
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequence-defined oligo(ortho-arylene) foldamers derived from the benzannulation of ortho(arylene ethynylene)s†
Dan
Lehnherr
 a,
Chen
Chen
b,
Zahra
Pedramrazi
b,
Catherine R.
DeBlase
a,
Joaquin M.
Alzola
a,
Ivan
Keresztes
c,
Emil B.
Lobkovsky
d,
Michael F.
Crommie
*b and
William R.
Dichtel
*ae
a,
Chen
Chen
b,
Zahra
Pedramrazi
b,
Catherine R.
DeBlase
a,
Joaquin M.
Alzola
a,
Ivan
Keresztes
c,
Emil B.
Lobkovsky
d,
Michael F.
Crommie
*b and
William R.
Dichtel
*ae
aDepartment of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, New York 14853-1301, USA. E-mail: wdichtel@cornell.edu
bDepartment of Physics, University of California at Berkeley, Berkeley, California 94720, USA. E-mail: crommie@berkeley.edu
cNuclear Magnetic Resonance Laboratory, Cornell University, Ithaca, New York 14853-1301, USA
dX-ray Crystallography Laboratory, Cornell University, Ithaca, New York 14853-1301, USA
eDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA
First published on 8th July 2016
Abstract
A Cu-catalyzed benzannulation reaction transforms ortho(arylene ethynylene) oligomers into ortho-arylenes. This approach circumvents iterative Suzuki cross-coupling reactions previously used to assemble hindered ortho-arylene backbones. These derivatives form helical folded structures in the solid-state and in solution, as demonstrated by X-ray crystallography and solution-state NMR analysis. DFT calculations of misfolded conformations are correlated with variable-temperature 1H and EXSY NMR to reveal that folding is cooperative and more favorable in halide-substituted naphthalenes. Helical ortho-arylene foldamers with specific aromatic sequences organize functional π-electron systems into arrangements ideal for ambipolar charge transport and show preliminary promise for the surface-mediated synthesis of structurally defined graphene nanoribbons.
Introduction
Oligomers and polymers based on linking arenes are ubiquitous in materials chemistry, with applications that include photovoltaics, field effect transistors, light emitting diodes, sensing, dielectrics, dyes, and coatings.1 Many of these applications achieve optimal performance by controlling the relative distance and orientation between key aromatic systems. From a structural standpoint, oligo- and polyphenylenes are among the most simple motifs, and high molecular weight poly(p-phenylene)s and poly(m-phenylene)s have been studied extensively.2p-Phenylenes have served as precursors to graphene nanoribbons, while m-phenylenes provide improved solubility3 and have found use in display technologies, including electrophosphorescent devices and liquid crystal displays.4Oligo and poly(o-arylene)s are much less explored because they are more difficult to access (Scheme 1). Hartley5 prepared oligo(o-phenylene)s using iterative Suzuki cross-couplings, while Aida6 oxidized Lipshutz cuprates to prepare longer oligomeric mixtures (Scheme 1). Ito and Nozaki synthesized poly(o-arylene)s via formal aryne polymerizations,7 and most recently Mikami and Uchiyama developed a copper-mediated polymerization of aryne intermediates.8 These pioneering studies revealed that o-phenylenes adopt specific folded structures, which might organize functional aromatic systems if more powerful synthetic protocols are developed.
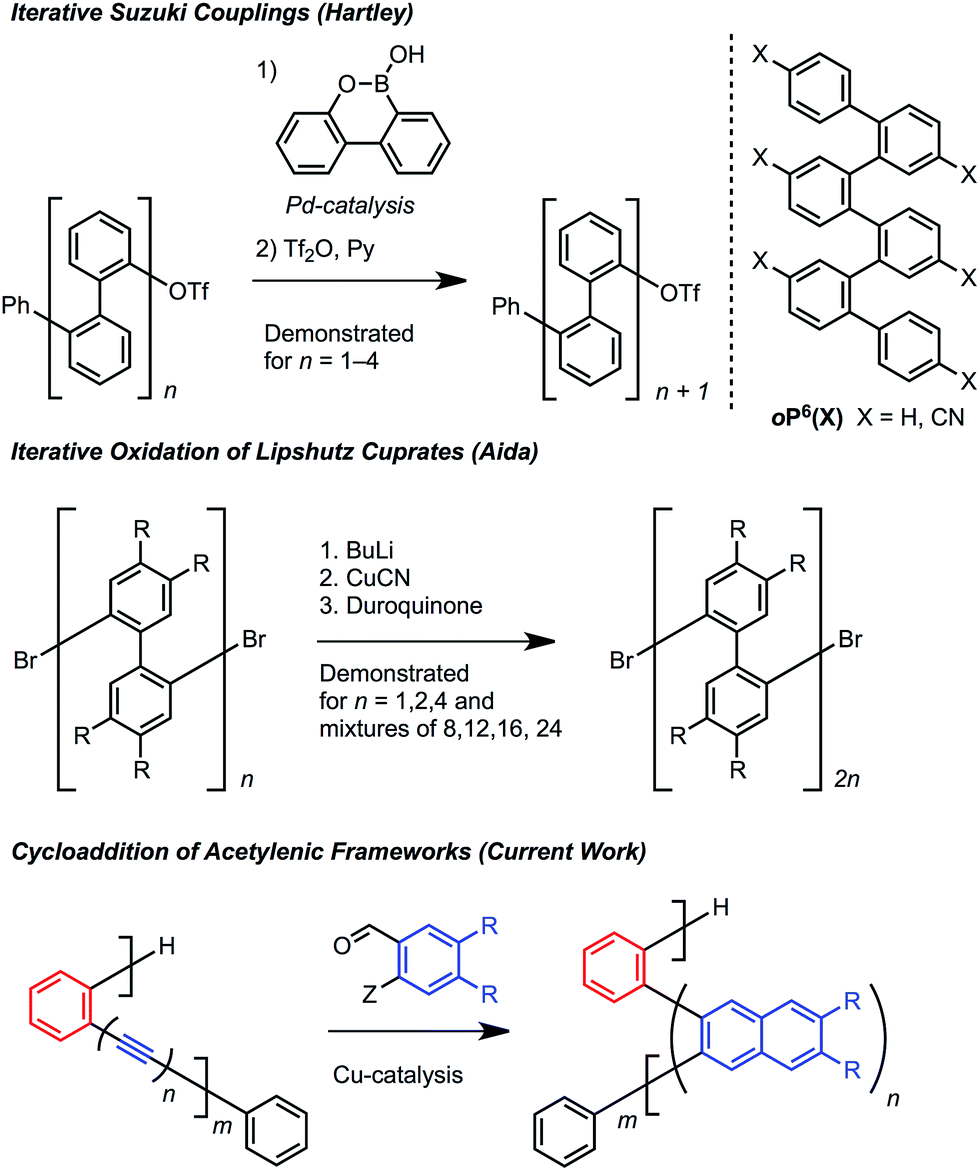 | ||
| Scheme 1 Synthetic approaches to o-arylenes. | ||
Cycloaddition reactions that convert oligo(o-phenyl ethynylene)s to oligo(o-arylene)s offer a means to simultaneously avoid challenging cross-coupling reactions and provide access to novel oligo-arene sequences. We recently expanded the scope of the Asao and Yamamoto9 benzannulation, which transforms alkynes into 2,3-disubstituted naphthalenes, to provide heavily substituted naphthalenes as single regioisomers.10,11 The reaction tolerates congested aromatic structures and is applicable to both small molecules12 and polymers.13 Here we apply this transformation to o-(arylene ethynylene)s and related structures containing strategically placed diynes. The resulting o-arylene structures adopt helical conformations in solution and the solid state, such that the specific pattern of alkynes and diynes in the starting material is translated into predictive arrangement of stacked aryl groups in the product. The naphthalene groups incorporated into these structures provide a stronger preference for folding, often >95%, whereas o-phenylene oligomers range from 49–96% folded, with electron withdrawing substituents needed to achieve higher values. These o-arylenes are also precursors to graphene-like nanostructures, which we demonstrate through surface mediated synthesis.
Results and discussion
Synthesis
Various hydrocarbon and halogenated benzaldehyde reagents convert o-(arylene ethynylene)s to o-arylenes with different sequences and halogenation patterns. Oligo(o-arylene)s of varying length and an alternating phenylene/ethynylene repeating pattern [(ab)n] were reacted with three different benzaldehydes, to access hydrocarbon, fluorine-, or chlorine-substituted structures. Despite observing complete conversion of the starting material in all cases, isolated yields decrease for precursors bearing more alkyne functions, which we attribute to their lower stability under the acidic reaction conditions. In these cases, the degradation products are easily removed during chromatography. In the future, alternative benzannulation conditions14 with reduced acid concentrations might provide improved yields. Sequence-defined o-arylene oligomers are available from o-phenylene ethynylene precursors containing mono- and diyne groups. For example, complex sequences such as “(abb)2a” and “(ab)2(ba)2” (Scheme 2) are achieved via cycloadditions starting from either diyne-based precursor 5, or mixed systems containing both mono- and diynes, such as precursor 6.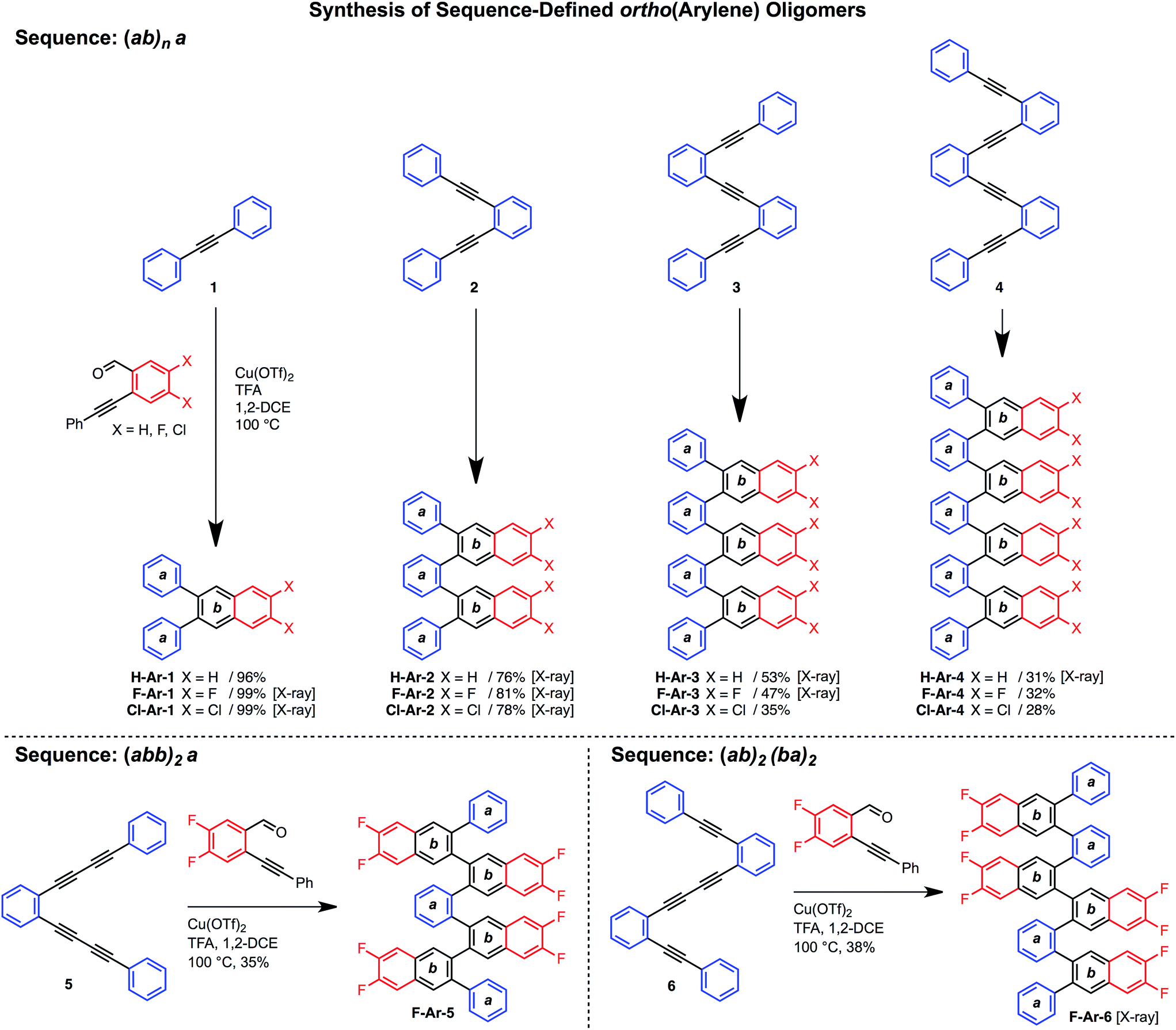 | ||
| Scheme 2 Synthesis of sequence-defined o-arylene oligomers from o-arylethynylenes via a cycloaddition strategy. | ||
Structural characterization
One of the defining characteristics of foldamers is that they have more than one conformation accessible, which includes a folded state and one or more unfolded, or partially unfolded, state.15 We used density functional theory (DFT) to investigate the conformational behaviour of our foldamers, including calculating their relative energies and the energy barrier for the conformational interconversion between the two lowest energy structures. 1D and 2D NMR experiments provided experimental validation of the solution-state structure and the folding dynamics (see ESI for details†).Many of the o-arylenes illustrated in Scheme 1 were characterized using single-crystal X-ray diffraction. These structures reveal their ability to fold in a “closed” helical conformation, enabling analysis of dihedral angles and distances between aromatic systems (Fig. 1 and Tables S5–S6†).16,17 The dihedral angles (ϕi, see Fig. 2) associated with the biaryl linkages that define the conformation of H-Ar-2, H-Ar-3 and H-Ar-4 consistently fall between −42° and −64° (or +42° and +64° for its enantiomer; these interconvert rapidly in solution at room temperature). The intramolecular π-stacking between the ith and (i + 3)th arene is evident in the crystal structures, with non-covalent C⋯C distances ranging from 3.1–3.5 Å. The solution-state structures of each foldamer were determined using 2D NMR techniques (COSY, NOSEY, ROSEY, HSQC, and HMBC) and support a folded structure analogous to that observed in the solid-state. The 1H NMR spectra of the X-Ar-3 foldamers (X = H, F, Cl) reveal one major set of signals in CDCl3, corresponding to the folded structure. An additional set of signals in the 1H NMR spectra are observed, which correspond to a minor, partially unfolded conformer (vide infra). The ratio between the folded and partially unfolded conformers is influenced by halogen substituents introduced during the benzannulation reaction. The minor conformation is most prominent in the spectra of hydrocarbon H-Ar-3, and decreases as X changes from H to Cl to F in the X-Ar-3 series (Fig. 2). DOSY NMR was inconsistent with these signals corresponding to different aggregation states, as the self-diffusion coefficients measured for both species were nearly identical. Furthermore, EXSY cross peaks between these two sets of signals establishes the ability for these species to interconvert in solution (see ESI for details†).
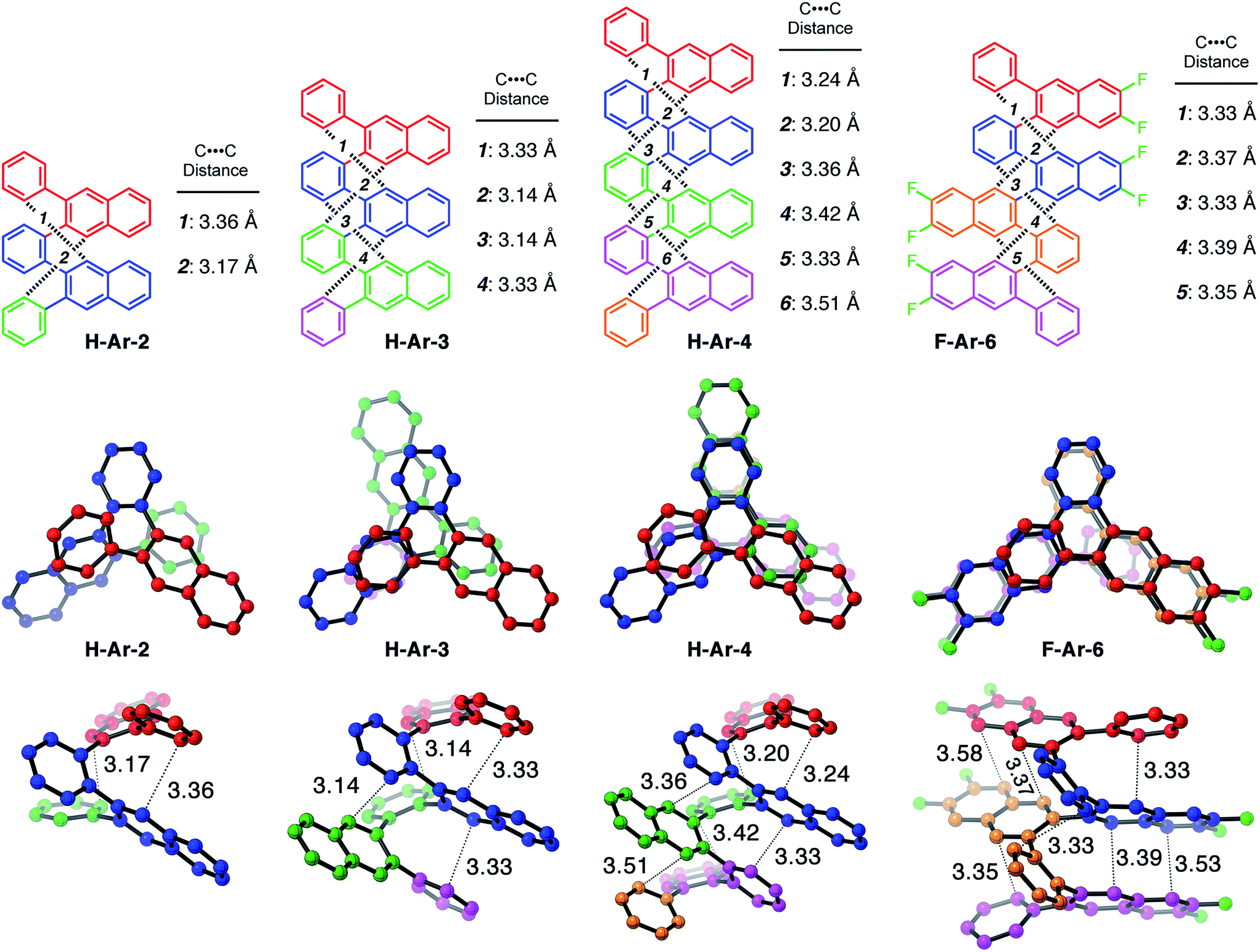 | ||
| Fig. 1 X-ray crystallographic structures of foldamers (left to right): H-Ar-2, H-Ar-3, H-Ar-4 and F-Ar-6. (Top) Line representation with selected carbon–carbon distances, (middle) top view and (bottom) side view with each repeat unit colored differently. | ||
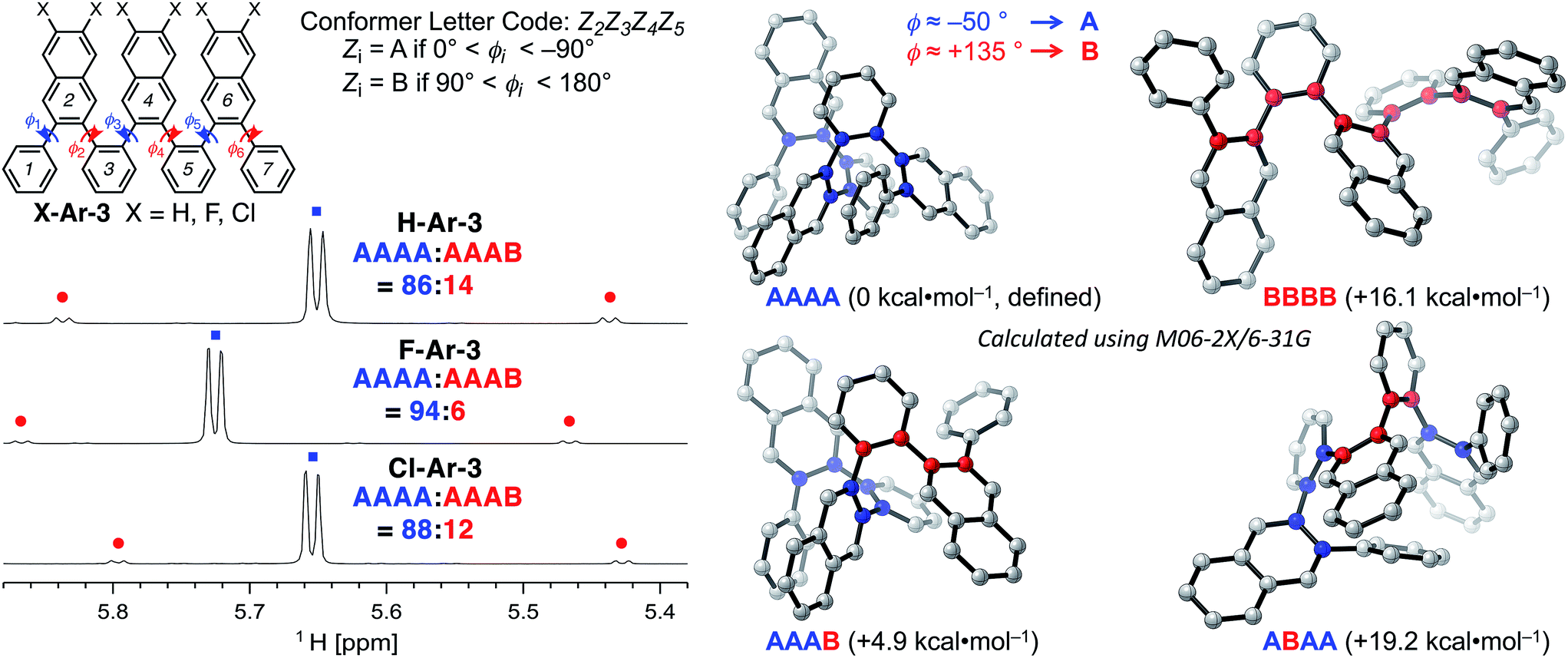 | ||
| Fig. 2 (Top left) Definition of dihedral angles ϕi and associated conformer letter code for o-arylenes using H-Ar-3 as an example. (Bottom left) Partial 1H NMR spectra (800 MHz, CDCl3, 0 °C) of X-Ar-3 illustrate the hydrogen signal used for determining the ratio and kinetics for the equilibrium and exchange between conformer AAAA and AAAB. (Right) DFT structures of various conformations of H-Ar-3 calculated using M06-2X/6-31G. | ||
DFT calculations performed using the M06-2X functional18 and the 6-31G basis set provide additional insight into the conformational structure of the oligo(o-arylene)s. This functional was chosen because it accurately describes non-covalent interactions, including aromatic stacking interactions. The conformation of the o-arylene foldamer is defined by the dihedral angles of aryl–aryl linkages (see Fig. 2 for the definition of ϕi). Within the context of foldamers X-Ar-[n], the relaxed potential energy surface analysis associated with C–C bond rotation of between the outermost naphthalene and its neighbouring phenylene ring provides two local minima, defined as A and B, corresponding to dihedral angles of ca. −50° and +135° (or +50° and −135° for its enantiomer), respectively (see ESI for details†). The series of all the aryl–aryl linkages of the foldamer defines the conformation, which we classify with a letter (A or B) and the series of these letters are arranged in order to provide an n-letter code for an oligomer of (n + 3)-arenes using the following definition for ϕ2 to ϕn−1. Following Hartley's naming convention,19 dihedral angle ϕi is classified as: A if (0° < ϕi < −90°), B (90° < ϕi < 180°), or C if (−90° ≤ ϕi < −180°); the opposite signs correspond to the analogous enantiomeric conformation. The “C” state emerges due to intramolecular edge-to-face π-stacking interactions between the ith and (i + 4)th arene, although these are often significantly less stable conformers than those that do not contain such C states.20 For example, the folded X-ray crystallographic geometries of trimeric and tetrameric foldamers H-Ar-3 and H-Ar-4 shown in Fig. 1 correspond to conformer AAAA and AAAAA, respectively.
The most stable conformations of the X-Ar-3o-arylenes (X = H, F, or Cl, ESI, Table S5†) have folded forms AAAA, which are significantly more stable than the unfolded forms BBBB for all substitution patterns. Conformation BBBB is 16.1 (X = H), 17.2 (X = F), and 17.6 kcal mol−1 (X = Cl) higher in energy relative to the corresponding AAAA conformer (Fig. 2). Solution-state NMR analysis supports AAAA being the most stable conformation, which is also the conformation observed by X-ray crystallography. Defect states are least energetically costly at the ends of the foldamer instead of the middle, as conformation AAAB is more stable than AABA. The AAAB structures are 4.9 (X = H), 5.3 (X = F), and 5.4 kcal mol−1 (X = Cl) higher in energy than AAAA, compared to 19.2 (X = H), 19.8 (X = F), and 19.8 kcal mol−1 (X = Cl) for AABA relative to AAAA.
The energy differences between AAAB and AAAA are larger for the halogenated foldamers (+5.3 and +5.4 kcal mol−1 for X = F and Cl, respectively, relative to their AAAA conformation) compared to the hydrocarbon foldamer (+4.9 kcal mol−1). This stabilization suggests that halogenation of the foldamer will lead to a higher population of AAAA-folded molecules, indeed this is observed in solution. NMR spectroscopy of X-Ar-3 in CDCl3 indicates that AAAB is the structure of the minor conformer observed in Fig. 2. The population of folded state AAAA increases from 86% to 88–94% as X changes from H to Cl to F, respectively, for CDCl3 solutions at 0 °C (Table 1). The population of the folded conformer increases as a function of oligomer length, specifically, the folded population for X-Ar-4 are 91% for X = H, 96% for X = Cl, and 97% for X = F. The modified sequence of arenes in foldamers F-Ar-5 and F-Ar-6 leads to naphthalene–naphthalene π-stacking, in contrast to phenylene–naphthalene π-stacking present in X-Ar-3 and X-Ar-4. Increased stabilization of the folded state derives from the larger cofacial π-stacking surface area provided by the naphthalene–naphthalene interactions in F-Ar-5 and F-Ar-6, resulting in nearly quantitative folding (99% and ≥99% folded, respectively) as determined by 1H NMR spectroscopy. DFT calculations correctly predict increased folding population for F-Ar-6 compared to F-Ar-5, with both superior to F-Ar-4 (see ESI for details†). These observations highlight one of the benefits of sequence-defined oligomers, as a simple change in sequence can influence folding behaviour and optical/electronic properties (vide infra). Additionally, the consistently high population of the folded conformer regardless of the substituents or sequence in these o-arylenes illustrates the benefit of incorporating naphthalenes into o-arylene foldamers (Table 1). Additional details regarding thermodynamic and kinetic parameters (rate & activation barriers) associated with the folding/unfolding equilibrium for these o-arylenes, including van 't Hoff and Eyring analyses is provided in the ESI.†
| Compound class | Number of arenes in the oligomer (n) & compound number | Percentage of population in the folded conformation An−3 in CDCl3 solutions at 0 °C | ||
|---|---|---|---|---|
| X = H | X = Cl | X = F | ||
| a ND = not determined. | ||||
| o-Phenylene-alt-o-naphthalene | 7 (X-Ar-3) | 86% | 88% | 94% |
| 9 (X-Ar-4) | 91% | 96% | 97% | |
| o-Phenylene | 6 (oP6(H)) | 49% (−5 °C) (ref. 19) | — | — |
| 6 (oP6(CN)) | >96% (−5 °C) (ref. 19) | — | — | |
| o-Phenylene-mix-o-naphthalene | 7 (F-Ar-5) | ND | ND | 99% |
| 9 (F-Ar-6) | ND | ND | ≥99% | |
Optical & electrochemical properties
The foldamers show UV-vis absorption and emission characteristics that are sensitive to the presence of naphthalene–naphthalene intramolecular π-stacking (Fig. 3). Longer oligomers of the X-Ar-1 to X-Ar-4 series show increased molar absorptivity associated with their larger number of chromophores but no major shift of λmax to lower energy associated with coplanar conjugated systems. Instead a slight blue-shift in λmax and a narrowing of the absorption band is observed. The photoemission spectra of F-Ar-4 to F-Ar-6 reflect differences in the helical structures. F-Ar-4 contains only phenylene–naphthalene through-space interactions and exhibits the most blue-shifted photoemission (λmax,em = 370 nm).21 Both F-Ar-5 and F-Ar-6 have two naphthalene–naphthalene interactions, which leads to 12 and 10 nm emission red shifts, respectively, relative to F-Ar-4. It is unclear whether the small differences in λmax,abs and λmax,em between F-Ar-5 and F-Ar-6 originate from differences in their arylene sequences or subtle differences of their through-space interactions.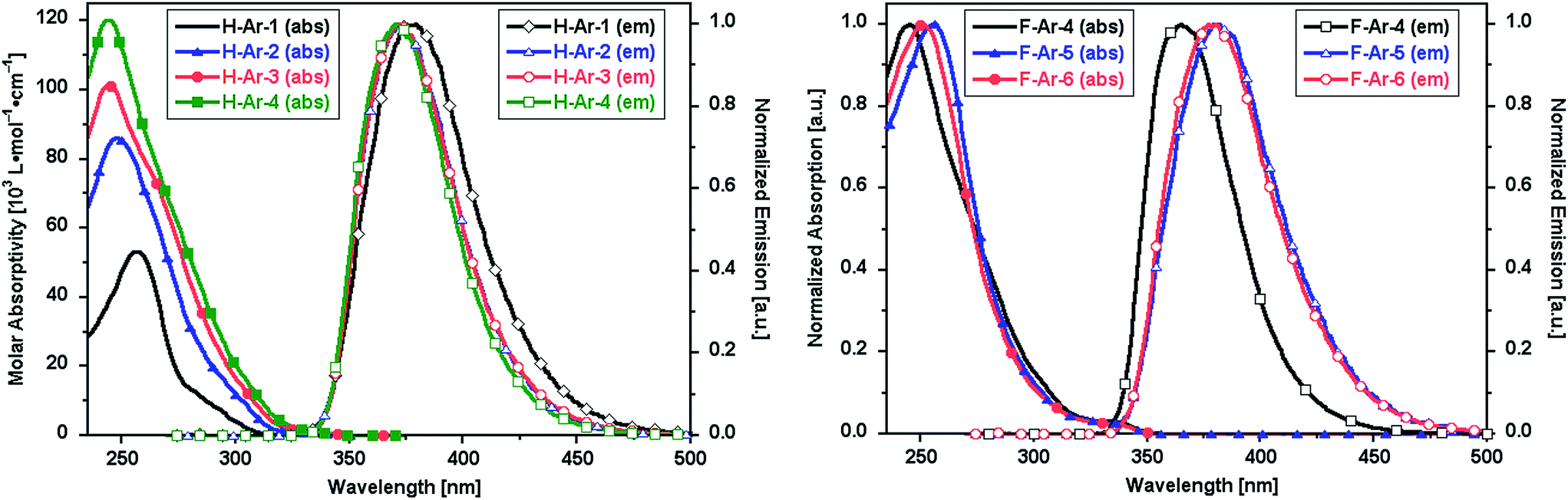 | ||
| Fig. 3 UV-vis absorption and emission spectra (λexc = 260 nm) of oligomeric series in CH2Cl2: (left) H-Ar-1 to H-Ar-4, (right) F-Ar-4 to F-Ar-6. | ||
Cyclic voltammetry (CV) experiments (CH3CN, 0.1 M Bu4NClO4 supporting electrolyte) reveal progressively more facile oxidation with increased oligomer length (Fig. 4). Namely, the onset of oxidation (vs. Ag/AgClO4) decreases monotonically from 1.184 V (H-Ar-1) to 1.090 V (H-Ar-2), 1.006 V (H-Ar-3) and finally 0.979 V (H-Ar-4), indicative of a lowering of the HOMO energy for longer oligomers.22,23 This trend is likely to arise from intramolecular through-space interactions that stabilize the electrochemically generated radical cation. It is also possible that oxidation occurs from an unfolded conformation that is a more planar, delocalized structure, but we judge this possibility less likely given the high energetic penalty for this process in the neutral form. The oxidation process is chemically irreversible as shown from the mismatch in current density between the anodic and cathodic peak for the oxidation wave in the CV. Electrochemical oxidation might induce full or partial oxidative cyclodehydrogenation to form larger aromatic structures (vide infra). This possibility led us to explore the conversion of foldamer structures into defined larger aromatics (e.g. nanographenes).
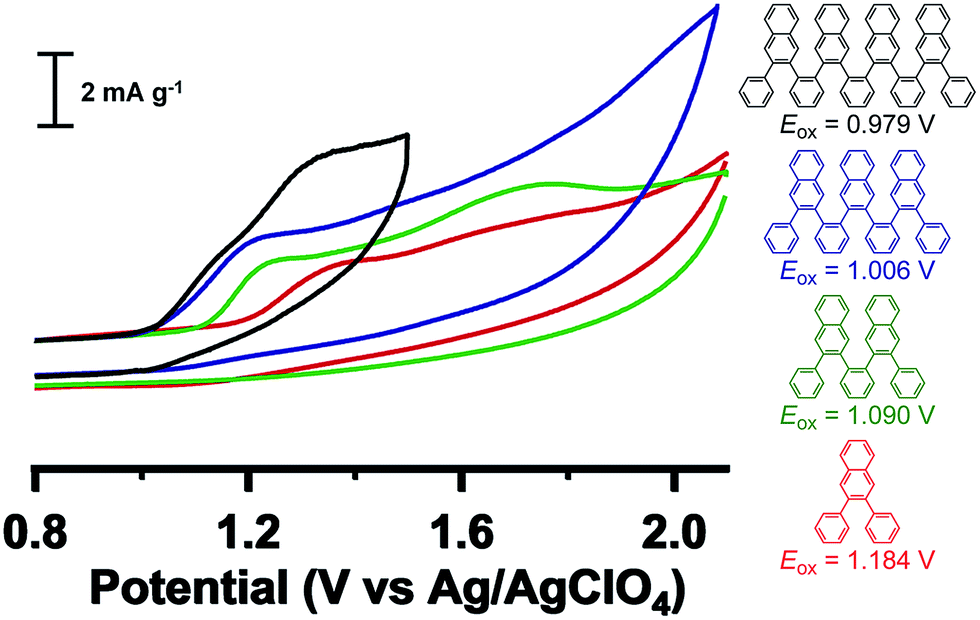 | ||
| Fig. 4 Cyclic voltammetry of foldamers H-Ar-1 to H-Ar-4 in CH3CN containing 0.1 M Bu4NClO4 measured at 50 mV s−1. | ||
On-surface nanographene synthesis
The ability to access o-arylenes and introduce functionality, such as halides, on the arenes in the final step opens up new opportunities. For example, polymeric derivatives of these foldamers might serve as precursors to graphene nanoribbons, in which this bottom-up approach enables the control of width, length, and edge structure (e.g., hydrogen vs. halogen edge termination).24,25 The oxidative cyclodehydrogenation of X-Ar-[n] could enable exquisite control in forming armchair graphene nanoribbons of 8 carbons in width and varying length up to 20 carbons with the potential for one edge of the ribbon being halogenated while the opposing edge is hydrogen terminated (Scheme 3). As a proof of principle, herein we report the conversion of foldamer H-Ar-3 to polycyclic aromatic hydrocarbon H-8via a surface-mediated process.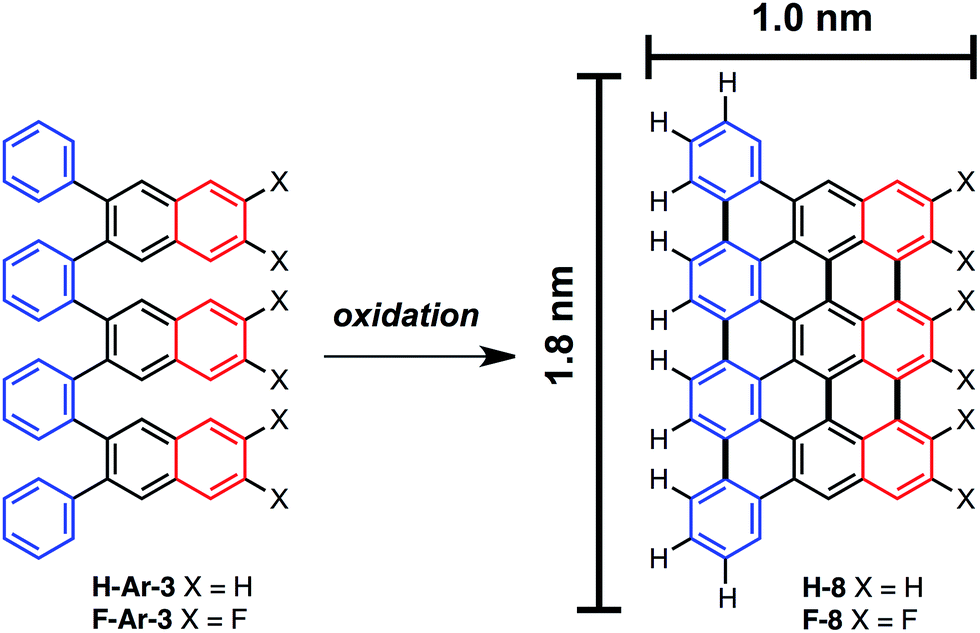 | ||
| Scheme 3 o-Arylenes as precursors to nanographenes H-8 and F-8 with control of width and length dimensions in addition to potential control of edge-functionalization. Dimensions shown are estimated for H-8 based on DFT calculations and measurements between the furthest hydrogen atoms. | ||
Evaporation of H-Ar-3 in ultra high vacuum enabled its deposition onto Au(111). The STM image in Fig. 5a illustrates a surface coverage of 0.8 monolayers of H-Ar-3 on Au(111) obtained after deposition of the H-Ar-3 molecules. The molecules are seen to form ordered crystalline domains approaching ca. 20 nm (a diagonal grain boundary separating two such domains is visible in Fig. 5a). At lower surface coverage, H-Ar-3 selectively attaches to step edges of Au(111) (Fig. 5b). A close up of several molecules (Fig. 5c) clearly shows the non-planarity of H-Ar-3, which exhibits an apparent height of 3–5 Å depending on molecular conformation.
 | ||
| Fig. 5 STM images of H-Ar-3 and H-8 molecules on Au(111). (a) 0.8 monolayer coverage of H-Ar-3 as deposited shows well-ordered crystalline domains. (b) Lower surface coverage of H-Ar-3 as deposited shows molecular adhesion to step edges of Au(111) crystal. (c) Close-up of several molecules of H-Ar-3 before annealing reveals non-planarity. (d) Individual nanographene H-8 obtained after annealing to 286 °C. (e) DFT optimized structure of H-8 overlaid on top of STM image of H-8 shown in (d). Scanning parameters: (a) sample bias: 1.0 V, tunnelling current: 10 pA; (b) 1.0 V, 2 pA; (c) 1.0 V, 2 pA; and (d) 0.1 V, 200 pA. All images were obtained at T = 7 K. | ||
Upon annealing the H-Ar-3/Au(111) to 286 °C, a change in the shape of the molecules is observed consistent with conversion to H-8.26Fig. 5d shows the dominant product (60%): a planar, trapezoidal molecule with measured height of 1.9 ± 0.1 Å. The dimensions of this structure (1.9 ± 0.2 nm × 1.3 ± 0.2 nm) are consistent with DFT predicted dimensions of H-8 (Fig. 5e). It should be noted that an intermediate annealing temperature of 250 °C is enough to convert some molecules (∼10%) to the desired H-8 product, but with the remaining sample still consisting of non-planar molecules.
Conclusions
We have developed a direct synthetic approach to oligomeric o-arylenes based on a cycloaddition strategy using acetylenic motifs to control arylene sequence. Characterization of these oligomers reveals they are able to fold into helical structures due to intramolecular non-covalent interactions, which are influenced by sequences of naphthalene and phenylene units as well as by halogen substituents. The folded structures are observed in the solid state by X-ray crystallography and in the solution state by NMR spectroscopy. DFT calculations shed light into the various conformations available, their relative energies and selected energy barriers for their interconversion. The sequence of the o-arylenes influences both folding properties and optical properties. These foldamers hold potential for sensing guests capable of disrupting intramolecular π-stacking or intercalating between arenes. Extending these methods to longer oligomers or polymers will provide structurally defined graphene nanoribbons with unsymmetrical edge functionalization (i.e., H vs. F edge termination). These studies highlight the utility of this cycloaddition approach for the realization of functional carbon nanostructures via a bottom up approach.Acknowledgements
This research was supported by the Army Research Office Multidisciplinary University Research Initiative (MURI) program under grant no. W911NF-15-1-0447 (molecular synthesis and STM imaging), a Beckman Young Investigator Award (to W. R. D.), the NSF (CHE-1124754, structural characterization), and by the US Department of Energy, Office of Basic Energy Sciences Nanomachine Program under contract no. DE-AC02-05CH11231 (molecular deposition). C. R. D. was supported by an NSF GRFP (DGE-1144153). This work made use of the Cornell Center for Materials Research Shared Facilities which are supported through the NSF MRSEC program (DMR-1120296), an NMR spectrometer at Cornell supported by the NSF-MRI program (CHE-1531632), and an 800 MHz NMR instrument at the State University of New York College of Environmental Science and Forestry in Syracuse facilities purchased using NIH grant no. 1S10OD012254-01A1. We thank David Kiemle for his assistance in acquiring the 800 MHz NMR data and Samantha N. MacMillan (X-ray Laboratory, Cornell) for additional X-crystallography presented in the ESI.†Notes and references
- (a) T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed; (b) A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS PubMed; (c) B. Lucas, T. Trigaud and C. Videlot-Ackermann, Polym. Int., 2012, 61, 374–389 CrossRef CAS; (d) P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS PubMed; (e) Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed; (f) N. Mártin, L. Sánchez, M. Á. Herranz, B. Illescas and D. M. Guldi, Acc. Chem. Res., 2007, 40, 1015–1024 CrossRef PubMed; (g) M. T. Dang, L. Hirsch, G. Wantz and J. D. Wuest, Chem. Rev., 2013, 113, 3734–3765 CrossRef CAS PubMed; (h) A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed; (i) A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS PubMed; (j) J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- B. A. G. Hammer and K. Müllen, Chem. Rev., 2016, 116, 2103–2140 CrossRef CAS PubMed.
- V. Percec, S. Okita and R. Weiss, Macromolecules, 1992, 25, 1816–1823 CrossRef CAS.
- (a) J. Liu, C. Gong, Z. Yu and Q. Pei, J. Mater. Chem., 2011, 21, 9772–9777 RSC; (b) K. Suda and K. Akagi, Macromolecules, 2011, 44, 9473–9488 CrossRef CAS.
- (a) J. He, J. J. L. Crase, S. H. Wadumethrige, K. Thakur, L. Dai, S. Zou, R. Rathore and C. S. Hartley, J. Am. Chem. Soc., 2010, 132, 13848–13857 CrossRef CAS PubMed; (b) C. S. Hartley and J. He, J. Org. Chem., 2010, 75, 8627–8636 CrossRef CAS PubMed; (c) S. M. Mathew and C. S. Hartley, Macromolecules, 2011, 44, 8425–8432 CrossRef CAS; (d) S. M. Mathew, J. T. Engle, C. J. Ziegler and C. S. Hartley, J. Am. Chem. Soc., 2013, 135, 6714–6722 CrossRef CAS PubMed; (e) S. M. Mathew, L. A. Crandall, C. J. Ziegler and C. S. Hartley, J. Am. Chem. Soc., 2014, 136, 16666–16675 CrossRef CAS PubMed; (f) C. S. Hartley, Acc. Chem. Res., 2016, 49, 646–654 CrossRef CAS PubMed; (g) For related oligo(o-naphthalene)s, see, T. Motomura, H. Nakamura, M. Suginome, M. Murakami and Y. Ito, Bull. Chem. Soc. Jpn., 2005, 78, 142–146 CrossRef CAS.
- E. Ohta, H. Sato, S. Ando, A. Kosaka, T. Fukushima, D. Hashizume, M. Yamasaki, K. Hasegawa, A. Muraoka, H. Ushiyama, K. Yamashita and T. Aida, Nat. Chem., 2011, 3, 68–73 CrossRef CAS PubMed.
- S. Ito, K. Takahashi and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 7547–7550 CrossRef CAS PubMed.
- Y. Mizukoshi, K. Mikami and M. Uchiyama, J. Am. Chem. Soc., 2015, 137, 74–77 CrossRef CAS PubMed.
- N. Asao, T. Nogami, S. Lee and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 10921–10925 CrossRef CAS PubMed.
- D. Lehnherr, J. M. Alzola, E. B. Lobkovsky and W. R. Dichtel, Chem.–Eur. J., 2015, 21, 18122–18127 CrossRef CAS PubMed.
- H. Arslan, K. L. Walker and W. R. Dichtel, Org. Lett., 2014, 16, 5926–5929 CrossRef CAS PubMed.
- S. Hein, H. Arslan, I. Keresztes and W. R. Dichtel, Org. Lett., 2014, 16, 4416–4419 CrossRef CAS PubMed.
- H. Arslan, J. D. Saathoff, D. N. Bunck, P. Clancy and W. R. Dichtel, Angew. Chem., Int. Ed., 2012, 51, 12051–12054 CrossRef CAS PubMed.
- X.-L. Fang, R.-Y. Tang, X.-G. Zhang, P. Zong, C.-L. Deng and J.-H. Li, J. Organomet. Chem., 2011, 696, 352–356 CrossRef CAS.
- (a) J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Science, 1997, 277, 1793–1796 CrossRef CAS PubMed; (b) D.-W. Zhang, X. Zhao and J.-L. Hou, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- It should be noted that both o-phenylenes and o-(phenylene ethynylene)s can adopt helical conformation, see ref. 2 and 17, respectively.
- (a) R. H. Grubbs and D. Kratz, Chem. Ber., 1993, 126, 149–157 CrossRef CAS; (b) T. V. Jones, R. A. Blatchly and G. N. Tew, Org. Lett., 2003, 5, 3297–3299 CrossRef CAS PubMed; (c) J. Jiang, M. M. Slutsky, T. V. Jones and G. N. Tew, New J. Chem., 2010, 34, 307–312 RSC; (d) T. V. Jones, M. M. Slutsky and G. N. Tew, New J. Chem., 2008, 32, 676–679 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- S. M. Mathew, J. T. Engle, C. J. Ziegler and C. S. Hartley, J. Am. Chem. Soc., 2013, 135, 6714–6722 CrossRef CAS PubMed.
- The A and B states in oligo(o-arylene)s, such as oligo(o-phenylene)s, is well documented, whereas the C state described herein is unprecedented and it is unclear whether it is unique to our system or if it is also attainable with oligo(o-phenylene)s.
- The photoemission spectra of o-phenylenes is known to exhibit a blue shift with increasing oligomer length, similar to that observed with [X]-Ar-1 to [X]-Ar-4, a phenomenon associated with folding, see: (a) ref. 5c ; (b) P. Guiglion and M. A. Zwijnenburg, Phys. Chem. Chem. Phys., 2015, 17, 17854–17863 RSC.
- A similar observation is made in the series comparing F-Ar-[n] (with n = 1–4). The onset of oxidation is generally less electropositive for longer oligomers, namely, 1.320 V (F-Ar-1), 1.279 V (F-Ar-2), 1.265 V (F-Ar-3), 1.180 V (F-Ar-4) vs. Ag/AgClO4 measured in MeCN containing Bu4NClO4.
- The sequence of the arenes within the o-arylene can also affect the onset of oxidation, specifically, 1.180 V (F-Ar-4), 1.172 V (F-Ar-6), 1.212 V (F-Ar-5) vs. Ag/AgClO4 measured in MeCN containing Bu4NClO4.
- (a) Y.-W. Son, M. L. Cohen and S. G. Louie, Phys. Rev. Lett., 2006, 97, 216803 CrossRef PubMed; (b) S. Dutta and S. K. Pati, J. Mater. Chem., 2010, 20, 8207–8223 RSC.
- (a) J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed; (b) L. Lafferentz, V. Eberhardt, C. Dri, C. Africh, G. Comelli, F. Esch, S. Hecht and L. Grill, Nat. Chem., 2012, 4, 215–220 CrossRef CAS PubMed; (c) Y.-C. Chen, T. Cao, C. Chen, Z. Pedramrazi, D. Haberer, D. G. de Oteyza, F. R. Fischer, S. G. Louie and M. F. Crommie, Nat. Nanotechnol., 2015, 10, 156–160 CrossRef CAS PubMed.
- Other metallic surfaces such as Cu(111) were explored but Au(111) provide the best results.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1483959–1483967. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02520j |
| This journal is © The Royal Society of Chemistry 2016 |