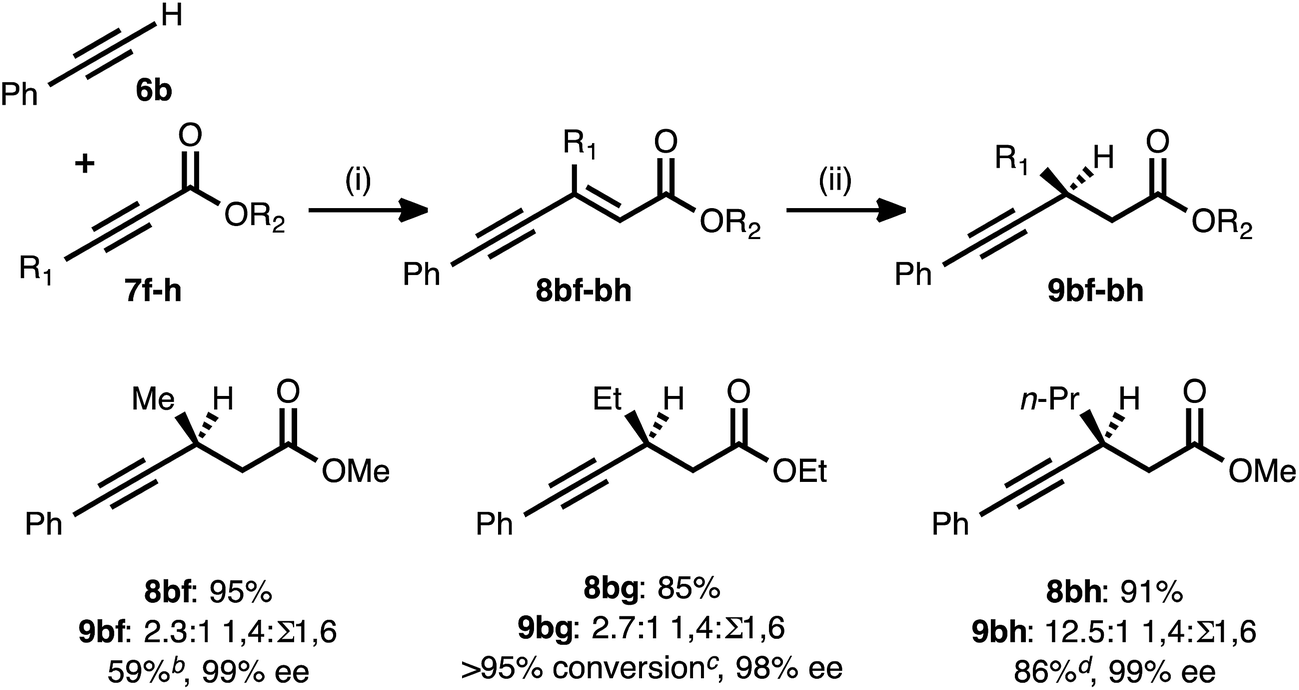
The fact that the conjugate reduction of β-cyclopropyl-substituted ynenoate 8ad proceeded with slight decreases in efficiency and regioselectivity can be understood in terms of the increased steric clash associated with the coordination of the Cu catalyst to this more sterically congested olefin. These interactions could be expected to retard the rate of hydride addition, and they also could cause hydride addition across the less sterically demanding alkyne unit to become more favorable.27 A more surprising change in regiochemical outcome came during the examination of ynenoates bearing alkyl groups of different chain lengths at their β-positions (Table 4). When methyl substituted propiolate 7f was coupled with phenylacetylene (6b) and the resulting ynenoate was subjected to the standard reduction conditions, 2.3:1 1,4:Σ1,6 regioselectivity was observed. The conjugate reduction of the β-ethyl congener (8bg) led to a similar outcome (2.7:1 1,4:Σ1,6). However, a marked increase in regioselectivity (12.5:1 1,4:Σ1,6) was observed in the reaction of the β-n-propyl analogue (8bh). As previously described, excellent regioselectivity (>19:1) was observed upon moving to longer chain lengths (e.g., the reactions of β-chloropropyl substrate 8ae or β-hexyl compound 8aa). While a detailed explanation for this phenomenon remains elusive, a trend of increasing 1,4-selectivity with increasing chain length clearly emerges from these data.
|
a Reaction conditions: (i) 6b:7f–h ratio = 1.25:1.0, 3 mol% Pd(OAc)2/TDMPP, 1.0 M in PhMe at 23 °C for 1–6 h. (ii) 5 mol% Cu(OAc)2·H2O/17, (EtO)2MeSiH (2 equiv.), t-BuOH (2 equiv.), 0.20 M in PhMe at 4 °C for 14–16 h. Yields are of isolated products after chromatography. Ratios of 1,4:1,6 selectivity were determined by 1H NMR analysis of the crude reaction mixture. Enantiomeric excesses were determined by chiral HPLC analysis.
b Isolated yield of analytically pure 1,4-reduction product 9bf obtained from the crude 2.3:1 mixture of regioisomeric products.
c Quantitative reaction conversion and yield were obtained, but 1,4-reduction product 9bg could not be separated from the various 1,6-reduction products.
d Isolated yield of 9bh (>90% purity) from the crude 12.5:1 mixture of regioisomeric products.
|
|---|
|
As shown in Table 2, the conjugate reduction of an ynenoate derived from an enynyl alkyne donor maintained excellent 1,4-selectivity. To further study the behavior of extended π-systems in the method, enynyl alkyne acceptor 7i was prepared (Scheme 3). This compound reacted smoothly with 4-phenyl-1-butyne (6a), delivering ynenoate 8ai. To our surprise, the reduction of the latter substrate using ligand 17 generated, exclusively and in high yield, (Z)-enyne 20. The same product was obtained when the reaction was performed using 1,2-bis(diphenylphosphino)benzene (10) as the ligand. The outstanding regio- and stereoselectivity observed in this case is remarkable, and it may be the result of a multi-dentate coordination event involving the Cu catalyst and both the alkyne and olefin subunits of this unique, cross-conjugated substrate.
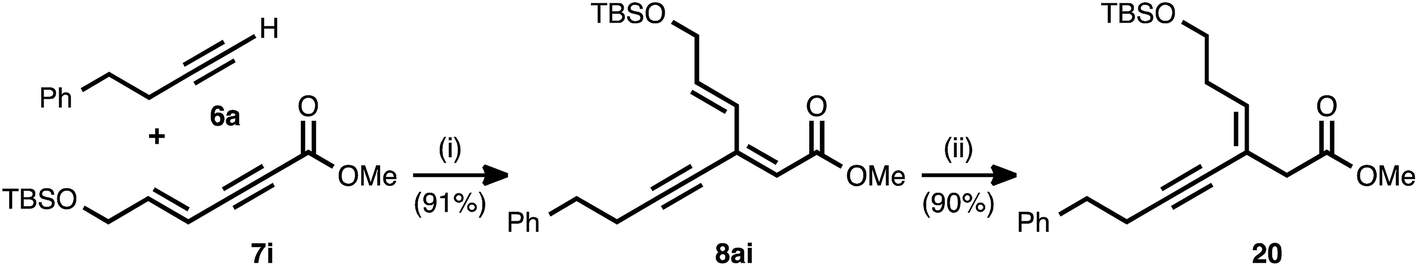 | ||
| Scheme 3 Regio- and stereoselective reduction of a substrate featuring an extended π-system. Conditions: (i) 3 mol% Pd(OAc)2/TDMPP, 1.0 M in PhMe at 23 °C for 16 h. (ii) 5 mol% Cu(OAc)2·H2O/17, (EtO)2MeSiH (2 equiv.), t-BuOH (2 equiv.), 0.20 M in PhMe at 4 °C for 16 h. | ||
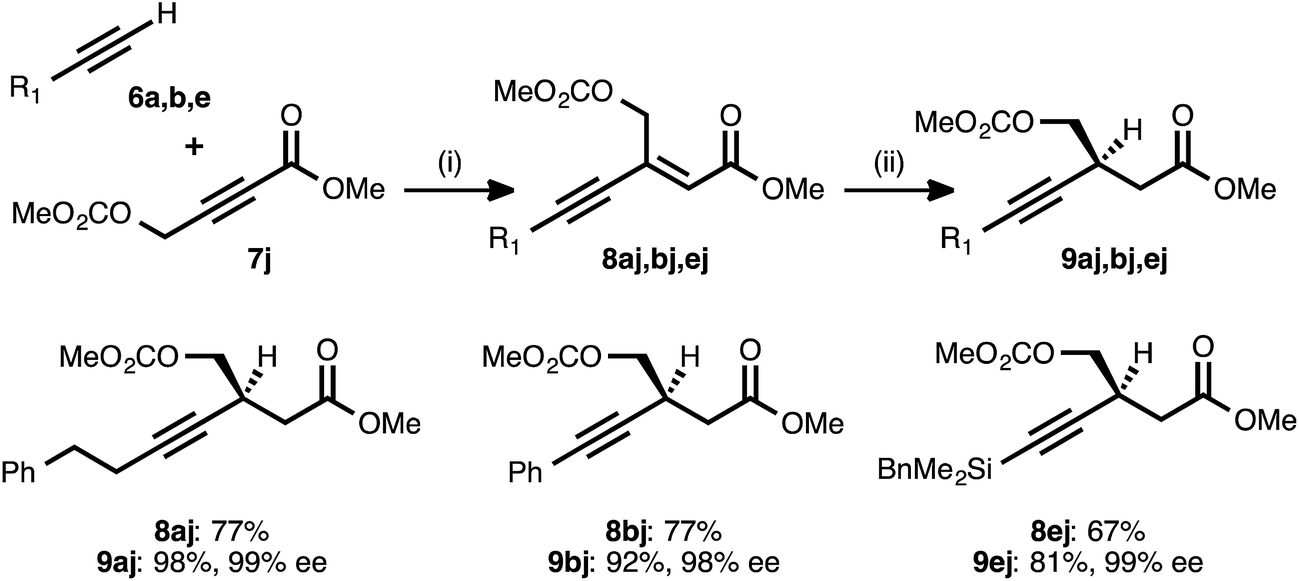
Propiolate acceptors bearing heteroatom substitution were next examined. Initial efforts focused on the reaction of carbonate-bearing propiolate 7j with different donor alkynes (Table 5). Cross couplings between this acceptor and 4-phenyl-1-butyne (6a), phenylacetylene (6b), and benzyldimethylsilylacetylene (6e) all proceeded in good to very good yields. The ynenoates so obtained were smoothly reduced to the corresponding β-alkynyl esters in high yield, with >19:1 regioselectivity, and in 99% ee. Notably, there was no indication that competitive ionization or reduction of the propargylic or allylic carbonates occurred during either of the two transition metal-catalyzed reaction stages.
|
a Reaction conditions: (i) 6:7j ratio = 1.25–1.75:1.0, 3 mol% Pd(OAc)2/TDMPP, 1.0 M in PhMe at 23 °C for 5–18 h. (ii) 5 mol% Cu(OAc)2·H2O/17, (EtO)2MeSiH (2 equiv.), t-BuOH (2 equiv.), 0.20 M in PhMe at 4 °C for 16–20 h. Yields are of isolated products after chromatography. Enantiomeric excesses were determined by chiral HPLC analysis.
|
|---|
|
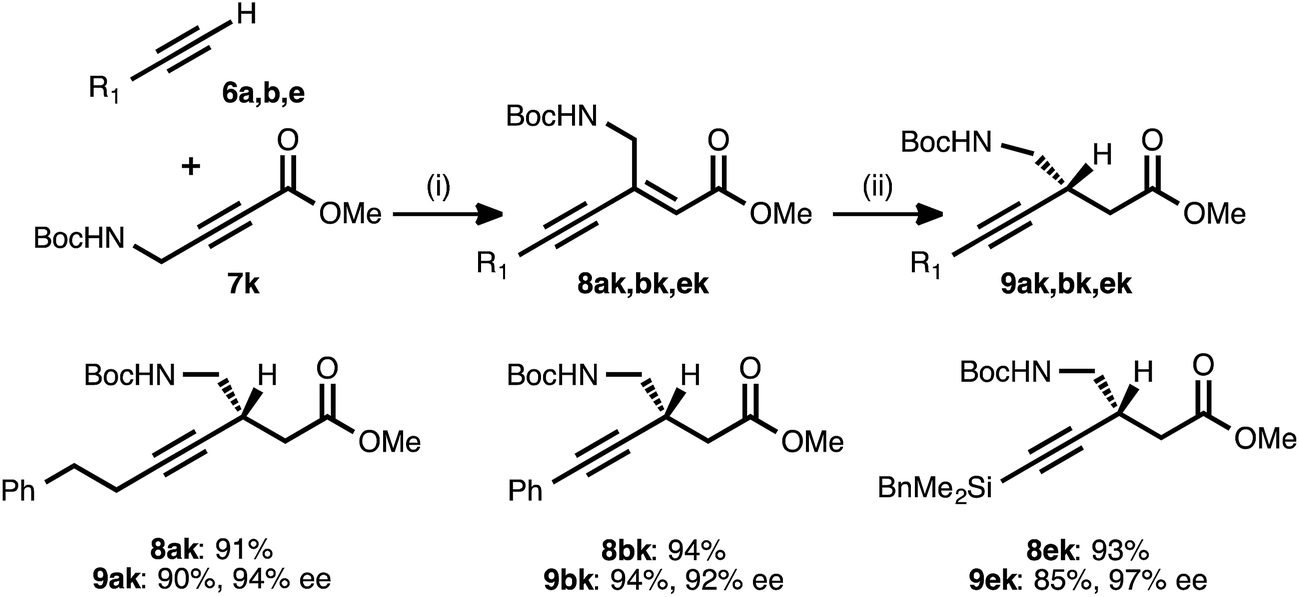
To explore the use of nitrogen-bearing acceptors in the method, propargylamine derivative 7k was prepared. Coupling reactions between this acceptor and donor alkynes 6a, 6b, and 6e all proceeded smoothly, even when performed with a lower catalyst loading (1.5 mol%) and under shorter reaction times (1–3 h, Table 6).28 However, the conjugate reduction of the resulting ynenoates using the standard conditions (with ligand 17) furnished β-alkynyl esters with more modest regio- and enantioselectivity. As a representative example, the reduction of ynenoate 8ak yielded ester 9ak with 9:1 1,4:Σ1,6 selectivity and 70% ee. A survey of different ligands led to the discovery that JOSIPHOS ligand 13 was particularly well matched for the reduction of these ynenoates: when it was used in place of ligand 17, excellent regioselectivity (>19:1) and enantioselectivity (≥92% ee) were obtained for the range of substrates. The decreased selectivity associated with the electron-deficient WALPHOS ligand 17 may have resulted from secondary interactions between the copper catalyst and the Lewis basic carbamate group, interactions that may have been disfavored when the more electron-rich and sterically encumbered JOSIPHOS ligand 13 was used.
|
a Reaction conditions: (i) 6:7k ratio = 1.25–1.75:1.0, 1.5 mol% Pd(OAc)2/TDMPP, 1.0 M in PhMe at 23 °C for 1–3 h. (ii) 2 mol% Cu(OAc)2·H2O/13, (EtO)2MeSiH (1.5 equiv.), t-BuOH (1.5 equiv.), 0.20 M in PhMe at 4 °C for 4 h. Yields are of isolated products after chromatography. Enantiomeric excesses were determined by chiral HPLC analysis.
|
|---|
|
Synthesis of chiral β-alkynyl esters via the Pd-catalyzed allylic alkylation of an ynenoate
As noted, carbonate-substituted ynenoate 8aj underwent both the PdII- and CuI-catalyzed reaction stages without competitive ionization. However, we questioned whether this ionization event could be intentionally induced under Pd0 catalysis, with the concomitant formation of a π-allylpalladium complex. Subsequent trapping of this complex with a heteroatom nucleophilic would generate a new ynenoate, itself a candidate for asymmetric reduction. This catalytic allylic alkylation process would thus provide a convenient means for the convergent synthesis of additional, valuable ynenoates and chiral β-alkynyl esters.29In pursuit of this, compound 8aj was reacted with pyrrole nucleophile 21a in the presence of Cs2CO3 and catalytic amounts of [(η3-C3H5)PdCl]2 and diphenylphosphinoferrocene (dppf) in 1,2-dichloroethane (Scheme 4).30 To our delight, desired allylic alkylation product 22a was successfully formed as a mixture of olefin isomers (9:1 Z:E), and pure (Z)-22a was isolated in very good yield after chromatography. Following conjugate reduction using ligand 17, β-alkynyl ester 23a was obtained in high yield and enantioselectivity. Allylic substitution with phenol nucleophile 21b likewise yielded oxygen-substituted ynenoate 22b, in this case as a single olefin isomer (>19:1 Z:E). The asymmetric conjugate reduction of 22b was similarly effective, giving rise to oxygen-substituted β-alkynyl ester 23b.
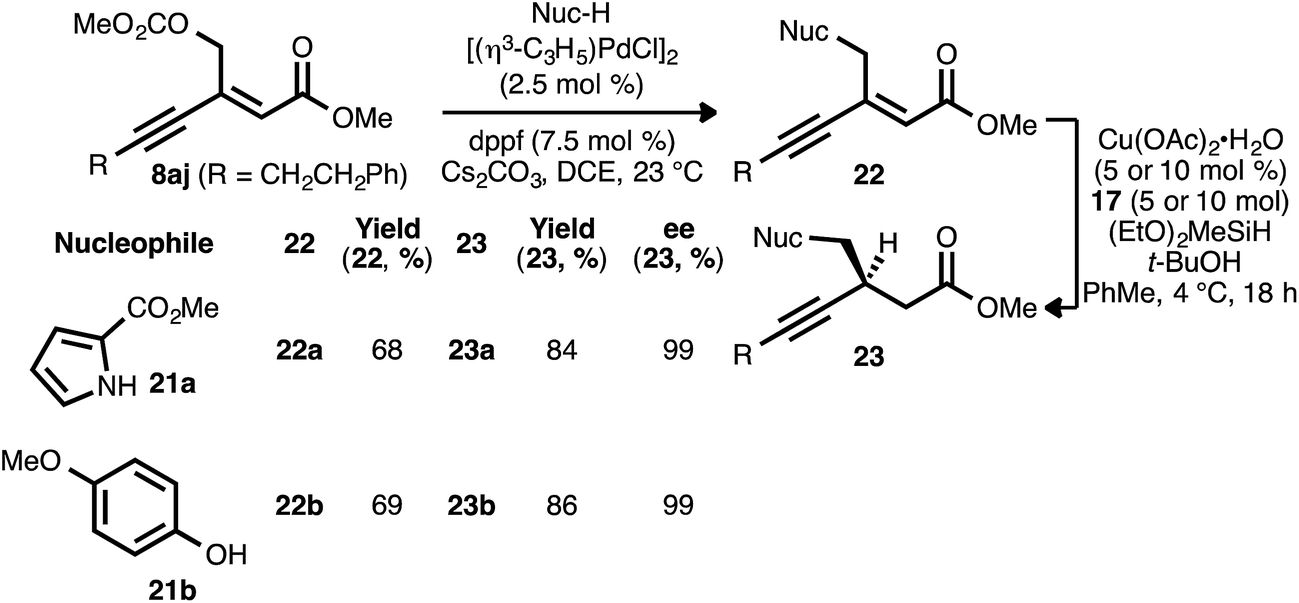 | ||
| Scheme 4 A Pd-AA approach to heteroatom-substituted β-alkynyl esters. | ||
Synthesis of chiral β-aryl, β-alkynyl esters: total synthesis of AMG 837
Recognizing the biological relevance of, specifically, β-aryl, β-alkynyl carboxylic acid derivatives, we were motivated to apply the present method to the synthesis of a representative compound within this subclass. As a target of interest, we selected AMG 837 (24), a GPR40 receptor agonist developed by Amgen, Inc.17c,31 The requisite acceptor alkyne was expediently prepared via the alkylation of known phenol 25 (ref. 32) with known bromide 26 (ref. 31a) (Scheme 5). Silyl-substituted acetylenes were chosen as alkyne coupling partners, as it was anticipated that the ethynylsilane moiety could be converted to the propynyl group of the target via desilylation and alkylation. To this end, the alkyne–alkyne coupling between trimethylsilylacetylene and propiolate 27 was performed, and this furnished ynenoate 28a with >95% conversion. However, the conjugate reduction of this intermediate using ligand ent-17 delivered predominantly 1,6-reduction products (1:1.5 1,4:Σ1,6 selectivity, entry 1). We suspected that this might have been a consequence of the increased steric hindrance around the β-carbon of this substrate, analogous to that observed in the reaction of β-cyclopropyl substrate 8ad. We questioned whether a corresponding increase in steric demand around the alkyne unit might serve to disfavor 1,6-reduction and redirect reactivity back toward the 1,4-pathway. Thus, the alkyne coupling reaction was performed using triisopropylsilylacetylene, delivering ynenoate 28b in 92% isolated yield. Gratifyingly, the reduction of this compound using ligand ent-17 proceeded with 13:1 regioselectivity in favor of the 1,4-pathway and with 86% ee, albeit with only 26% conversion (entry 2). Further ligand evaluation led us to discover that reactions promoted by JOSIPHOS ligands 13 and 14 proceeded with >95% conversion, maintained high regioselectivity, and, in the latter case, provided satisfying enantioselectivity (82% ee, entries 3 and 4).
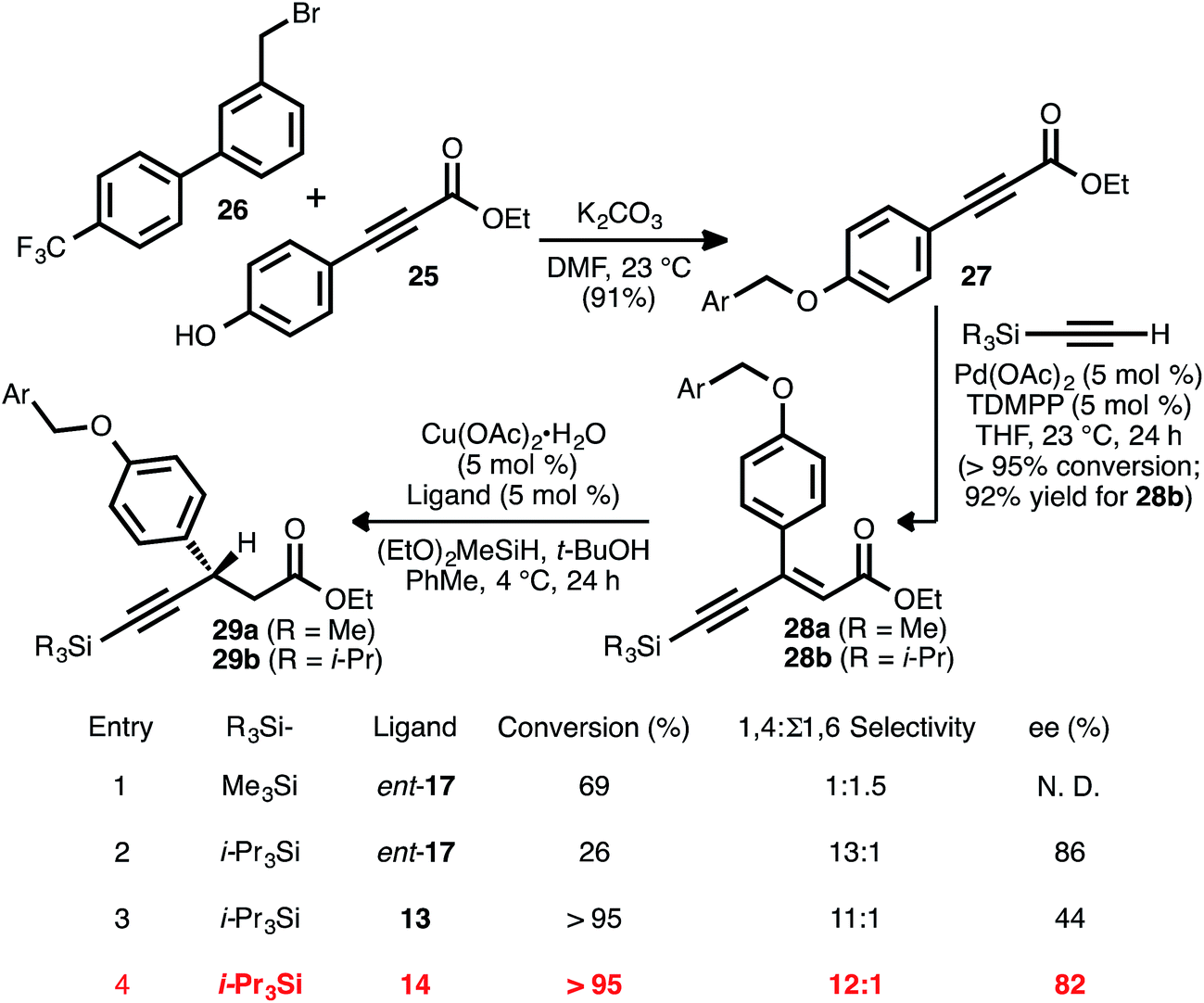 | ||
| Scheme 5 Synthesis of β-alkynyl esters 29a and 29ben route to AMG 837 (24). | ||
The reaction conditions of Scheme 5, entry 4 were incorporated into a one-pot conjugate reduction-alkyne desilylation process (Scheme 6). The conjugate reduction was carried out to completion, and then the reaction mixture was directly treated with tetrabutylammonium fluoride to effect alkyne desilylation. This sequence delivered terminal alkyne 30 in 74% yield from 28b (82% ee). Alkyne methylation using Pd and Cu co-catalysis in the presence of NHC ligand 31 (ref. 8b and 33) furnished β-alkynyl ester 32. Hydrolysis of the ethyl ester moiety of 32 proceeded smoothly, and AMG 837 (24) was obtained in 83% yield.
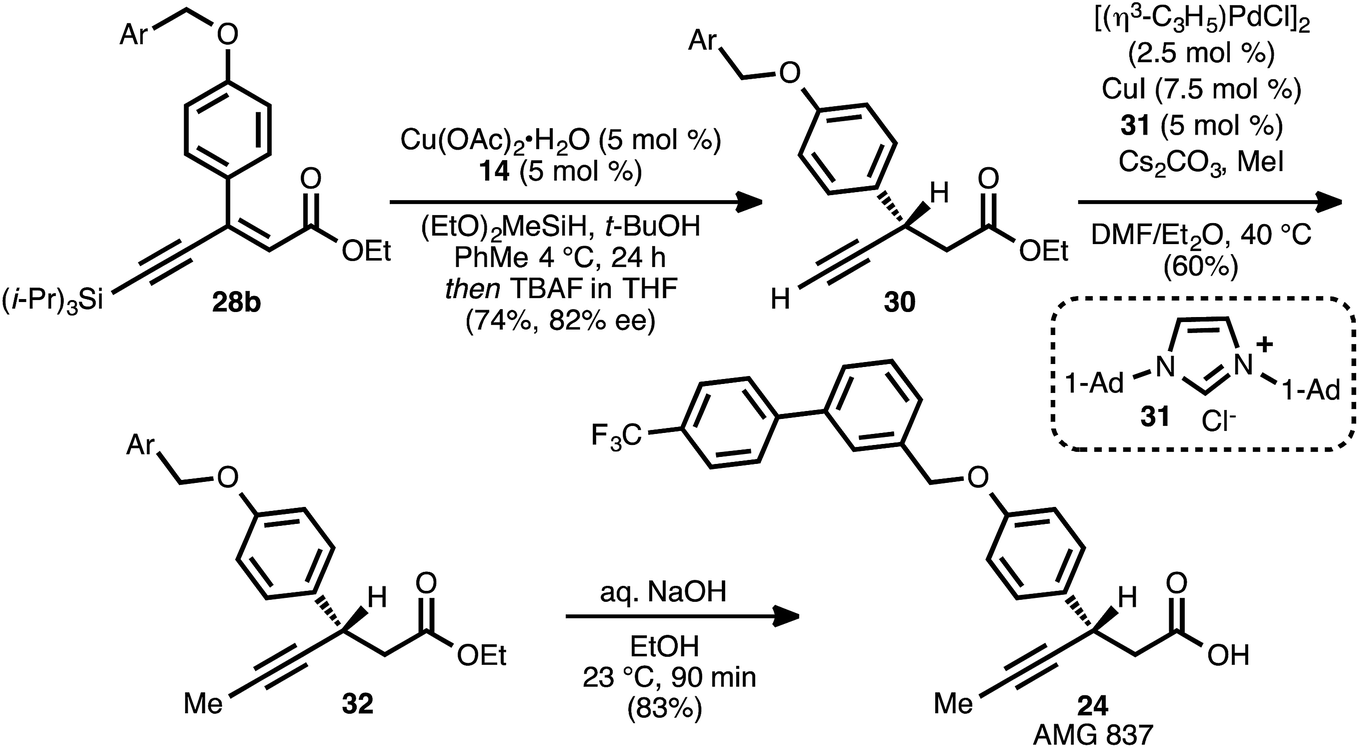 | ||
| Scheme 6 Completion of the synthesis of AMG 837 (24). | ||
The brevity and efficiency of this synthesis, which was completed in five steps and 31% yield from compound 25, stemmed from the use of a propiolate as a coupling partner. By directly employing an ester-derived acceptor, the need to unmask an ester equivalent (e.g., a Meldrum's acid alkylidene) during the synthetic sequence was avoided. Moreover, the successful synthesis of this target highlights the applicability of the present method toward the specific subclass of β-aryl, β-alkynyl esters.
Synthesis of chiral β-alkynyl ketones
The success achieved in the synthesis of chiral β-alkynyl esters from terminal alkynes and propiolates prompted us to examine other classes of activated alkynes as acceptors. Ketone-derived acceptors (ynones) were readily engaged as partners in the alkyne–alkyne coupling, giving rise to a range of ynenone products (Table 7). These compounds were then exposed to the reduction conditions developed for ynenoates, with one modification: tert-butanol was omitted as an additive, as it has been shown to promote 1,2-reduction in reactions of enones.22e Under these modified conditions, asymmetric hydrosilylation of the ynenone substrates occurred, giving rise to silyl enol ether intermediates. The addition of either tetrabutylammonium fluoride or methanolic HCl rapidly effected desilylation, and the corresponding β-alkynyl ketones were isolated in good yields and with excellent levels of enantioselectivity. Alkyl, aryl, and silyl donor alkynes were all compatible with this process, as were ynone acceptors bearing methyl, ethyl, and cyclohexyl substituents.|
a Reaction conditions: (i) 7:6 ratio = 1.5–2.0:1.0, 5 mol% Pd(OAc)2/TDMPP, 1.0 M in PhMe at 23 °C for 14 or 24 h. (ii) 5 mol% Cu(OAc)2·H2O/17, (EtO)2MeSiH (2 equiv.), 0.20 M in PhMe at 23 °C for 0.25–2 h, followed by quenching with TBAF (1.0 M in THF, 3.5 equiv.). Yields are of isolated products after chromatography. Enantiomeric excesses were determined by chiral HPLC analysis.
b The coupling was performed using a 2.0:1.0 ratio of 6i:7l.
c The reduction was conducted at 0 °C, and the silyl enol ether was quenched using 1% HCl in MeOH instead of TBAF.
|
|---|
|
Synthesis of chiral β-alkynyl sulfones
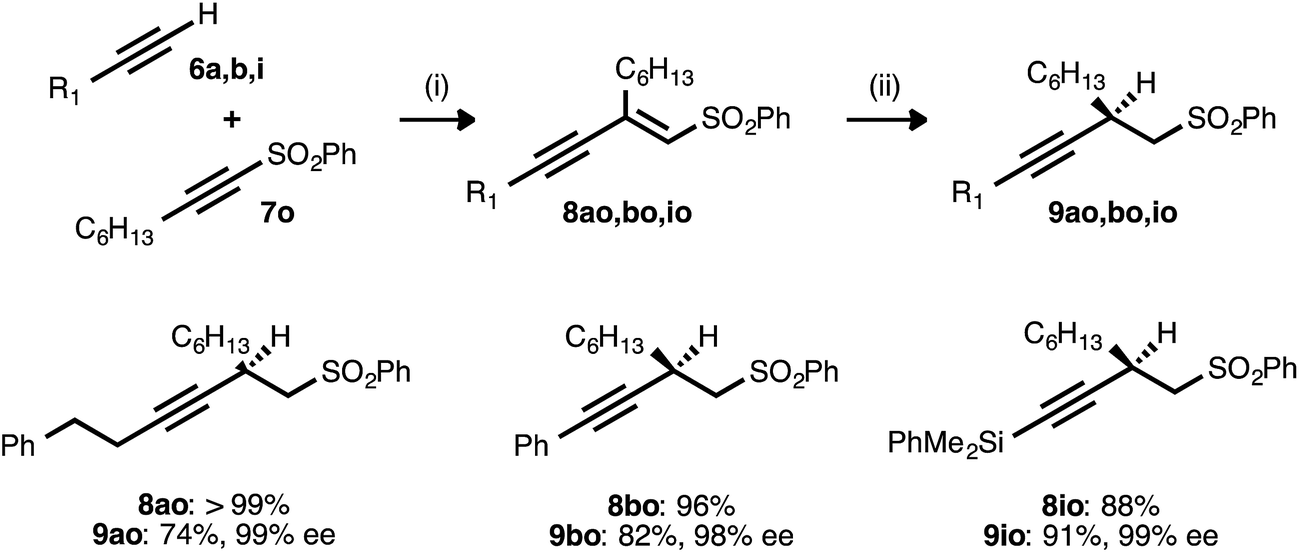
Alkyne–alkyne couplings employing acetylenic sulfones as acceptors were particularly successful. Reactions between terminal alkynes 6a, 6b, and 6i and sulfone 7o proceeded in high yields, setting the stage for the conversion of the resulting enynyl sulfones to synthetically valuable β-alkynyl sulfones via conjugate reduction (Table 8). This process was initially investigated using the conditions developed for ynenoate reduction: namely, the use of catalytic Cu(OAc)2·H2O, ligand 17, and diethoxymethylsilane in toluene with added tert-butanol. Although the desired β-alkynyl sulfones were obtained with complete regio- and stereoselectivity, reaction conversion was limited (<20% conversion after 24 h at room temperature). A report from Charette and Desrosiers22g describing the asymmetric conjugate reduction of α,β-unsaturated sulfones in mixed organic/aqueous media encouraged us to examine water as a reaction additive. We soon discovered that reactions performed using water in place of the tert-butanol additive proceeded to high conversion. When conducted at 50 °C, these transformations consistently proceeded to completion within 6 h. The corresponding alkyl-, aryl-, and silyl-substituted β-alkynyl sulfones were isolated in high yields. Excellent enantioselectivity (98–99% ee) was still uniformly observed, despite this introduction of water and this increase in reaction temperature.|
a Reaction conditions: (i) 6:7o ratio = 1.25:1.0, 3 mol% Pd(OAc)2/TDMPP, 1.0 M in PhMe at 23 °C for 17–22 h. (ii) 5 mol% Cu(OAc)2·H2O/17, (EtO)2MeSiH (2.0 equiv.), H2O (5.0 equiv.), 0.20 M in PhMe at 50 °C for 2.5–6 h. Yields are of isolated products after chromatography. Enantiomeric excesses were determined by chiral HPLC analysis.
|
|---|
|
One-pot β-alkynyl ester synthesis
To further assess the robustness and operationally simplicity of the method, a one-pot, sequential alkyne–alkyne coupling/conjugate reduction process was investigated (Scheme 7). In this event, the coupling was carried out using donor 6a and a slight excess of acceptor 7k. Following complete consumption of the donor alkyne, the reaction vessel was cooled to 0 °C. A pre-formed solution of the copper catalyst was introduced, and then the silane and tert-butanol were added. Under these conditions, asymmetric conjugate reduction occurred smoothly, and ester 9ak was obtained in 78% isolated yield and 92% ee.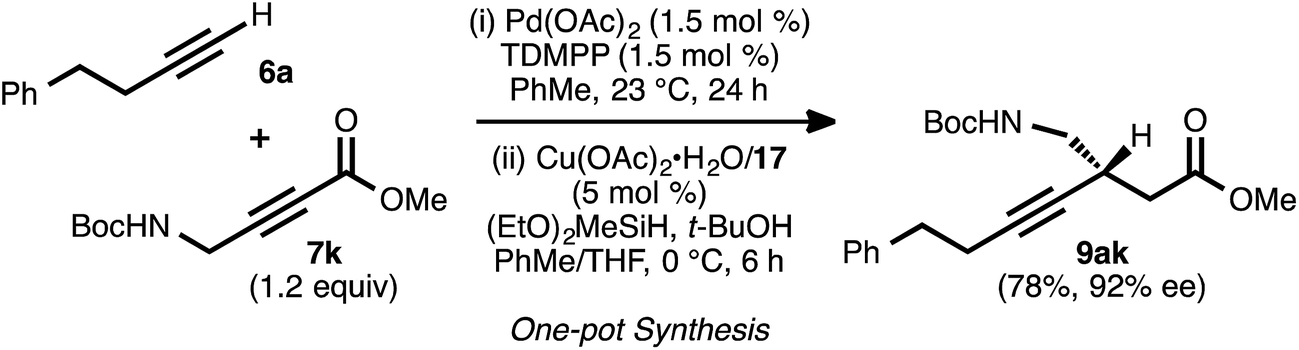 | ||
| Scheme 7 One-pot sequential alkyne–alkyne coupling/conjugate reduction. | ||
Synthetic utility
The β-alkynyl compounds so produced were readily converted into a range of other chiral small molecules through chemoselective transformations. Carbonate-bearing compound 9aj was hydrolyzed to furnish alcohol 33 (Scheme 8, eqn (A)). Treatment of the latter compound with Otera's esterification catalyst34 effected cyclization of the oxygen atom onto the ester group, furnishing chiral lactone 34 in 78% yield. Alternatively, cyclization onto the alkyne group was possible using gold and Brønsted acid co-catalysis, leading to chiral tetrahydrofuran 35(ref. 35). Nitrogen-bearing substrate 9bk was similarly converted to pyrrolidinone 36via trimethylaluminum-mediated lactamization (eqn (B)). Upon exposure to Pd(OAc)2, LiBr, and acrolein, compound 9bk underwent a tandem 5-endo-dig cyclization/reductive Heck-type addition, generating chiral dihydropyrrole 37.36 It was also possible to engage both the carboxylate group and the alkyne moiety of a substrate in a single bond-forming event: when β-alkynyl ester 9ca was hydrolyzed to acid 38 and the latter compound was treated with catalytic Pd(PhCN)2Cl2 and Et3N in refluxing MeCN, cycloisomerization occurred to generate chiral lactone 39 (eqn (C)).37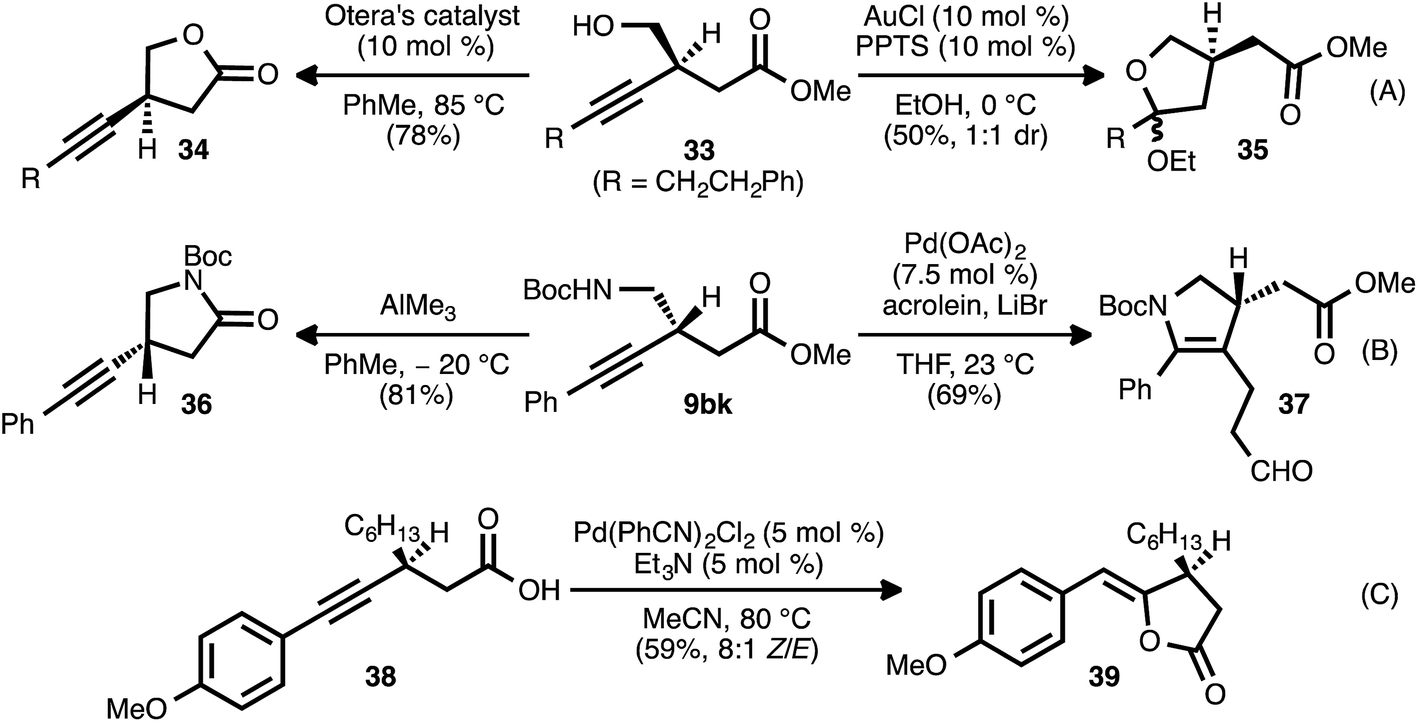 | ||
| Scheme 8 Chemoselective elaboration of β-alkynyl esters. | ||
In addition to these cycloisomerization processes, addition reactions involving β-alkynyl carbonyl and sulfonyl derivatives were also possible (Scheme 9). In the presence of a catalytic amount of copper thiophene-2-carboxylate (CuTC), the terminal alkyne group of ester 9fa underwent cycloaddition with tosyl azide (40) to deliver 1,2,3-triazole 41 in quantitative yield (eqn (A)).38 Fokin and co-workers have described such tosyl-substituted triazoles as efficient precursors to azavinyl carbenes,39 and this high-yielding, catalytic asymmetric synthesis of 41 provides an efficient entry into a new series of these compounds. The asymmetric addition of the terminal alkyne group of 9fa to trans-cinnamaldehyde (42) under Zn-ProPhenol catalysis40 likewise proceeded smoothly and with complete diastereocontrol, delivering tertiary alcohol 43 in high yield (85%).
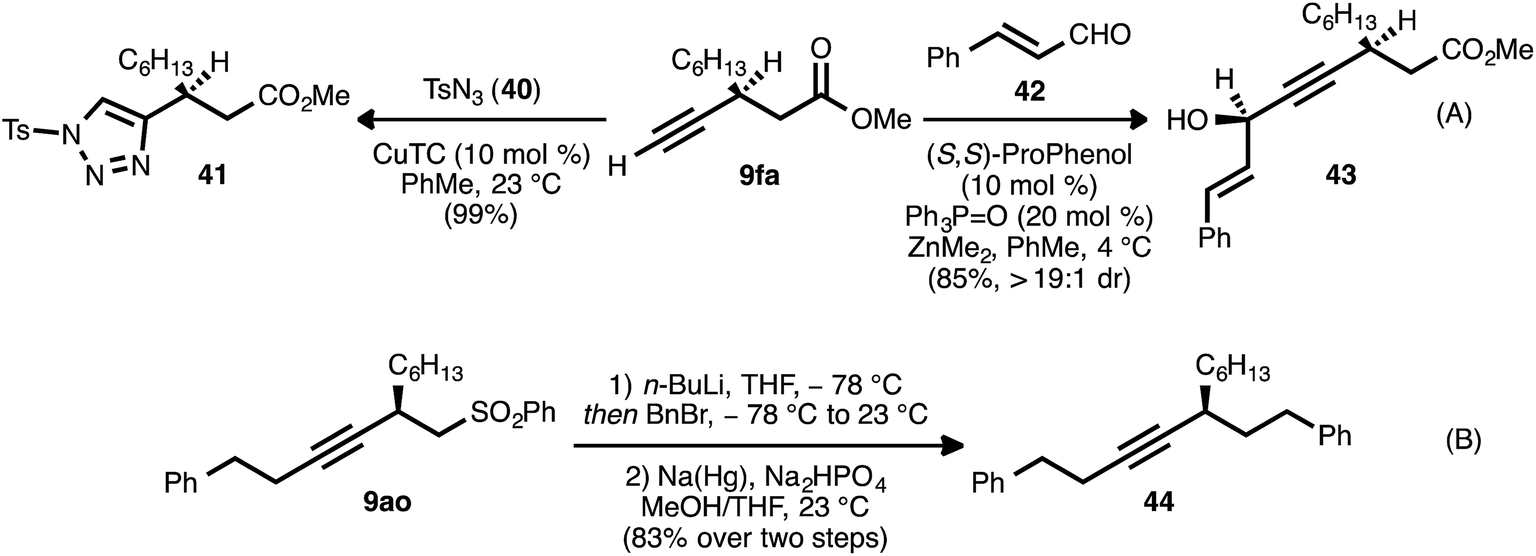 | ||
| Scheme 9 Addition reactions of β-alkynyl carbonyl and sulfonyl derivatives. | ||
In a further display of chemoselectivity, β-alkynyl sulfone 9ao was engaged in an alkylation/reductive desulfonylation sequence (eqn (B)). Following deprotonation of this substrate with n-butyllithium, alkylation with benzyl bromide occurred cleanly. Treatment of the crude product with sodium–mercury amalgam in buffered MeOH/THF22g delivered chiral hydrocarbon 44 in high yield (83% for two steps). Notably, competitive reduction of the alkyne moiety was not observed during this process.
Determination of the absolute stereochemistry
β-Alkynyl ester 9bf, which was prepared from ynenoate 8bfvia conjugate reduction using WALPHOS ligand 17, was hydrogenated to aliphatic compound 45 (Scheme 10). The optical rotation exhibited by this product was of comparable magnitude but opposite sign to that reported by Feringa and co-workers for the known (S)-enantiomer.41 The product is therefore assigned as the (R)-enantiomer, and the stereochemical assignments of the remaining β-alkynyl esters, ketones, and sulfones prepared using ligand 17 are assigned by analogy.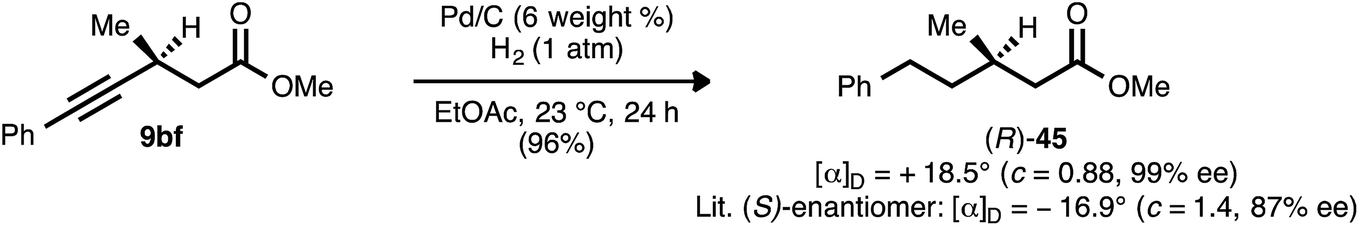 | ||
| Scheme 10 Catalytic hydrogenation of β-alkynyl ester 9bf. | ||
Asymmetric conjugate reductions performed using JOSIPHOS ligand 13 have been reported to proceed with the opposite sense of absolute stereoinduction relative to those performed using WALPHOS ligand 17.22f,j As a result, the β-alkynyl esters prepared using ligand 13 (e.g., compounds 9ak, 9bk, and 9ek) are assigned as depicted: enantiomeric to the set of products obtained using ligand 17. Entirely consistent with these reports, the conjugate reduction of ynenoate 28b using WALPHOS ligand ent-17 furnished the same major enantiomer of product as did the reduction of 28b using either JOSIPHOS ligand 13 or JOSIPHOS ligand 14 (Cf.Scheme 5). The preceding data suggest that the product, β-alkynyl ester 29b, would bear (S)-stereochemistry. This assignment was verified upon completion of the synthesis of AMG 837 (24) from 29b: the optical rotation data obtained for the latter were in agreement with prior reports (observed [α]25D = +4.4° (c = 0.9, CHCl3); lit. [(S)-enantiomer]: +7.2° (c = 0.5, CHCl3)31b).
Conclusions
We present a full account describing a sequential catalysis strategy for the asymmetric synthesis of chiral β-alkynyl carbonyl and sulfonyl derivatives from simple alkyne precursors. A Pd-catalyzed alkyne–alkyne cross coupling generates, with complete regio- and stereoselectivity, enyne intermediates that are reduced under CuH catalysis. A detailed assessment of ligand effects in the latter stage led to the identification of WALPHOS ligand 17 and JOSIPHOS ligands 13 and 14 as highly effective for the regio- and enantioselective reduction of a broad range of activated enynes.Relative to our initial report in this area, the substrate scope has been significantly expanded. Fourteen new, sterically and electronically diverse chiral β-alkynyl esters have been prepared. Additionally, the scope of the sequential catalysis approach has been expanded to encompass ynones and acetylenic sulfones as acceptor alkynes. To this end, new and previously unreported syntheses of chiral β-alkynyl ketones and sulfones are described.
The transformations described herein demonstrated high levels of robustness. Excellent results (>90% yield and 99% ee) were obtained when the reactions were executed on gram scale. A one-pot β-alkynyl ester synthesis was also developed, one that furnished the target in high yield without sacrificing selectivity.
In contrast to other methods for the synthesis of these targets, such as the direct addition of an alkyne to an activated olefin, the process described herein displays a significant breadth of scope. Alkyl, silyl, and aryl-substituted alkynes were all engaged as donors, and ester, ketone, and sulfone-derived acceptors all proved compatible. This ability to directly employ ester-based acceptors is especially noteworthy, given the paucity of methods for the catalytic asymmetric alkynylation of acrylates.
The method tolerates oxygen and nitrogen substitution, and this enabled the preparation of six different heterocyclic scaffolds from the corresponding β-alkynyl compounds. In addition, a new Pd-catalyzed allylic alkylation reaction was developed to enable the convergent introduction of heteroatom functionality to the ynenoate intermediate. This expanded the scope of the method even further, as the ynenoates so prepared responded well to the conjugate reduction conditions. Upon further reaction optimization, the substrate scope was expanded to include β-aryl ynenoates. This facilitated a concise synthesis of AMG 837, a chiral β-alkynyl carboxylic acid of pharmaceutical interest.
In a further demonstration of the synthetic utility of the process, the β-alkynyl products were engaged in efficient addition reactions. These included a catalytic, stereocontrolled addition of a terminal alkyne to an aldehyde. The versatility of the sulfone substituent in the product via an alkylation–desulfonylation sequence involving the β-alkynyl sulfone provides tertiary propargylic all carbon stereocenters. Further, given the flexibility of the alkyne beyond semi-hydrogenation both to Z- and E-olefins as well as full saturation truly becomes enabling in creating structural diversity. The ability to access such diverse products from simple alkyne precursors highlights the utility of the method in complex molecule synthesis.
Acknowledgements
We thank the National Science Foundation (CHE-1360634) and the National Institutes of Health (GM033049) for their generous support of our programs. J. T. M. acknowledges support from an Abbott Laboratories Stanford Graduate Fellowship. B. R. T. and J.-P. L. are grateful for NIH postdoctoral fellowships (CA137999 and GM083428, respectively). We warmly thank Solvias for gifts of chiral bisphosphine ligands and Johnson-Matthey for gifts of palladium salts.Notes and references
- (a) P. Perlmutter, Conjugate Addition Reactions in Organic Synthesis, Pergamon Press, Oxford, 1992 Search PubMed; (b) Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, Springer-Verlag, Berlin, Heidelberg, 1999 Search PubMed.
- (a) M. P. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033–8061 CrossRef CAS; (b) M. Kanai and M. Shibasaki, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, New York, 2000, pp. 569–592 Search PubMed.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- (a) T. R. Wu and J. M. Chong, J. Am. Chem. Soc., 2005, 127, 3244–3245 CrossRef CAS PubMed; (b) S. C. Pellegrinet and J. M. Goodman, J. Am. Chem. Soc., 2006, 128, 3116–3117 CrossRef CAS PubMed.
- (a) Y.-S. Kwak and E. J. Corey, Org. Lett., 2004, 6, 3385–3388 CrossRef CAS PubMed; (b) O. V. Larionov and E. J. Corey, Org. Lett., 2010, 12, 300–302 CrossRef CAS PubMed.
- (a) M. Yamashita, K. Yamada and K. Tomioka, Org. Lett., 2005, 7, 2369–2371 CrossRef CAS PubMed; (b) S. Cui, S. D. Walker, J. C. S. Woo, C. J. Borths, H. Mukherjee, M. J. Chen and M. M. Faul, J. Am. Chem. Soc., 2010, 132, 436–437 CrossRef CAS PubMed.
- (a) S. Fujimori, T. F. Knöpfel, P. Zarotti, T. Ichikawa, D. Boyall and E. M. Carreira, Bull. Chem. Soc. Jpn., 2007, 80, 1635–1657 CrossRef CAS; (b) S. Fujimori and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 4964–4967 CrossRef CAS PubMed; (c) T. F. Knöpfel and E. M. Carreira, J. Am. Chem. Soc., 2003, 125, 6054–6055 CrossRef PubMed; (d) T. F. Knöpfel, P. Zarotti, T. Ichikawa and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 9682–9683 CrossRef PubMed.
- (a) R. Yazaki, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 10275–10277 CrossRef CAS PubMed; (b) R. Yazaki, N. Kumagai and M. Shibasaki, Org. Lett., 2011, 13, 952–955 CrossRef CAS PubMed; (c) R. Yazaki, N. Kumagai and M. Shibasaki, Chem.–Asian. J., 2011, 6, 1778–1790 CrossRef CAS PubMed.
- (a) T. Nishimura, T. Sawano and T. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 8057–8059 CrossRef CAS PubMed; (b) T. Nishimura, X.-X. Guo, N. Uchiyama, T. Katoh and T. Hayashi, J. Am. Chem. Soc., 2008, 130, 1576–1577 CrossRef CAS PubMed; (c) T. Nishimura, S. Tokuji, T. Sawano and T. Hayashi, Org. Lett., 2009, 11, 3222–3225 CrossRef CAS PubMed; (d) E. Fillion and A. K. Zorzitto, J. Am. Chem. Soc., 2009, 131, 14608–14609 CrossRef CAS PubMed; (e) X. Dou, Y. Huang and T. Hayashi, Angew. Chem., Int. Ed., 2016, 55, 1133–1137 CrossRef CAS PubMed.
- T. Nishimura, T. Sawano, K. Ou and T. Hayashi, Chem. Commun., 2011, 47, 10142–10144 RSC.
- G. Blay, L. Cardona, J. R. Pedro and A. Sanz-Marco, Chem.–Eur. J., 2012, 18, 12966–12969 CrossRef CAS PubMed.
- G. Blay, M. C. Muñoz, J. R. Pedro and A. Sanz-Marco, Adv. Synth. Catal., 2013, 355, 1071–1076 CrossRef CAS.
- A. Sanz-Marco, A. García-Ortiz, G. Blay and J. R. Pedro, Chem. Commun., 2014, 50, 2275–2278 RSC.
- A. Sanz-Marco, A. García-Ortiz, G. Blay, I. Fernández and J. R. Pedro, Chem.–Eur. J., 2014, 20, 668–672 CrossRef CAS PubMed.
- L. Villarino, R. García-Fandiño, F. López and J. L. Mascareñas, Org. Lett., 2012, 14, 2996–2999 CrossRef CAS PubMed.
- J.-I. Ito, K. Fujii and H. Nishiyama, Chem.–Eur. J., 2013, 19, 601–605 CrossRef CAS PubMed.
- (a) T. Shimada, H. Ueno, K. Tsutsumi, K. Aoyagi, T. Manabe, S.-Y. Sasaki and S. Katoh, Spiro compounds and pharmaceutical use thereof, US 8299296, October 30, 2012; (b) D. C.-H. Lin, et al. , PLoS One, 2011, 6, e27270 CrossRef CAS PubMed, and references therein; (c) J. D. Houze, L. Zhu, Y. Sun, M. Akerman, W. Qiu, A. J. Zhang, R. Sharma, M. Schmitt, Y. Wang, J. Liu, J. Liu, J. C. Medina, J. D. Reagan, J. Luo, G. Tonn, J. Zhang, J. Y.-L. Lu, M. Chen, E. Lopez, K. Nguyen, L. Yang, L. Tang, H. Tian, S. J. Shuttleworth and D. C.-H. Lin, Bioorg. Med. Chem. Lett., 2012, 22, 1267–1270 CrossRef CAS PubMed; (d) S. B. Bharate, K. V. S. Nemmani and R. A. Vishwakarma, Expert Opin. Ther. Pat., 2009, 19, 237 CrossRef CAS PubMed , and references therein.
- B. M. Trost, Science, 1983, 219, 245–250 CrossRef CAS PubMed.
- (a) B. M. Trost, C. Chan and G. Rühter, J. Am. Chem. Soc., 1987, 109, 3486–3487 CrossRef CAS; (b) B. M. Trost, M. T. Sorum, C. Chan, A. E. Harms and G. Rühter, J. Am. Chem. Soc., 1997, 119, 698–708 CrossRef CAS.
- B. M. Trost, J. L. Gunzner and T. Yasukata, Tetrahedron Lett., 2001, 42, 3775–3778 CrossRef CAS.
- A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932–7934 CrossRef CAS.
- (a) W. S. Mahoney, D. M. Brestensky and J. M. Stryker, J. Am. Chem. Soc., 1988, 110, 291–293 CrossRef CAS; (b) D. H. Appella, Y. Moritani, R. Shintani, E. M. Ferreira and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9473–9474 CrossRef CAS; (c) G. Hughes, M. Kimura and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 11253–11258 CrossRef CAS PubMed; (d) B. H. Lipshutz, J. M. Servesko and B. R. Taft, J. Am. Chem. Soc., 2004, 126, 8352–8353 CrossRef CAS PubMed; (e) B. H. Lipshutz, J. M. Servesko, T. B. Petersen, P. P. Papa and A. A. Lover, Org. Lett., 2004, 6, 1273–1275 CrossRef CAS PubMed; (f) B. H. Lipshutz and J. M. Servesko, Angew. Chem., Int. Ed., 2003, 42, 4789–4792 CrossRef CAS PubMed; (g) J.-N. Desrosiers and A. B. Charette, Angew. Chem., Int. Ed., 2007, 46, 5955–5957 CrossRef CAS PubMed; (h) T. Llamas, R. G. Arrayás and J. C. Carretero, Angew. Chem., 2007, 119, 3393–3396 CrossRef, For reviews of CuH-catalyzed conjugate reductions, see: (i) C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed; (j) B. H. Lipshutz, Synlett, 2009, 509–524 CrossRef CAS.
- See (a) N. Krause, Chem. Ber., 1990, 123, 2173–2180 CrossRef CAS; (b) J. Canisius, A. Gerold and N. Krause, Angew. Chem., Int. Ed., 1999, 38, 1644–1646 CrossRef CAS; (c) N. Krause and G. Handke, Tetrahedron Lett., 1991, 32, 7229–7232 CrossRef CAS; (d) N. Krause and A. Hoffmann-Roder, in Modern Organocopper Chemistry, ed. N. Krause, Wiley-VCH, Weinheim, 2002, pp. 145–166, and references therein CrossRef.
- A portion of these results were disclosed in a preliminary Communication: B. M. Trost, B. R. Taft, J. T. Masters and J.-P. Lumb, J. Am. Chem. Soc., 2011, 133, 8502–8505 CrossRef CAS PubMed.
- B. A. Baker, Ž. V. Bošković and B. H. Lipshutz, Org. Lett., 2008, 10, 289–292 CrossRef CAS PubMed.
- H.-U. Blaser, W. Brieden, B. Pugin, F. Spindler, M. Studer and A. Togni, Top. Catal., 2002, 19, 3–16 CrossRef CAS.
- The impact of subtle catalyst–substrate interactions on regioselectivity has previously been discussed, in the context of the related process of CuH-catalyzed enone conjugate reduction. See: K. R. Voigtritter, N. A. Isley, R. Moser, D. H. Aue and B. H. Lipshutz, Tetrahedron, 2012, 68, 3410–3416 CrossRef CAS.
- B. M. Trost, J.-P. Lumb and J. M. Azzarelli, J. Am. Chem. Soc., 2011, 133, 740–743 CrossRef CAS PubMed.
- For a review of allylic alkylation reactions involving heteroatom nucleophiles, see: B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427–440 RSC.
- B. M. Trost, M. Osipov and G. Dong, J. Am. Chem. Soc., 2010, 132, 15800–15807 CrossRef CAS PubMed.
- For previous syntheses, see: (a) S. D. Walker, C. J. Borths, E. DiVirgilio, L. Huang, P. Liu, H. Morrison, K. Sugi, M. Tanaka, J. C. S. Woo and M. M. Faul, Org. Process Res. Dev., 2011, 15, 570–580 CrossRef CAS; (b) J. Y. Hamilton, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 7532–7535 CrossRef CAS PubMed , and ref. 8b.
- M. Ishikawa, T. Haketa, C. Nakama, T. Nishimura, J. Shibata, T. Shimamura and T. Yamakawa, US201165739 A1, March 17, 2011.
- M. Eckhardt and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 13642–13643 CrossRef CAS PubMed.
- Otera’s catalyst is 1,1,3,3-tetrabutyl-1,3-diisothiocyanatodistannoxane. See (a) J. Otera, N. Danoh and H. Nozaki, J. Org. Chem., 1991, 56, 5307–5311 CrossRef CAS; (b) J. Otera, Chem. Rev., 1993, 93, 1449–1470 CrossRef CAS.
- V. Belting and N. Krause, Org. Lett., 2006, 8, 4489–4492 CrossRef CAS PubMed.
- Z. Shen and X. Lu, Tetrahedron, 2006, 62, 10896–10899 CrossRef CAS.
- C. Lambert, K. Utimoto and H. Nozaki, Tetrahedron Lett., 1984, 25, 5323–5326 CrossRef CAS.
- J. Raushel and V. V. Fokin, Org. Lett., 2010, 12, 4952–4955 CrossRef CAS PubMed.
- (a) T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef CAS PubMed; (b) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034–18035 CrossRef CAS PubMed; (c) N. P. Grimster, L. Zhang and V. V. Fokin, J. Am. Chem. Soc., 2010, 132, 2510–2511 CrossRef CAS PubMed.
- B. M. Trost, M. J. Bartlett, A. H. Weiss, A. J. von Wangelin and V. S. Chan, Chem.–Eur. J., 2012, 18, 16498–16509 CrossRef CAS PubMed.
- F. López, S. R. Harutyunyan, A. Meetsma, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 2752–2756 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc01724j |
| This journal is © The Royal Society of Chemistry 2016 |