
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Face and edge directed self-assembly of Pd12 tetrahedral nano-cages and their self-sorting†
Prodip
Howlader
and
Partha Sarathi
Mukherjee
*
Inorganic and Physical Chemistry Department, Indian Institute of Science, Bangalore-560012, India. E-mail: psm@ipc.iisc.ernet.in; Fax: +91-80-23601552; Tel: +91-80-22933352
First published on 20th May 2016
Abstract
Reactions of a cis-blocked Pd(II) 90° acceptor [cis-(tmeda)Pd(NO3)2] (M) with 1,4-di(1H-tetrazol-5-yl)benzene (H2L1) and [1,3,5-tri(1H-tetrazol-5-yl)benzene] (H3L2) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 3:2 molar ratios respectively, yielded soft metallogels G1 and G2 [tmeda = N,N,N′,N′-tetramethylethane-1,2-diamine]. Post-metalation of the gels G1 and G2 with M yielded highly water-soluble edge and face directed self-assembled Pd12 tetrahedral nano-cages T1 and T2, respectively. Such facile conversion of Pd(II) gels to discrete tetrahedral metallocages is unprecedented. Moreover, distinct self-sorting of these two tetrahedral cages of similar sizes was observed in the self-assembly of M with a mixture of H2L1 and H3L2 in aqueous medium. The edge directed tetrahedral cage (T1) was successfully used to perform Michael reactions of a series of water insoluble nitro-olefins assisted by encapsulation into the cage in aqueous medium.
:1 and 3:2 molar ratios respectively, yielded soft metallogels G1 and G2 [tmeda = N,N,N′,N′-tetramethylethane-1,2-diamine]. Post-metalation of the gels G1 and G2 with M yielded highly water-soluble edge and face directed self-assembled Pd12 tetrahedral nano-cages T1 and T2, respectively. Such facile conversion of Pd(II) gels to discrete tetrahedral metallocages is unprecedented. Moreover, distinct self-sorting of these two tetrahedral cages of similar sizes was observed in the self-assembly of M with a mixture of H2L1 and H3L2 in aqueous medium. The edge directed tetrahedral cage (T1) was successfully used to perform Michael reactions of a series of water insoluble nitro-olefins assisted by encapsulation into the cage in aqueous medium.
Introduction
Nature, especially in biological systems, has an extraordinary ability to develop complex and functional molecular assemblies employing reversible non-covalent interactions.1 These natural aesthetic examples have enticed synthetic chemists over the past several years to develop potential synthetic protocols to produce myriad complex assemblies employing non-covalent interactions like H-bonding, π–π interactions and stronger metal-ligand coordination.2–4 In principle, by tuning these directional non-covalent driving forces one can construct supramolecular polymers as well as discrete molecular assemblies. Such ordered and self-organized polymers may go through gelation in the presence of strong inter-molecular interactions (network formation).5 The three-dimensional fibrous network structure generated by the self-assembly of the gelator molecules may impart solid-like properties in the gels, which make them worthy candidates for substrate recognition, catalysis and biomedical applications.6 Meanwhile, discrete molecular architectures are also potential candidates for stabilizing reactive intermediates, sensing, and host-guest chemistry, as well as cavity-induced ‘organic transformations’.7Introducing multiple interactions in a single system and tuning them by varying different parameters such as solvent type, temperature and stoichiometry to construct desired molecular architectures (discrete or polymeric), including their transformation, is interesting and provides diverse functional materials. Discrete supramolecular metallocages have been designed, mainly employing pyridyl, imidazole and carboxylate linkers.8 Polytetrazoles are not preferred linkers in designing molecular cages due to difficulty in predicting the final structure because of the multiple nitrogen atoms. The N–H moiety in the tetrazole ring promotes many metal complexes of polytetrazoles to form metallogels by intermolecular H-bonding.9
The coordination-driven self-assembly strategy has inspired chemists in the past two decades to design basic 3D structures of high symmetry and well defined shapes and sizes.7b,10 A tetrahedron is one of the common 3D geometries. Several tetrahedral molecular cages have been reported using mainly octahedral metal ions with a few examples of analogous cages of lanthanides with a higher coordination number, though their solubility is restricted to organic solvents in the majority of cases.11,12 Due to geometric restriction, square planar metal ions like Pd(II) have not been explored much for the design of regular tetrahedral cages.
Herein, we report the formation of a supramolecular Pd(II) metallogel (G1) upon 1:1 treatment of H2L1 [1,4-di(1H-tetrazol-5-yl)benzene] with cis-(tmeda)Pd(NO3)2 (M) in water or DMSO (Scheme 1). Post-metalation of G1 with M in water/or DMSO caused deprotonation of the N–H moieties and transformed the gel into a highly water-soluble unusual tetrahedral M12L16 (T1) cage where the donors (L1) occupy the six edges of the tetrahedron. Such post-metalation was expected to form an open 2D square M8L14 as observed using pyrazole linkers instead of a closed 3D tetrahedral cage.13 This unusual outcome enticed our attention towards a face directed tetrahedron to examine the generality of designing tetrahedral cages of square planar metal ions employing polytetrazole donors. Replacement of H2L1 with H3L2 [1,3,5-tri(1H-tetrazol-5-yl)benzene] in the above-mentioned two-step reaction afforded a Pd(II) gel (G2) followed by a water soluble chiral tetrahedral cage M12L24 (T2) (Scheme 1), where the four triangular faces of the tetrahedron were occupied by L2. T1 and T2 are unusual from a symmetry point of view.11b,14 The spatial orientations of the linkers L1 and L2 enable the final assemblies (T1 and T2) to adopt a specific geometry with a loss of symmetry to become chiral cages of square planar Pd(II) using achiral building blocks.
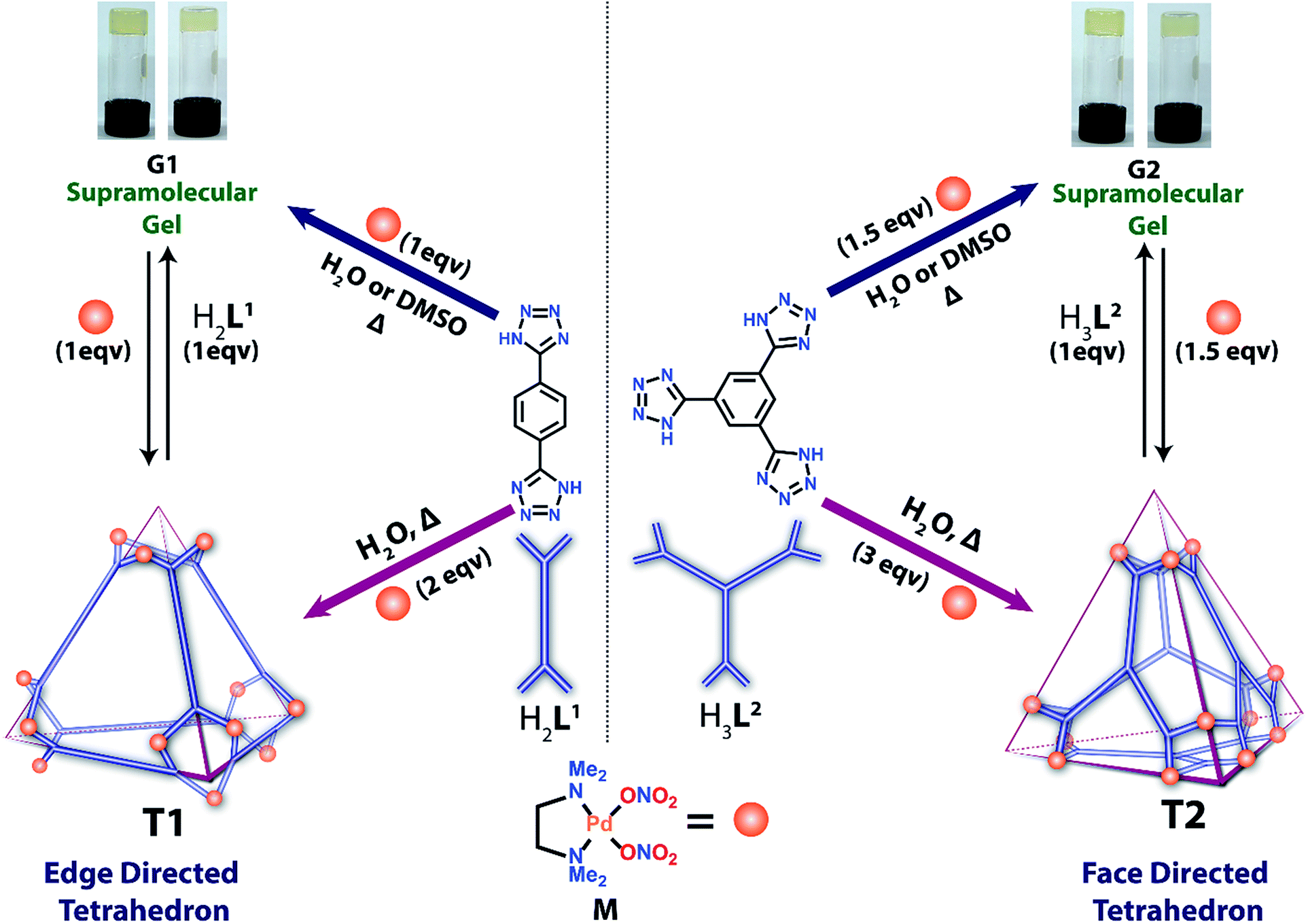 | ||
| Scheme 1 Schematic representation of the synthesis of metallogels and their facile conversion to 3D tetrahedral nano-cages. | ||
Results and discussion
Synthesis and characterization of metallogels
Both the ligands (H2L1 and H3L2) were prepared from their cyano derivatives following the reported procedure.9 Supramolecular hydrogel G1 was prepared by adding an aqueous yellow solution of the acceptor M to H2L1 in a 1:1 molar ratio with a total weight percentage of 2. The mixture was heated with stirring at 60 °C for 2 h to give a transparent solution, and subsequent cooling to room temperature yielded the gel G1 (Scheme 1). A similar gel was also obtained when DMSO was used as a solvent in the above reaction. The viscoelastic nature of the gel was characterized by two types of dynamic rheology15 experiments: (a) frequency sweep at a constant stress of 1.0 Pa and (b) stress sweep at a constant frequency of 1.0 Hz. The stiffness of the gel (G′/G′′) can be measured from the first experiment whereas fragility (yield stress) can be measured from the second one. For G1 and G2 these experimental values are quite small (ESI†), which suggests the formation of a weak gel (Fig. 1). Finally, field emission scanning electron microscopy of the xerogels revealed that the formation of a fibrous nanostructure (tertiary structure) is responsible for gelation (ESI†).
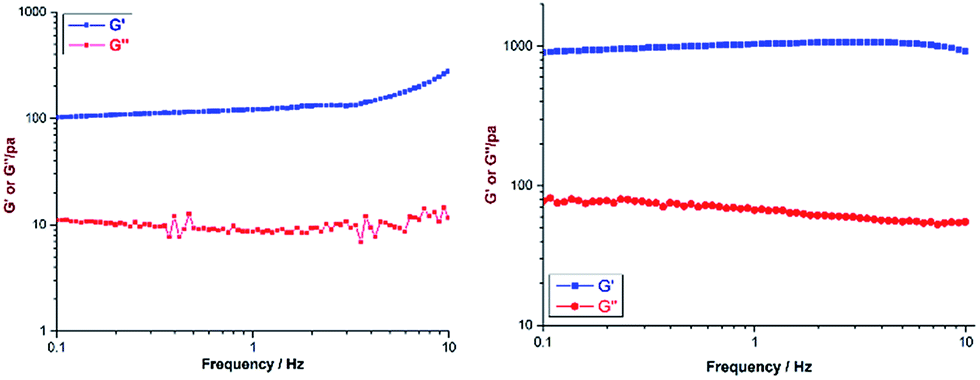 | ||
| Fig. 1 G′′ and G′ vs. frequency sweep for G1 (left) and G2 (right). | ||
Synthesis and characterization of nano-cages
Treatment of the metallogel G1 with an aqueous solution of one molar equivalent of M at 60 °C for 3 h yielded T1. To investigate the extent of the deprotonation of the ligand H2L1 during the self-assembly process, a pH monitored self-assembly reaction was performed in water with 2 molar equivalents of M and one molar equivalent of H2L1. The change in pH of the reaction mixture after the consumption of all the ligand H2L1 (ESI†) clearly suggests deprotonation of both of the N–H protons of H2L1. Evaporation of the solvent under vacuum and subsequent washing of the resulting solid with acetone yielded the final product in pure form. In the case of the DMSO gel, a similar procedure was followed with a DMSO solution of M, and finally, T1 was obtained as a white solid upon treating the solution with ethyl acetate. 1H NMR analysis of the product (T1) in D2O displayed a single peak at 9.1 ppm, which is significantly downfield shifted compared to the peak for free H2L1 (ESI†). ESI-MS analysis in water indicated a [12 + 6] composition of M and L1 in the final product (T1) by the appearance of two major peaks at m/z = 1110 and 1500 with isotropic distribution patterns corresponding to the fragments [T1(NO3)8]4+ and [T1(NO3)9]3+, respectively (ESI†).
T2 was synthesized following the above-mentioned procedure using H3L2 instead of H2L1. The metallogel G2 was obtained by treating H3L2 and M in a 2:3 molar ratio in DMSO or water. G2 was converted to T2 by post-metalation treatment with M. The presence of a singlet at 10.05 ppm in 1H NMR indicated the formation of a single and symmetrical product (ESI†). ESI-MS analysis displayed two major peaks at m/z = 1070.64 and m/z = 1048.53 with the isotropic distribution patterns corresponding to the fragments [T2(NO3)8]4+ and [T2(NO3)9]3+, which confirmed the [12 + 4] composition of M and L2 in the final assembly T2 (ESI†).
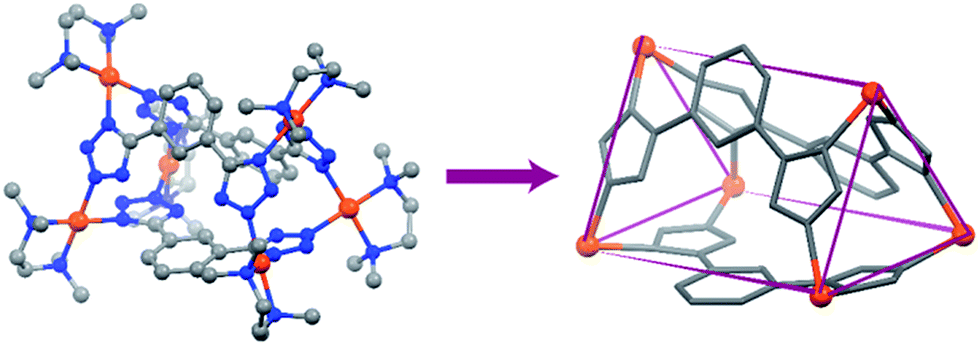
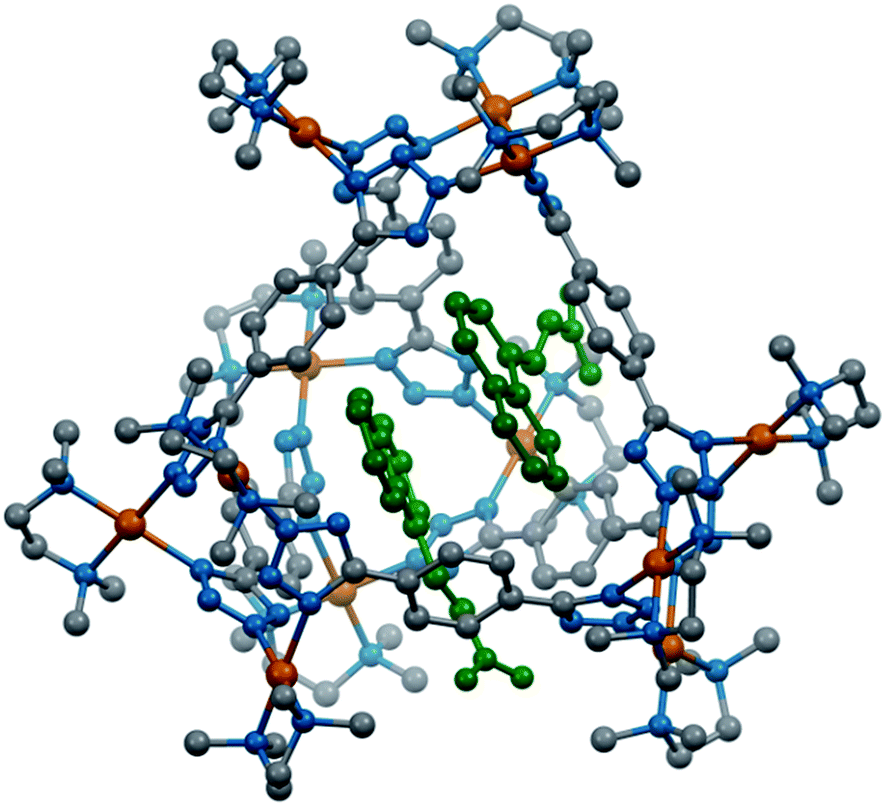
Finally, both the cages were successfully crystallized by diffusion of acetone vapour into the aqueous solutions of the cages. Single crystal XRD analysis of both T1 and T2 unequivocally confirmed the formation of 3D tetrahedral cages (Fig. 2). T1 was crystallized in the C2/c space group and each vertex corner of the tetrahedron contains 3 Pd(II) acceptors connected by the nitrogens of the tetrazole moieties of the linkers to form a Pd3 triangle and the ligands L1 are fastened by two corners along the edges. Whereas T2 was crystallized in the I222 space group and each vertex corner of the tetrahedron contains three acceptors and the linker L2 is tied by three corners along the face. So, in the case of T1 the ligands occupy the edges of the tetrahedron while in T2 the donors occupy the triangular faces of the tetrahedron. L1 and L2 bind the Pd(II) centers through the nitrogens of the 1 and 3 positions of the tetrazole rings (Fig. 3). Such binding modes of the tetrazole ligands (for L1 and L2) make them special in terms of “symmetry breaking”,11b because L1 and L2 have no plane of symmetry when they are coordinated to metal centers, except in the molecular plane, which no longer exists in the 3D cage. Now if we consider the 1,3 binding mode, the tetrazole moiety is residing on the right with respect to the phenyl ring (Fig. 3). If we consider this geometry as a Δ configuration; the 2,5 binding mode brings the tetrazole moiety over to the left side, which may be represented as a Λ configuration. Crystal structures of T1 and T2 (CCDC 1471523 and 1471434†) clearly displayed that, in a particular nano-cage, all the linkers have a similar type of coordination mode with the Pd(II) acceptors as shown in Fig. 4. Hence, the handedness of all the Pd3 triangles (vertices of each tetrahedron) is the same, either ΔΔΔΔ or ΛΛΛΛ. Such spatial arrangements of the donors and acceptors make the resulting cages chiral even without using any chiral building blocks. As the cage T1 was crystallized in a centrosymmetric space group (C2/c), the crystal packing exposed the presence of both enantiomers along the c glide plane in the unit cell.
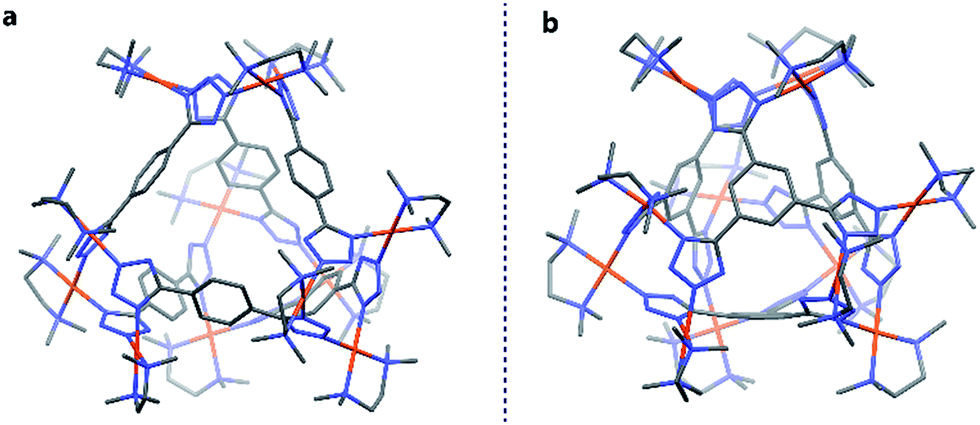 | ||
| Fig. 2 Crystal structures of the tetrahedra T1 and T2 (all the hydrogen atoms are omitted for clarity). Colour codes: grey: carbon, blue: nitrogen and brown: palladium. | ||
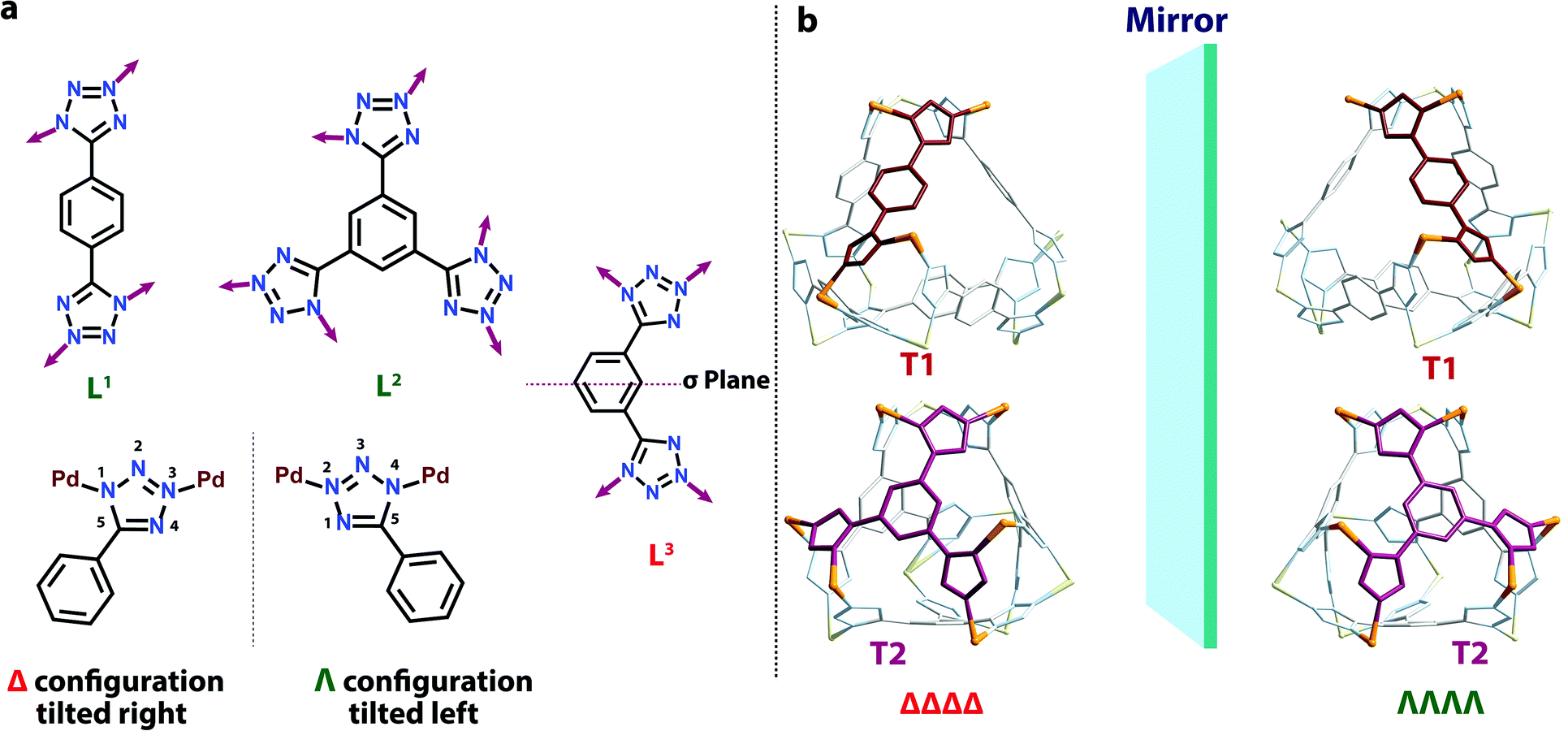 | ||
| Fig. 3 (a) Coordination modes of L1L2 and L3 in the final assemblies. (b) Representations of the enantiomeric forms of T1 (top) and T2 (bottom). | ||
 | ||
| Fig. 4 Crystal structure of the achiral molecular prism P. Colour codes: grey, carbon; blue, nitrogen; and brown, palladium. | ||
Although T2 was crystallized in a chiral (I222) space group, the Flack parameter was evaluated to be 0.5, which is due to the formation of an inversion twin i.e. a racemic mixture. Nonetheless, linker L3 forms a prismatic structure M6L33 (P) (CCDC 1471478†).This was synthesized in a similar way as followed for T1 and T2. The binding mode of L3 is shown in Fig. 3. It forms two Pd3 triangles with opposing handedness yielding an achiral geometry (Fig. 4). So, the achiral or chiral nature of the final assemblies is controlled by the handedness of the Pd3 triangular units.
Variable-temperature 1H NMR study of T1
Interestingly, the 1H NMR study of T1 at room temperature does not support the asymmetric nature of the solid-state XRD structure. The single peak at 9.1 ppm is due to the possibility of rotation of the phenyl ring with a frequency higher than the frequency of the NMR technique at room temperature. Variable temperature NMR spectra at low temperature were needed to confirm this phenomenon. Although the cage was synthesized in water or DMSO, it is soluble in MeOH. This allowed us to study 1H NMR of the cage at −50 °C (Fig. 5), which displayed two different peaks at 8.5 and 9.8 ppm. Moreover, a DOSY NMR study at that temperature showed the presence of a single product. Upon increasing the temperature, the peaks started to merge together and at around −20 °C these peaks disappeared and the original peak at 9.1 ppm appeared.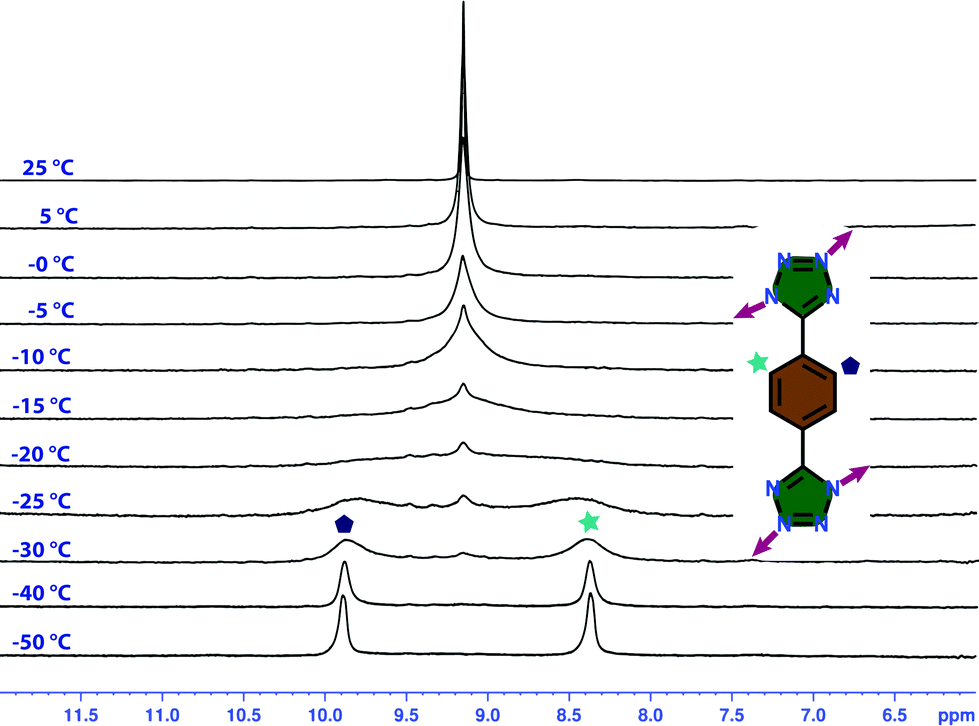 | ||
| Fig. 5 Temperature dependent 1H NMR spectra of T1 in CD3OD showing the presence of two different sets of phenyl protons at low temperature. | ||
Self-sorting experiment of T1 and T2
As both the linkers L1 and L2 form tetrahedral cages of almost equivalent shape and size with complementary building units (edge directed and face directed respectively), we were curious to know whether the presence of both the donors (L1 and L2) in a single reaction mixture would result in these two individual tetrahedral cages by a self-sorting process or a complicated multicomponent product. To investigate this, a self-assembly experiment was carried out in water taking M, H2L1 and H3L2 in a 24:6:4 molar ratio. The final product was isolated and characterized by 1H NMR and mass spectroscopy. The presence of two sharp singlets at 10.07 and 9.13 ppm in the 1H NMR spectra clearly indicates the formation of T2 and T1 in pure form without the presence of any byproduct (Fig. 6).
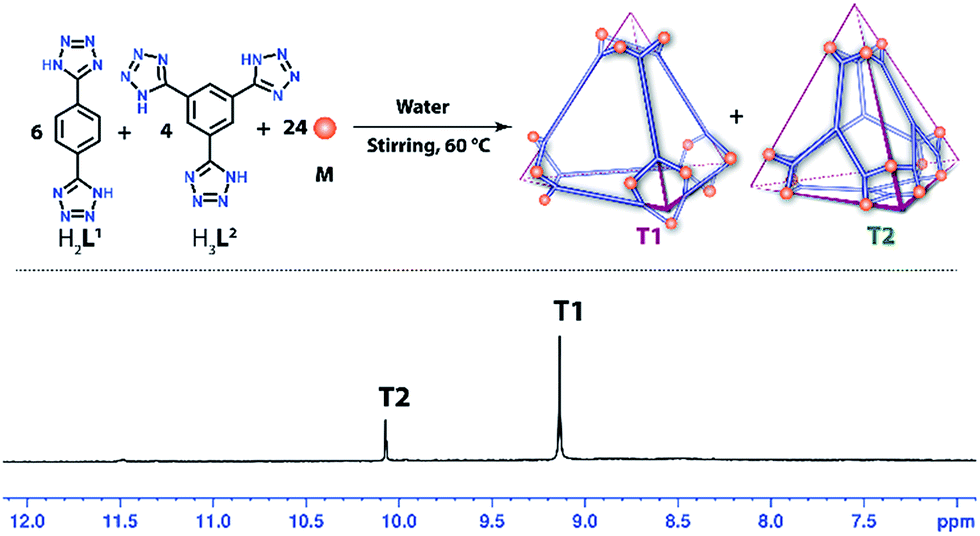 | ||
| Fig. 6 Self-sorting of T1 and T2 from a mixture of M, H2L1 and H3L2 in water (top) and the 1H NMR spectrum of the resulting mixture. | ||
Catalytic Michael addition reaction
The donors (L1) in the edge directed cage T1 occupy the edges of the tetrahedron by joining two Pd3 triangles, keeping four triangular faces of the tetrahedron open, having dimensions of 6.8 Å × 6.9 Å, which may allow one or more aromatic guests to enter. Cage T2 has complementary geometry, where all four faces of the tetrahedron are occupied by the linker L2 leaving no open windows for aromatic guest encapsulation. The edges of this cage are also blocked by the methyl groups of the acceptor leaving no pathway for the entry of guests.This structural nature of the cages motivated us to study the possibility of encapsulating a water insoluble aromatic guest. To verify our observation the aqueous solutions of the cages were treated with 1-(2-nitrovinyl)naphthalene (1). As anticipated, the aromatic guest (1) was encapsulated in T1 which was identified by a change in the colour of the cage solution (colourless to light yellow) and was finally confirmed by 1H NMR spectroscopy (Fig. 7) where significant up-field shifts of the guest peaks (1) were noticed. The stoichiometry of cage vs. guest was evaluated to be 1:2 from the 1H NMR spectra (ESI†). The broad 1H NMR peaks for the encapsulated cage can be explained in terms of the rotational movement of the phenyl ring of the cage T1, which is restricted by the guest molecules present inside the cage. This host-guest binding was further confirmed by 1H DOSY NMR (ESI†), which showed identical diffusion coefficients for the guest (1) and the host cage.
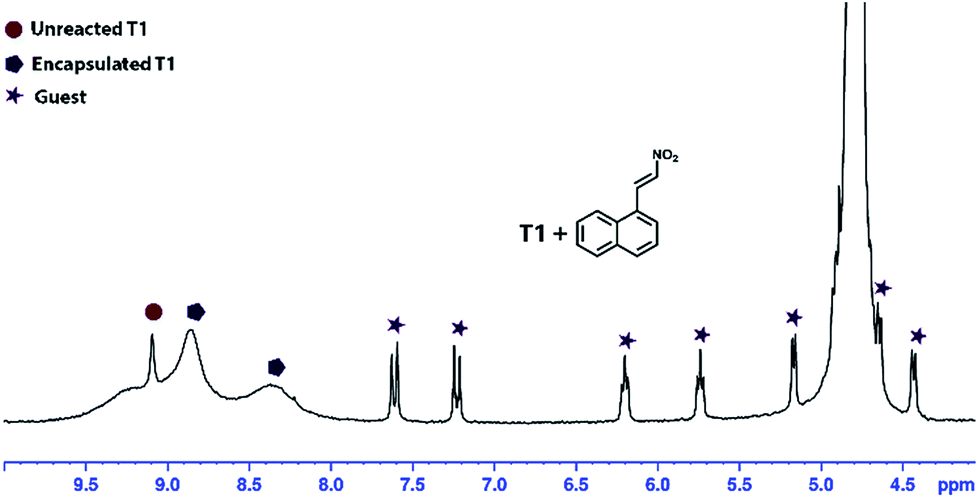 | ||
| Fig. 7 1H NMR spectrum of guest (1) encapsulated in T1 (1 ⊂ T1) in D2O. | ||
Several attempts to crystallize the guest encapsulated cage 1 ⊂ T1 have so far been unsuccessful. The host-guest (1:2) complex was modeled and the structure was optimized by a semi-empirical method with a PM6 basis set. As the nitro group is more polar than the naphthyl group, it may lean outside the cavity. However, the naphthyl moieties are stabilized inside the hydrophobic cavity of the cage by π–π stacking interactions (Fig. 8).
 | ||
| Fig. 8 PM6 energy optimized structure of the guest encapsulated cage 1 ⊂ T1. | ||
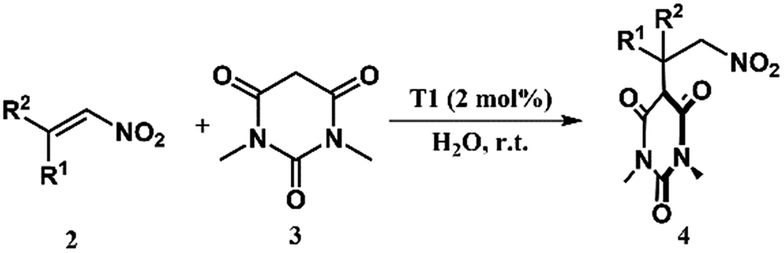
This successful encapsulation of the aromatic nitro-alkene enabled it to be soluble in an aqueous medium and it was possible to carry out further organic transformation utilizing the cage T1 in a phase transfer catalytic manner. Several reactions were studied using 1,3-dimethylbarbituric acid (3) and different nitro-alkenes (2) (Table 1). Although similar reactions were studied by us using a urea decorated functional self-assembled molecular prism in a heterogeneous manner,7i the present study was done in a homogeneous manner. The initial results presented in Table 1 indicate an enhancement of the yield in the presence of the cage T1. Cage T2 didn't show any such encapsulation of nitro-olefins due to the absence of large open windows. Control reactions were carried out with T2, which indicated almost no catalytic effect on these reactions.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Time (min) | Yieldb (%) | ||
| Blank | With T2 | With T1 | |||||
| a Nitro-olefins 2 (0.02 mmol), 1,3-dimethylbarbituric acid 3 (0.02 mmol), catalyst T1 (2 mol%), and water (1 mL), at r.t. with stirring. b Crude yields were determined by 1H NMR studies. | |||||||
| 1 | 1-Pyrenyl | H | 4a | 72 h | 14 | — | 20 |
| 2 | 1-Naphthyl | H | 4b | 90 | 15 | 22 | 41 |
| 3 | Ph | Me | 4c | 60 | 18 | 27 | 59 |
| 4 | 4-Me-Ph | H | 4d | 15 | 10 | — | 50 |
| 5 | 4-MeO-Ph | H | 4e | 30 | 13 | 20 | 54 |
| 6 | 2-Furanyl | H | 4f | 10 | 12 | — | 60 |
Conclusions
In conclusion, self-assembly of a Pd(II) acceptor M with polytetrazole donors H2L1 and H3L2 in 1:1 and 3:2 molar ratios yielded supramolecular metallogels G1 and G2, respectively. Post-metalation reactions of the gels with M transformed them to edge- and face-directed self-assembled water-soluble tetrahedral cages T1 and T2. Reversible transformation of the cages to their corresponding parent gels was also achieved upon treating them with respective tetrazoles. To the best of our knowledge, such facile and reversible transformation of metallogels to discrete metallocages is unusual. Although pyridine, imidazole and carboxylates have been widely employed to obtain metallosupramolecular discrete architectures, T1–T2 represent a unique class of tetrahedral architectures of square planar metal ions employing polytetrazoles as linkers. Interestingly, the coordination modes of the tetrazole units promote ‘symmetry breaking’ in these assemblies. Single crystal structures showed that the handedness of the Pd3 vertices formed by the tetrazole units is responsible for the ‘symmetry breaking’ which led to the formation of chiral nano-cages having T symmetry. However, the binding mode of L3 imparts a mirror symmetry leading to an achiral prismatic structure P. The mixed-ligand self-assembly with M produced self-sorted cages T1 and T2 without any by-product. This clean self-sorting between the iso-structural tetrahedral cages of similar sizes in a complex mixture of H2L1, H3L2 and M is reminiscent of the selectivity observed in nature. The open triangular windows in the edge directed tetrahedron (T1) enabled it to encapsulate water insoluble aromatic nitro-olefins in aqueous medium followed by Michael reactions of the nitro-olefins with 1,3-dimethybarbituric acid using phase transfer-type catalysis. The freshly prepared water-soluble tetrahedral cages provide a platform for the development of a new generation of functional molecular nanovessels employing polytetrazole donors for chemical reactions and encapsulation of various guests.
Acknowledgements
P. S. M. thanks the DST (New Delhi) for the research grant (EMR/2015/002353). P. H. is grateful to Mr Sayantan Chatterjee for the rheology experiments and also thankful to Mr Shaunak Chakraborty for fruitful discussion on the single crystal structure analysis.References
- B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS PubMed.
- (a) G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed; (b) M. C. T. Fyfe and J. F. Stoddart, Acc. Chem. Res., 1997, 30, 393–401 CrossRef CAS; (c) X.-M. Chen and G.-F. Liu, Chem.–Eur. J., 2002, 8, 4811–4817 CrossRef CAS PubMed; (d) A. P. H. J. Schenning, P. Jonkheijm, E. Peeters and E. W. Meijer, J. Am. Chem. Soc., 2001, 123, 409–416 CrossRef CAS; (e) L. Zhang, Y.-J. Lin, Z.-H. Li and G.-X. Jin, J. Am. Chem. Soc., 2015, 137, 13670–13678 CrossRef CAS PubMed; (f) Y.-F. Han and G.-X. Jin, Acc. Chem. Res., 2014, 47, 3571–3579 CrossRef CAS PubMed.
- (a) Y.-R. Zheng, H.-B. Yang, B. H. Northrop, K. Ghosh and P. J. Stang, Inorg. Chem., 2008, 47, 4706–4711 CrossRef CAS PubMed; (b) Z. Zhao, Y.-R. Zheng, M. Wang, J. B. Pollock and P. J. Stang, Inorg. Chem., 2010, 49, 8653–8655 CrossRef CAS PubMed; (c) D. Li, W. Zhou, K. Landskron, S. Sato, C. J. Kiely, M. Fujita and T. Liu, Angew. Chem., 2011, 123, 5288–5293 CrossRef; (d) W. Wang, Y. Zhang, B. Sun, L.-J. Chen, X.-D. Xu, M. Wang, X. Li, Y. Yu, W. Jiang and H.-B. Yang, Chem. Sci., 2014, 5, 4554–4560 RSC.
- (a) H. Kihara, T. Kato, T. Uryu and J. M. J. Fréchet, Chem. Mater., 1996, 8, 961–968 CrossRef CAS; (b) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed; (c) T. Yokoyama, S. Yokoyama, T. Kamikado, Y. Okuno and S. Mashiko, Nature, 2001, 413, 619–621 CrossRef CAS PubMed; (d) J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Chem. Soc. Rev., 2013, 42, 1700–1712 RSC; (e) W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC.
- (a) E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed; (b) T. Rossow and S. Seiffert, Polym. Chem., 2014, 5, 3018–3029 RSC; (c) J. A. Foster, R. M. Parker, A. M. Belenguer, N. Kishi, S. Sutton, C. Abell and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 9722–9729 CrossRef CAS PubMed; (d) P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC; (e) X.-Q. Wang, W. Wang, G.-Q. Yin, Y.-X. Wang, C.-W. Zhang, J.-M. Shi, Y. Yu and H.-B. Yang, Chem. Commun., 2015, 51, 16813–16816 RSC; (f) K. Pandurangan, J. A. Kitchen, S. Blasco, F. Paradisi and T. Gunnlaugsson, Chem. Commun., 2014, 50, 10819–10822 RSC.
- (a) S. Seiffert and J. Sprakel, Chem. Soc. Rev., 2012, 41, 909–930 RSC; (b) S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC; (c) A. Tamura and N. Yui, Chem. Commun., 2014, 50, 13433–13446 RSC; (d) J. F. Miravet and B. Escuder, in Supramolecular Systems in Biomedical Fields, The Royal Society of Chemistry, 2013, pp. 331–354, 10.1039/9781849737821-00331; (e) K. Ariga, H. Ito, J. P. Hill and H. Tsukube, Chem. Soc. Rev., 2012, 41, 5800–5835 RSC; (f) X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- (a) S. K. Samanta and M. Schmittel, Org. Biomol. Chem., 2013, 11, 3108–3115 RSC; (b) W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., 2011, 123, 3541–3545 CrossRef; (c) X. Bao, S. Rieth, S. Stojanović, C. M. Hadad and J. D. Badjić, Angew. Chem., 2010, 122, 4926–4929 CrossRef; (d) M. Porel, C.-H. Chuang, C. Burda and V. Ramamurthy, J. Am. Chem. Soc., 2012, 134, 14718–14721 CrossRef CAS PubMed; (e) C. J. Hastings, R. G. Bergman and K. N. Raymond, Chem.–Eur. J., 2014, 20, 3966–3973 CrossRef CAS PubMed; (f) D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746–2747 CrossRef CAS PubMed; (g) W. M. Hart-Cooper, K. N. Clary, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2012, 134, 17873–17876 CrossRef CAS PubMed; (h) D. Samanta and P. S. Mukherjee, Chem. Commun., 2013, 49, 4307–4309 RSC; (i) P. Howlader, P. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1668–1676 CrossRef CAS PubMed; (j) T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464–2474 CrossRef CAS PubMed; (k) S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476–8479 CrossRef CAS PubMed; (l) A. M. Castilla, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 2342–2351 CrossRef CAS PubMed; (m) C. Zhao, F. D. Toste, K. N. Raymond and R. G. Bergman, J. Am. Chem. Soc., 2014, 136, 14409–14412 CrossRef CAS PubMed; (n) C. Zhao, Q.-F. Sun, W. M. Hart-Cooper, A. G. DiPasquale, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 18802–18805 CrossRef CAS PubMed.
- (a) M. Fujita, Chem. Soc. Rev., 1998, 27, 417–425 RSC; (b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed; (c) S. Mukherjee and P. S. Mukherjee, Chem. Commun., 2014, 50, 2239–2248 RSC; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (e) M. Han, Y. Luo, B. Damaschke, L. Gómez, X. Ribas, A. Jose, P. Peretzki, M. Seibt and G. H. Clever, Angew. Chem., Int. Ed., 2016, 55, 445–449 CrossRef CAS PubMed; (f) Y. J. Park, J. Y. Ryu, H. Begum, M. H. Lee, P. J. Stang and J. Lee, J. Am. Chem. Soc., 2015, 137, 5863–5866 CrossRef CAS PubMed; (g) D. M. Engelhard, S. Freye, K. Grohe, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 4747–4750 CrossRef CAS PubMed; (h) G. H. Clever, W. Kawamura, S. Tashiro, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2012, 51, 2606–2609 CrossRef CAS PubMed; (i) M. L. Saha, N. Mittal, J. W. Bats and M. Schmittel, Chem. Commun., 2014, 50, 12189–12192 RSC; (j) C. Gütz, R. Hovorka, N. Struch, J. Bunzen, G. Meyer-Eppler, Z.-W. Qu, S. Grimme, F. Topić, K. Rissanen, M. Cetina, M. Engeser and A. Lützen, J. Am. Chem. Soc., 2014, 136, 11830–11838 CrossRef PubMed; (k) S. Shanmugaraju, S. A. Joshi and P. S. Mukherjee, Inorg. Chem., 2011, 50, 11736–11745 CrossRef CAS PubMed; (l) S. Ghosh, B. Gole, A. K. Bar and P. S. Mukherjee, Organometallics, 2009, 28, 4288–4296 CrossRef CAS.
- H. Lee, S. Kang, J. Y. Lee and J. H. Jung, Soft Matter, 2012, 8, 2950–2955 RSC.
- (a) Y. Yamanoi, Y. Sakamoto, T. Kusukawa, M. Fujita, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 2001, 123, 980–981 CrossRef CAS PubMed; (b) T. Nakamura, H. Ube, R. Miyake and M. Shionoya, J. Am. Chem. Soc., 2013, 135, 18790–18793 CrossRef CAS PubMed; (c) A. K. Bar, R. Chakrabarty, G. Mostafa and P. S. Mukherjee, Angew. Chem., Int. Ed., 2008, 47, 8455–8459 CrossRef CAS PubMed; (d) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, J. Am. Chem. Soc., 2012, 134, 9488–9497 CrossRef CAS PubMed; (e) K. Mahata, M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2010, 132, 15933–15935 CrossRef CAS PubMed; (f) R. Hovorka, G. Meyer-Eppler, T. Piehler, S. Hytteballe, M. Engeser, F. Topić, K. Rissanen and A. Lützen, Chem.–Eur. J., 2014, 20, 13253–13258 CrossRef CAS PubMed; (g) C. Gütz, R. Hovorka, C. Klein, Q.-Q. Jiang, C. Bannwarth, M. Engeser, C. Schmuck, W. Assenmacher, W. Mader, F. Topić, K. Rissanen, S. Grimme and A. Lützen, Angew. Chem., Int. Ed., 2014, 53, 1693–1698 CrossRef PubMed; (h) R. Zhu, J. Lübben, B. Dittrich and G. H. Clever, Angew. Chem., Int. Ed., 2015, 54, 2796–2800 CrossRef CAS PubMed; (i) J. E. Beves, C. J. Campbell, D. A. Leigh and R. G. Pritchard, Angew. Chem., Int. Ed., 2013, 52, 6464–6467 CrossRef CAS PubMed; (j) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed.
- (a) T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476–2490 RSC; (b) W. Meng, T. K. Ronson and J. R. Nitschke, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10531–10535 CrossRef CAS PubMed; (c) T. K. Ronson, D. A. Roberts, S. P. Black and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 14502–14512 CrossRef CAS PubMed.
- (a) Y.-R. Zheng, Z. Zhao, H. Kim, M. Wang, K. Ghosh, J. B. Pollock, K.-W. Chi and P. J. Stang, Inorg. Chem., 2010, 49, 10238–10240 CrossRef CAS PubMed; (b) L.-L. Yan, C.-H. Tan, G.-L. Zhang, L.-P. Zhou, J.-C. Bünzli and Q.-F. Sun, J. Am. Chem. Soc., 2015, 137, 8550–8555 CrossRef CAS PubMed.
- (a) S.-Y. Yu, H.-P. Huang, S.-H. Li, Q. Jiao, Y.-Z. Li, B. Wu, Y. Sei, K. Yamaguchi, Y.-J. Pan and H.-W. Ma, Inorg. Chem., 2005, 44, 9471–9488 CrossRef CAS PubMed; (b) H.-P. Huang, S.-H. Li, S.-Y. Yu, Y.-Z. Li, Q. Jiao and Y.-J. Pan, Inorg. Chem. Commun., 2005, 8, 656–660 CrossRef CAS; (c) K. Umakoshi, Y. Yamauchi, K. Nakamiya, T. Kojima, M. Yamasaki, H. Kawano and M. Onishi, Inorg. Chem., 2003, 42, 3907–3916 CrossRef CAS PubMed; (d) I. Ara, J. Forniés, R. Lasheras, A. Martín and V. Sicilia, Eur. J. Inorg. Chem., 2006, 2006, 948–957 CrossRef; (e) X.-F. Jiang, J.-H. Hu, J. Tong and S.-Y. Yu, Inorg. Chem. Commun., 2013, 36, 232–235 CrossRef CAS.
- (a) G. Seeber, B. E. F. Tiedemann and K. N. Raymond, in Supramolecular Chirality, ed. M. Crego-Calama and D. N. Reinhoudt, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 147–183, DOI:10.1007/128_033; (b) D. L. Caulder, R. E. Powers, T. N. Parac and K. N. Raymond, Angew. Chem., Int. Ed., 1998, 37, 1840–1843 CrossRef CAS; (c) D. L. Caulder, C. Brückner, R. E. Powers, S. König, T. N. Parac, J. A. Leary and K. N. Raymond, J. Am. Chem. Soc., 2001, 123, 8923–8938 CrossRef CAS PubMed; (d) M. Scherer, D. L. Caulder, D. W. Johnson and K. N. Raymond, Angew. Chem., Int. Ed., 1999, 38, 1587–1592 CrossRef; (e) W. M. Hart-Cooper, C. Zhao, R. M. Triano, P. Yaghoubi, H. L. Ozores, K. N. Burford, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Sci., 2015, 6, 1383–1393 RSC; (f) G. Zhang, G. Gil-Ramírez, A. Markevicius, C. Browne, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2015, 137, 10437–10442 CrossRef CAS PubMed.
- B. N. S. Thota, A. J. Savyasachi, N. Lukashev, I. Beletskaya and U. Maitra, Eur. J. Org. Chem., 2014, 2014, 1406–1415 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR, ESI-MS spectra and X-ray data in CIF format. CCDC 1471523, 1471434 and 1471478. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02012g |
| This journal is © The Royal Society of Chemistry 2016 |