
Phthalimide end-capped thienoisoindigo and diketopyrrolopyrrole as non-fullerene molecular acceptors for organic solar cells†
Pierre
Josse
a,
Clément
Dalinot
a,
Yue
Jiang
ab,
Sylvie
Dabos-Seignon
a,
Jean
Roncali
a,
Philippe
Blanchard
*a and
Clément
Cabanetos
*a
aCNRS UMR 6200, MOLTECH-Anjou, University of Angers, 2 Bd Lavoisier, 49045 Angers, France. E-mail: philippe.blanchard@univ-Angers.fr; clement.cabanetos@univ-Angers.fr
bResearch Institute of Materials Science, South China University of Technology, 381 Wushan Rd, Tianhe, Guangzhou, Guangdong, China
First published on 18th November 2015
Abstract
Two acetylene-bridged molecules, built by grafting phthalimides on thienoisoindigo (TII) and diketopyrrolopyrrole (DPP) blocks, have been synthesized, characterized and evaluated as electron acceptor materials in air-processed inverted organic solar cells. Once blended with poly(3-hexylthiophene), power conversion efficiencies (PCEs) of ca. 0.4% and 3.3% were achieved for TII and DPP based devices, respectively. To the best of our knowledge these PCEs (i) rank amongst the highest reported so far for diketopyrrolopyrrole based acceptors and (ii) make this contribution the very first example of thienoisoindigo based materials used as non-fullerene electron acceptors.
1. Introduction
The synthesis of active materials for organic solar cells (OSCs) is a focus of considerable attention.1–3 Multi-disciplinary research effort in device fabrication and design of donor materials has led to rapid progress.4–7 Actually, power conversion efficiencies (PCEs) exceeding 10.0% have been recently reported for cells based either on low band gap polymers or on molecular donors.8–10On the other hand, fullerene derivatives have served as standard electron-acceptor materials for almost two decades.11–14 However, despite large electron affinity, high electron-mobility and isotropic charge-transport, owing to their 3-dimensional structures,15–17 fullerene derivatives present several drawbacks such as low absorption in the visible spectrum, limited structural tunability of energy levels and relatively high cost and environmental impact of their soluble derivatives PC61BM and PC71BM.
In this context, polymeric and molecular non-fullerene acceptors have received increasing attention in recent years.18–21 In particular, it has been a surge in the development of well-defined small molecules due to their inherent advantages in terms of synthesis, purification and analysis of structure–property relationships. Various classes of molecular systems have been synthesized and evaluated as acceptors including vinazene,22–24 9,9′-bifluorenylidene,25,26 diketopyrrolopyrrole derivatives,27–30 dicyan-substituted quinacridone,31 functionalized isoindigo,32 BODIPYs,33 electron-deficient pentacenes,34 fluoranthene-fused imide,35,36 spiro-annulated derivatives,37,38 naphthalene-diimides39,40 or perylene-diimides.41–44 Rapid improvement of PCEs has been reported for OSCs combining extended molecular architectures,45–47 mainly based on PDIs,48–52 with high performance low band gap polymers. For instance, Nuckolls et al. recently achieved an impressive 8.3% power conversion efficiency by blending helical perylene diimide oligomers with the well-known PTB7-Th, thus competing with the best fullerene based OSCs.53 On the other hand, few examples of PCEs exceeding 2.5% have been recorded so far with the standard poly(3-hexylthiophene) (P3HT) donor material.22,54
In our continuing commitment towards simplified chemical structures of active OPV materials,55–58 we report here on the synthesis and preliminary evaluation of two electron-acceptors built, in a few steps, by attaching electron-withdrawing phthalimide groups on two central chromophore blocks namely thienoisoindigo (TII) and diketopyrrolopyrrole (DPP) (Scheme 1) through the use of acetylenic linkages. Such connectors indeed permit the extension of the effective conjugation while lowering the energy level of the frontier orbitals and reducing the steric interactions between thiophene units and phenyl rings.59,60
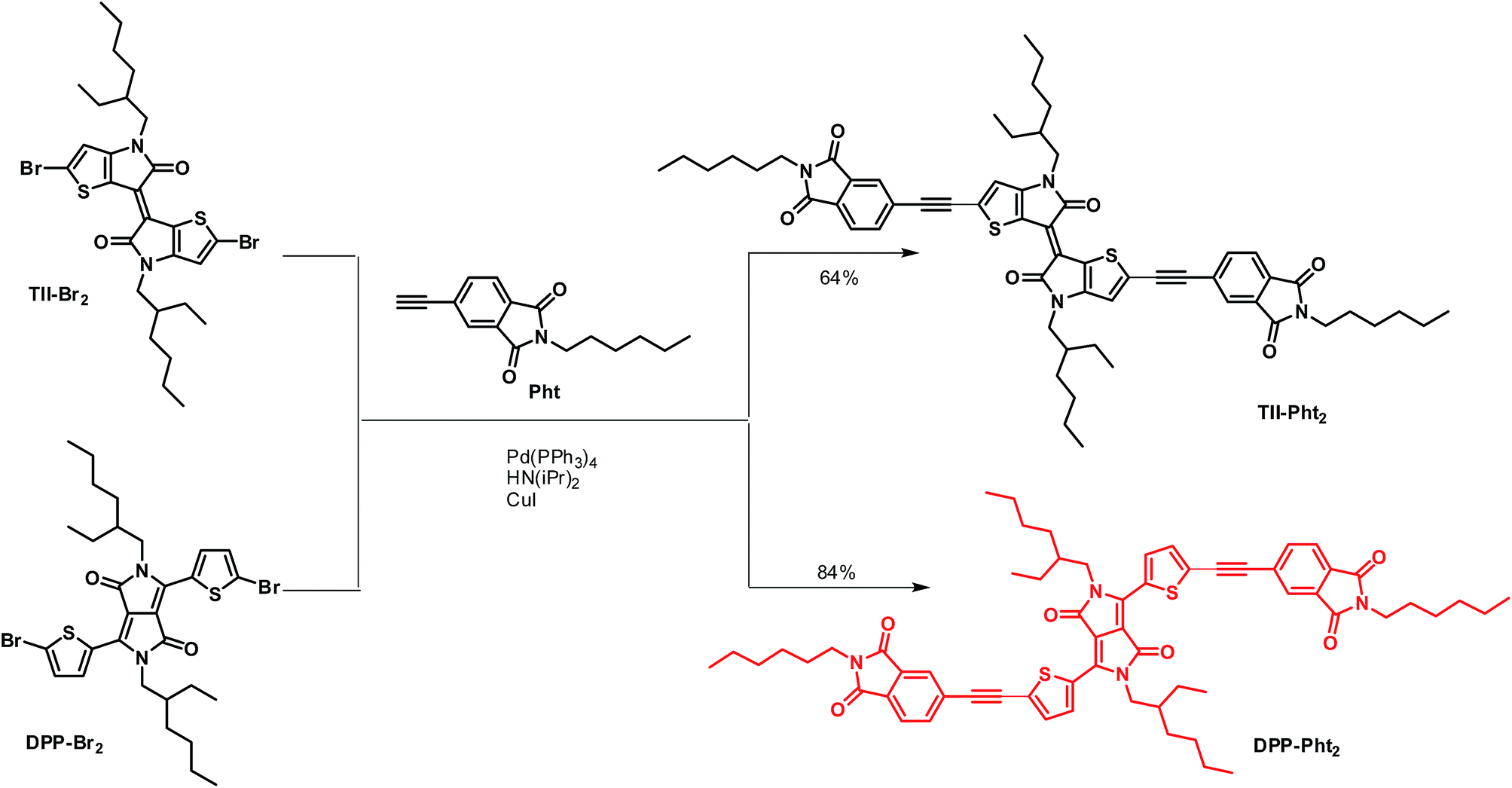 | ||
| Scheme 1 Synthetic route to TII-Pht2 (black) and DPP-Pht2 (red). | ||
2. Experimental section
Materials
All reagents and chemicals from commercial sources were used without further purification. Reactions were carried out under a neutral atmosphere unless otherwise stated. Solvents were dried and purified using standard techniques.Measurements and characterization
Flash chromatography was performed with analytical-grade solvents using Aldrich silica gel (technical grade, pore size 60 Å, 230–400 mesh particle size). Flexible plates ALUGRAM® Xtra SIL G UV254 from MACHEREY-NAGEL were used for TLC. Compounds were detected by UV irradiation (Bioblock Scientific) or staining with I2, unless stated otherwise. Melting point measurements were performed using a Reichert Jung THERMOVAR (ranging from room temperature to 250 °C). NMR spectra were recorded with a Bruker AVANCE III 300 (1H, 300 MHz and 13C, 75 MHz). Chemical shifts are given in ppm relative to TMS and coupling constant J in Hz. UV-vis spectra were recorded with a Perkin Elmer 950 spectrometer. Mass spectrometry was performed with a JEOL JMS-700 B/E. Cyclic voltammetry was performed in 0.1 M Bu4NPF6/CH2Cl2 (HPLC grade). Solutions were degassed by argon bubbling prior to each experiment. Experiments were carried out in a one-compartment cell equipped with platinum electrodes and a saturated calomel reference electrode (SCE) using a Biologic SP-150 potentiostat with positive feedback compensation. FTIR measurements were performed using a Bruker Vertex 70 instrument.Synthetic procedures
(E)-2,2′-dibromo-4,4′-bis(2-ethylhexyl)-[6,6′-bithieno[3,2-b]pyrrolylidene]-5,5′(4H,4′H)-dione TII-Br2,61 3,6-bis(5-bromothiophen-2-yl)-2,5-bis(2-ethylhexyl)pyrrolo[3,4-c]pyrrole-1,4-(2H,5H)-dione DPP-Br262 and 5-ethynyl-2-hexylisoindoline-1,3-dione Pht63 were synthesized according to previously reported methods.TII-Pht2 . (99 mg, 64% yield) 1H NMR (300 MHz, CDCl3, ppm) δ: 7.90 (s, 2H), 7.82 (d, J = 7.8 Hz, 2H), 7.76 (dd, 3J = 6.6 Hz, 4J = 1.2 Hz, 2H), 6.96 (s, 2H), 3.73–3.65 (m, 8H), 1.89–1.81 (m, 2H), 1.72–1.63 (m, 4H), 1.44–1.25 (m, 28H), and 0.97–0.86 (m, 18H). 13C NMR (75 MHz, CDCl3, ppm) δ: 170.68, 167.80, 167.70, 151.26, 136.47, 132.71, 131.27, 130.26, 128.66, 125.71, 123.41, 120.97, 117.37, 115.83, 96.90, 88.63, 46.05, 38.64, 38.44, 31.50, 28.79, 28.66, 26.68, 24.07, 23.20, 22.65, 14.21, 14.15, and 10.75. FAB + HRMS: calculated for C60H68N4O6S2 1004.4580, found 1004.4574. FTIR (cm−1): 2169 (νst(C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)); 1711 (νst(C
C)); 1711 (νst(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)). Mp: >250 °C.
O)). Mp: >250 °C.
DPP-Pht2 . (128 mg, 84% yield) 1H NMR (300 MHz, CDCl3, ppm) δ: 8.91 (d, J = 4.2 Hz, 2H), 7.96 (s, 2H), 7.84 (s, 4H), 7.45 (d, J = 4.2 Hz, 2H), 4.02 (d, J = 7.8 Hz, 4H), 3.68 (t, J = 7.2 Hz, 4H), 1.90 (m, 2H), 1.68 (m, 4H), 1.44–1.25 (m, 28H), and 0.94–0.86 (m, 18H). 13C NMR (75 MHz, CDCl3, ppm) δ: 167.80, 167.68, 161.61, 139.69, 136.70, 135.72, 134.22, 132.72, 131.82, 131.58, 129.18, 128.37, 127.28, 125.44, 123.44, 109.42, 95.96, 86.49, 46.31, 39.31, 38.47, 31.49, 30.27, 28.66, 28.44, 26.68, 23.70, 23.19, 22.65, 14.19, 14.14, and 10.61. FAB + HRMS: calculated for C62H70N4O6S2 1030.4737, found 1030.4754. FTIR (cm−1): 2192 (νst(C
C)); 1770 (νst(CO)). Mp: 219 °C.
3. Results and discussion
TII-Pht2 and DPP-Pht2 were obtained in 64 and 84% yields, respectively, by the Sonogashira cross-coupling reaction between the di-brominated dyes TII-Br2 or DPP-Br2 and 5-ethynyl-2-hexylisoindoline-1,3-dione (Pht) (Scheme 1).The targeted compounds were worked-up using conventional methods and purified by simple column chromatography (see ESI†).
Fig. 1 shows the UV-vis absorption spectra of the two acceptors in chloroform solutions and as spin-cast films while the corresponding data are listed in Table 1.
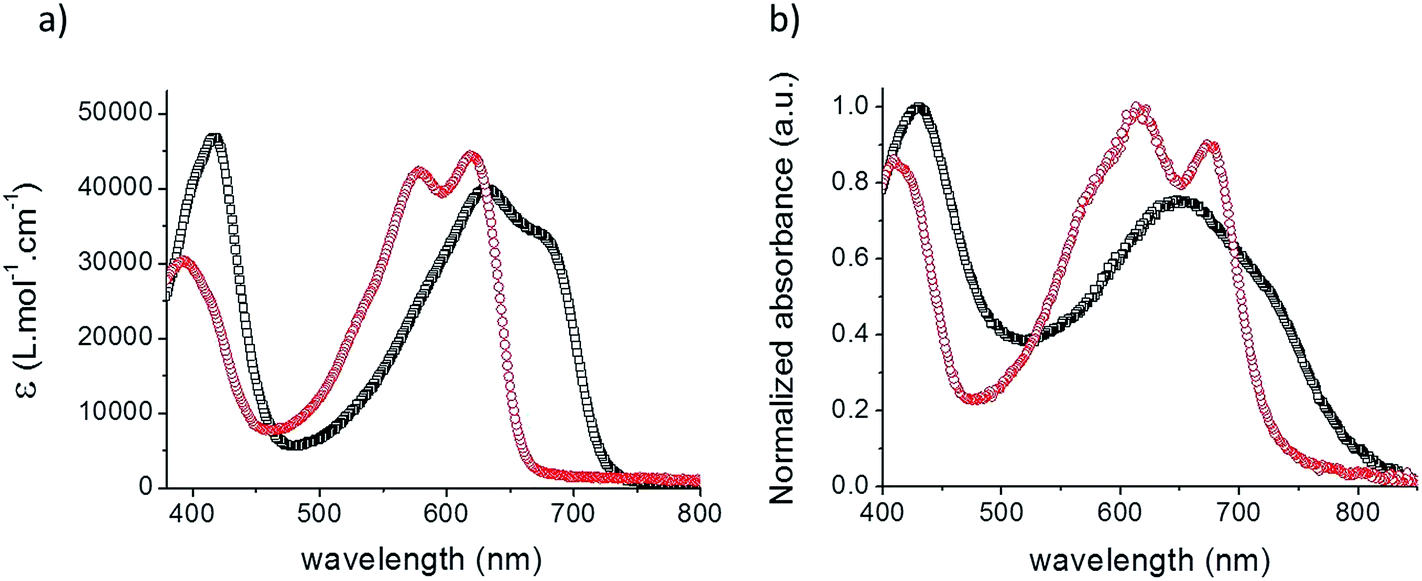 | ||
| Fig. 1 UV-vis absorption spectra of TII-Pht2 (black squares) and DPP-Pht2 (red circles) in CHCl3 solutions (a) and as a thin film on glass (b). | ||
| Compound | λ max (nm) in CHCl3 | ε (M−1 cm−1) | λ max (nm) film | λ onset (nm) film | E optg (eV) |
|---|---|---|---|---|---|
| TII-Pht2 | 417 | 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
432 | 800 | 1.55 |
| 631 | 40000 |
652 | |||
| DPP-Pht2 | 579 | 42000 |
614 | 751 | 1.65 |
| 620 | 44000 |
677 |
The spectrum of both materials displays two absorption bands attributed to π–π* (350–450 nm) and intramolecular charge transfer (ICT) transitions (500–750 nm), respectively. Although comparable extinction coefficients (ε) were estimated, TII-Pht2 is characterized by a broader and significantly red-shifted (of ca. 50 nm) absorption band compared to DPP-Pht2. As expected, the spectra of the films show broader absorption bands and red-shifted long wavelength absorption onsets at 751 and 800 nm corresponding to optical energy gaps (Eoptg) of 1.65 and 1.55 eV for DPP-Pht2 and TII-Pht2, respectively.
Cyclic voltammetry was performed in dichloromethane with Bu4NPF6 as the supporting electrolyte. The cyclic voltammogram (CV) shows a reversible oxidation wave with anodic peak potential (Epa) at ca. 1.0 V for both compounds (Fig. 2 and Table 2).
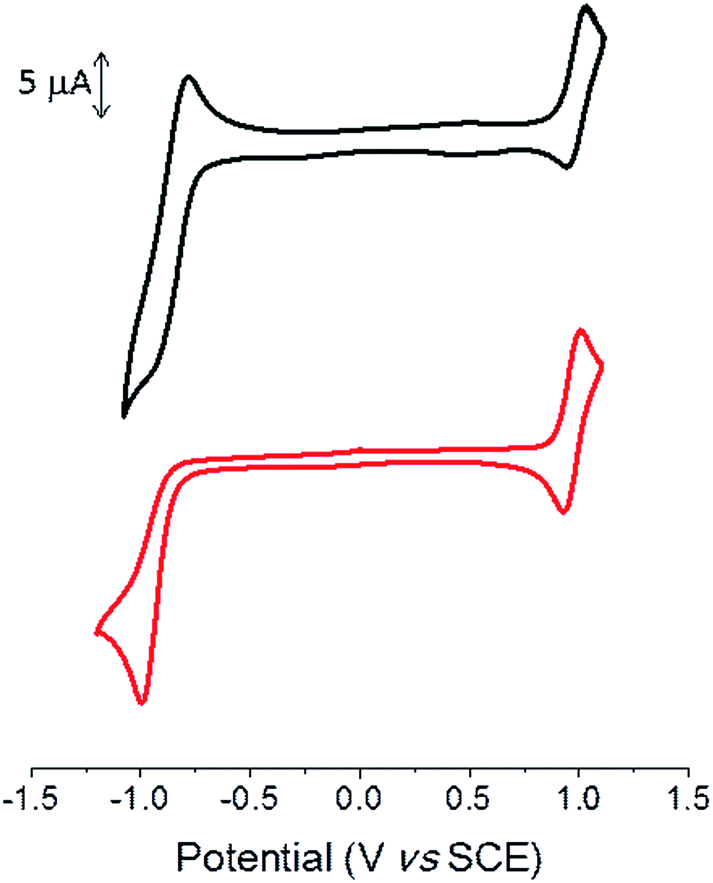 | ||
| Fig. 2 Cyclic voltammograms of TII-Pht2 (black) and DPP-Pht2 (red) 1 mM in 0.1 M Bu4NPF6/CH2Cl2, 100 mV s−1, Pt working electrode. | ||
The CV of the DPP derivative exhibits an irreversible reduction wave with a cathodic peak potential (Epc) of −0.99 V, whereas a quasi-reversible reduction process is observed for TII-Pht2 with Epc = −0.95 V. The potentials of the onsets of oxidation and reduction processes lead to estimated HOMO/LUMO levels of −5.89/−4.24 eV and −5.88/−4.13 eV for TII-Pht2 and DPP-Pht2, respectively (Table 2).
Density functional theory (DFT) calculations using Gaussian 9 program based on PBE1PBE 6-311++G(2df,2pd) were carried out in parallel. The branched alkyl chains of the phthalimide moieties were replaced by simple methyl groups in order to reduce the computation time. Fig. 3 shows the optimized geometries and distribution of the frontier orbitals for the two compounds.
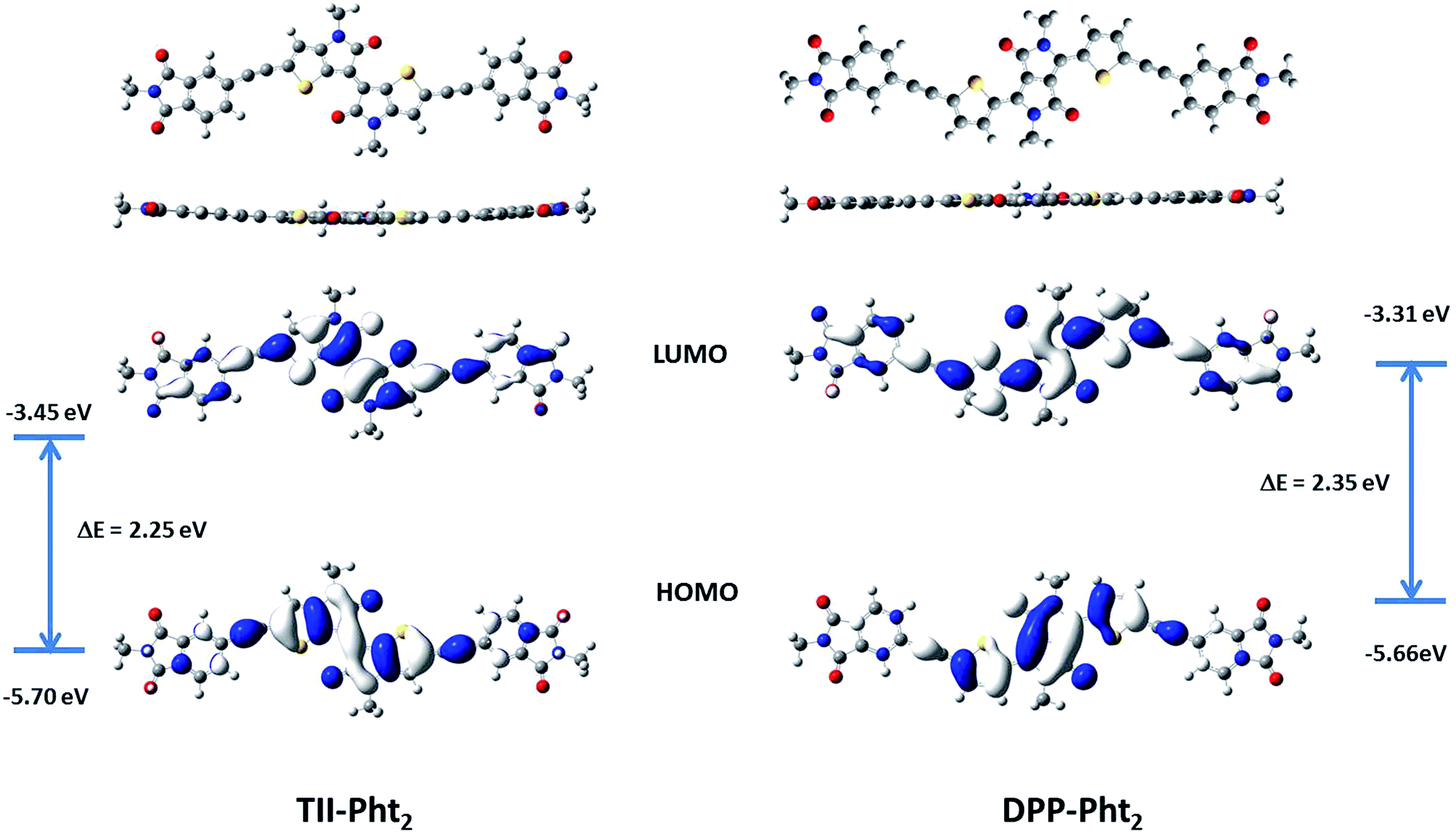 | ||
| Fig. 3 Optimized geometries and molecular orbital distributions of TII-Pht2 and DPP-Pht2. | ||
Both molecules exhibit a quasi-planar structure with a torsion angle between the phthalimide block and the central core of ca. 1.0 and 0.04° for TII-Pht2 and DPP-Pht2, respectively. For both compounds the HOMO is essentially localized on the central dye while the LUMO is more evenly distributed along the whole backbone. The calculated energy levels and energy gaps are in satisfactory agreement with the values estimated from UV-vis absorption and cyclic voltammetric data.
A preliminary evaluation of the potential of the two compounds as electron acceptor materials for OSCs has been carried out on solution-processed bulk heterojunction (BHJ) solar cells. Although higher PCE could probably be obtained with high performance low band gap polymers, the use of P3HT as a donor material allows a direct comparison with the large body of results published for the P3HT/PCBM standard system.65 To this end, inverted solar cells of configuration: ITO/ZnO/active layer/MoO3/Ag were fabricated (see ESI†). Device characteristics are summarized in Table 3 and the best current density–voltage (J–V) curves measured under AM. 1.5 simulated solar illumination (80 mW cm−2) are plotted in Fig. 4.
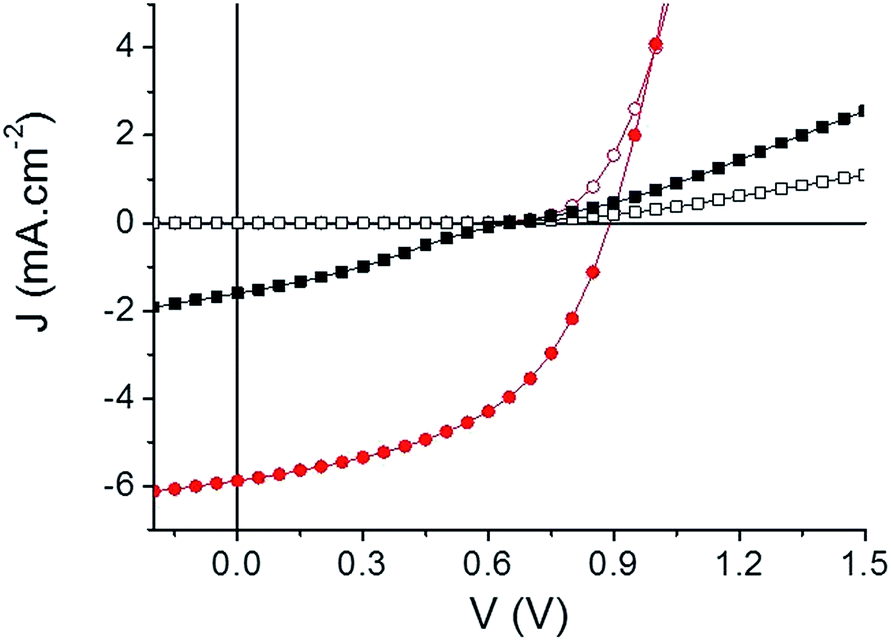 | ||
| Fig. 4 Current density–voltage characteristics of the best inverted cells. TII-Pht2 (black squares) and DPP-Pht2 (red circles) based devices in the dark (open) and under illumination (full). | ||
The best results were obtained with active layers spun-cast at 1000 rpm from chloroform solutions containing 20 mg mL−1 of donor and acceptor materials and annealed at 110 °C for 5 min. Optimal weight to weight P3HT/acceptor ratios were 1:1.5 and 1:2 for TII-Pht2 and DPP-Pht2, respectively. In addition, the impact of diiodooctane (DIO) as an additive for the processing of the active film was investigated.66,67
Comparison of the results clearly shows that DPP-Pht2 leads to better open-circuit voltage (Voc), short-circuit current density (Jsc) and PCE than TII-Pht2. Moreover, for both acceptors, DIO significantly improves efficiencies through a simultaneous increase of Voc, Jsc and fill factor (FF). These effects are particularly spectacular for the DPP-Pht2 cells for which a five-fold increase of PCE is observed.68,69 Thus, a promising PCE of 3.28% with a Voc of 0.89 V, a Jsc of 5.91 mA cm−2 and a FF of 50% were obtained. These results rank among the highest reported so far for DPP-based acceptors.18 On the other hand, despite a moderate efficiency of ca. 0.36%, it is noteworthy that we report herein the very first example of OSCs incorporating a thienoisoindigo-based molecular acceptor.
In order to assess the respective contribution of each component of the cells on the overall PCE, the external quantum efficiencies (EQE) of the best performing inverted solar cells have been recorded.
The spectra in Fig. 5 reveal a common peak around 560 nm attributed to the absorption of the P3HT donor material and broad shoulders in the 650–730 nm and 650–800 nm regions attributed to the contribution of DPP-Pht2 and TII-Pht2, respectively, to the photo-current. The large difference in intensity between the two EQE curves is in good agreement with the higher Jsc values obtained for the DPP based-devices. Finally, it is noteworthy that the current density integrated from the EQE of the best DPP-Pht2 and TII-Pht2 based devices are close to their corresponding Jsc values (5.85 and 1.42 mA cm−2vs. 5.91 and 1.62 mA cm−2).
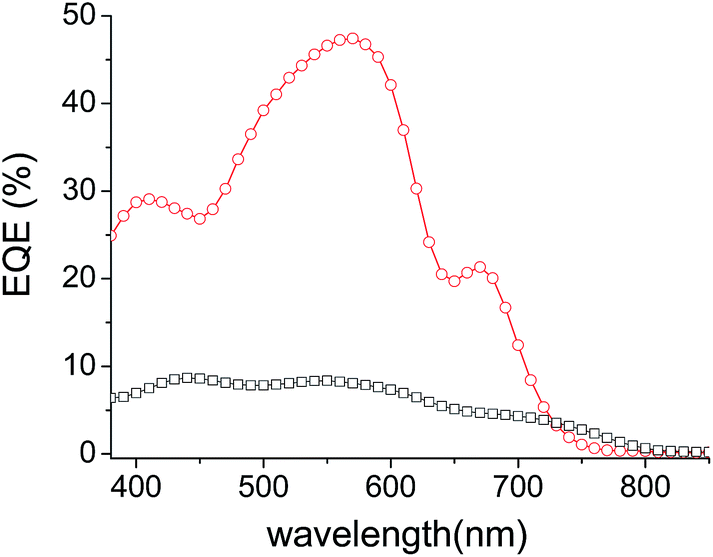 | ||
| Fig. 5 EQE curves of DPP-Pht2 (red circles) and TII-Pht2 (black squares) based OSCs. | ||
In order to complete these results, the electron mobilities (μe) of both acceptors were investigated using the space-charge limited current (SCLC) method with devices of structure ITO/DPP-Pht2 or TII-Pht2/LiF/Al (see ESI†).70μe values of 2.3 × 10−5 and 1.5 × 10−4 cm2 V−1 s−1 were obtained for TII-Pht2 and DPP-Pht2, respectively. This ca. one order of magnitude higher electron mobility of DPP-Pht2 probably contributes to the better Jsc, FF and consequently the PCE of OSCs based on this acceptor.
The nano-scale morphology of the optimized active layers has been investigated using atomic force microscopy (AFM). As shown in Fig. 6, the P3HT:TII-Pht2 blend exhibits smaller and more homogenous domains than those observed on the surface of DPP-Pht2-based cells with an average root mean square (RMS) roughness value of ca. 6 nm vs. ca. 32 nm for DPP-Pht2 based cells.
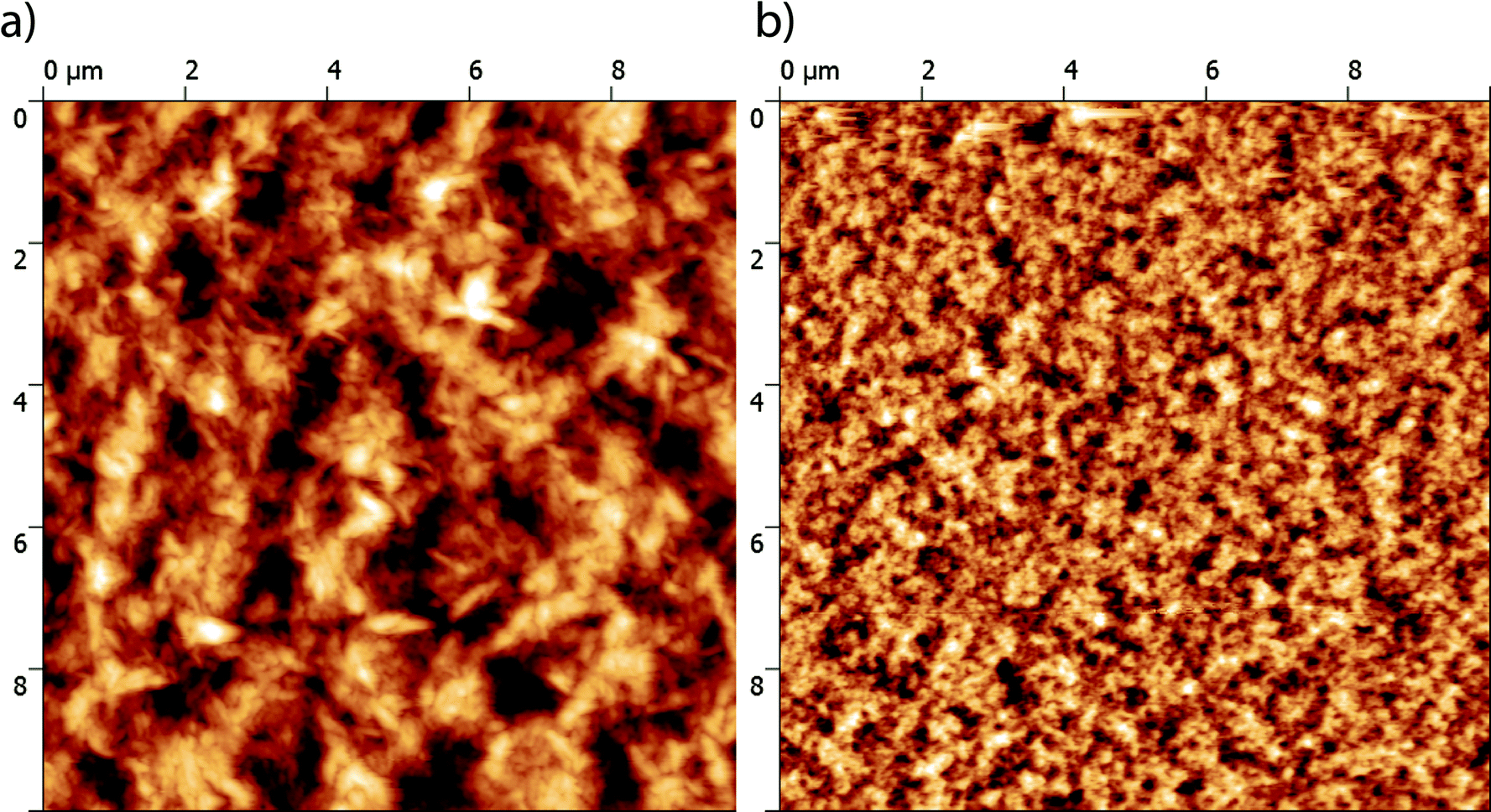 | ||
| Fig. 6 AFM phase images (10 × 10 μm) of optimized DPP-Pht2 (a) and TII-Pht2 (b) based active layers. | ||
Furthermore, this imaging technique also revealed the presence of fibrillar microcrystalline domains in the P3HT:DPP-Pht2 blend that can be associated with a better self-organization of the donor which is expected to be beneficial for effective charge transport.71
4. Conclusion
In summary, two simple acetylene-bridged n-type small molecules based on diketopyrrolopyrrole or thienoisoindigo core end-capped with phthalimide electron-accepting moieties have been synthesized. Their potential as electron-acceptor materials for OSCs was estimated in inverted BHJ solar cells. Once blended with P3HT, promising PCEs of ca. 0.36% and 3.28% for TII-Pht2 and DPP-Pht2 based devices, respectively, were reached. It is worth noting that it is, to the best of our knowledge, (i) the first example of thienoisoindigo based materials used as a non-fullerene electron acceptor and (ii) the highest efficiency reported so far for a diketopyrrolopyrrole based molecular acceptor material in BHJ solar cells. Further device optimizations and side chain engineering are ongoing to improve these performances.Acknowledgements
The University of Angers is acknowledged for financial support (AAP CS project SolarIs). The Chinese Government Scholarship (CGC) program is thanked for the Ph-D grant of Y. Jiang. The PIAM (Plateforme d’Ingénierie et Analyses Moléculaires) of the University of Angers is thanked for the characterization of organic compounds.References
- F. C. Krebs, N. Espinosa, M. Hösel, R. R. Søndergaard and M. Jørgensen, Adv. Mater., 2014, 26, 29–39 CrossRef CAS PubMed.
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2015, 44, 78–90 RSC.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766–4771 CrossRef CAS PubMed.
- Z. M. Beiley, M. G. Christoforo, P. Gratia, A. R. Bowring, P. Eberspacher, G. Y. Margulis, C. Cabanetos, P. M. Beaujuge, A. Salleo and M. D. McGehee, Adv. Mater., 2013, 25, 7020–7026 CrossRef CAS PubMed.
- Y.-H. Chen, L.-Y. Lin, C.-W. Lu, F. Lin, Z.-Y. Huang, H.-W. Lin, P.-H. Wang, Y.-H. Liu, K.-T. Wong, J. Wen, D. J. Miller and S. B. Darling, J. Am. Chem. Soc., 2012, 134, 13616–13623 CrossRef CAS PubMed.
- S.-H. Liao, H.-J. Jhuo, P.-N. Yeh, Y.-S. Cheng, Y.-L. Li, Y.-H. Lee, S. Sharma and S.-A. Chen, Sci. Rep., 2014, 4, 6813 CrossRef CAS PubMed.
- J. You, C.-C. Chen, Z. Hong, K. Yoshimura, K. Ohya, R. Xu, S. Ye, J. Gao, G. Li and Y. Yang, Adv. Mater., 2013, 25, 3973–3978 CrossRef CAS PubMed.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886–3893 CrossRef CAS PubMed.
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642–6671 CrossRef CAS PubMed.
- H.-Y. Lin, W.-C. Huang, Y.-C. Chen, H.-H. Chou, C.-Y. Hsu, J. T. Lin and H.-W. Lin, Chem. Commun., 2012, 48, 8913–8915 RSC.
- K. R. Graham, C. Cabanetos, J. P. Jahnke, M. N. Idso, A. El Labban, G. O. Ngongang Ndjawa, T. Heumueller, K. Vandewal, A. Salleo, B. F. Chmelka, A. Amassian, P. M. Beaujuge and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 9608–9618 CrossRef CAS.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed.
- J. C. Hummelen, B. W. Knight, F. LePeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532–538 CrossRef CAS.
- T. B. Singh, N. Marjanović, G. J. Matt, S. Günes, N. S. Sariciftci, A. Montaigne Ramil, A. Andreev, H. Sitter, R. Schwödiauer and S. Bauer, Org. Electron., 2005, 6, 105–110 CrossRef CAS.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC.
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- A. a. F. Eftaiha, J.-P. Sun, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 1201–1213 CAS.
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS.
- J. T. Bloking, X. Han, A. T. Higgs, J. P. Kastrop, L. Pandey, J. E. Norton, C. Risko, C. E. Chen, J.-L. Brédas, M. D. McGehee and A. Sellinger, Chem. Mater., 2011, 23, 5484–5490 CrossRef CAS.
- C. H. Woo, T. W. Holcombe, D. A. Unruh, A. Sellinger and J. M. J. Fréchet, Chem. Mater., 2010, 22, 1673–1679 CrossRef CAS.
- B. Walker, X. Han, C. Kim, A. Sellinger and T.-Q. Nguyen, ACS Appl. Mater. Interfaces, 2012, 4, 244–250 CAS.
- F. G. Brunetti, X. Gong, M. Tong, A. J. Heeger and F. Wudl, Angew. Chem., Int. Ed., 2010, 49, 532–536 CrossRef CAS PubMed.
- H. U. Kim, J.-H. Kim, H. Suh, J. Kwak, D. Kim, A. C. Grimsdale, S. C. Yoon and D.-H. Hwang, Chem. Commun., 2013, 49, 10950–10952 RSC.
- Y. Lin, Y. Li and X. Zhan, Adv. Energy Mater., 2013, 3, 724–728 CrossRef CAS.
- P. Sonar, G.-M. Ng, T. T. Lin, A. Dodabalapur and Z.-K. Chen, J. Mater. Chem., 2010, 20, 3626–3636 RSC.
- Y. Lin, P. Cheng, Y. Li and X. Zhan, Chem. Commun., 2012, 48, 4773–4775 RSC.
- A. D. Hendsbee, S. M. McAfee, J.-P. Sun, T. M. McCormick, I. G. Hill and G. C. Welch, J. Mater. Chem. C, 2015, 3, 8904–8915 RSC.
- T. Zhou, T. Jia, B. Kang, F. Li, M. Fahlman and Y. Wang, Adv. Energy Mater., 2011, 1, 431–439 CrossRef CAS.
- S. M. McAfee, J. M. Topple, J.-P. Sun, I. G. Hill and G. C. Welch, RSC Adv., 2015, 5, 80098–80109 RSC.
- A. M. Poe, A. M. Della Pelle, A. V. Subrahmanyam, W. White, G. Wantz and S. Thayumanavan, Chem. Commun., 2014, 50, 2913–2915 RSC.
- Y. Shu, Y.-F. Lim, Z. Li, B. Purushothaman, R. Hallani, J. E. Kim, S. R. Parkin, G. G. Malliaras and J. E. Anthony, Chem. Sci., 2011, 2, 363–368 RSC.
- Y. Zhou, Y.-Z. Dai, Y.-Q. Zheng, X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. Commun., 2013, 49, 5802–5804 RSC.
- Y. Zhou, L. Ding, K. Shi, Y.-Z. Dai, N. Ai, J. Wang and J. Pei, Adv. Mater., 2012, 24, 957–961 CrossRef CAS PubMed.
- D. Xia, D. Gehrig, X. Guo, M. Baumgarten, F. Laquai and K. Mullen, J. Mater. Chem. A, 2015, 3, 11086–11092 CAS.
- X.-F. Wu, W.-F. Fu, Z. Xu, M. Shi, F. Liu, H.-Z. Chen, J.-H. Wan and T. P. Russell, Adv. Funct. Mater., 2015, 25, 5954–5966 CrossRef CAS.
- G. Ren, E. Ahmed and S. A. Jenekhe, Adv. Energy Mater., 2011, 1, 946–953 CrossRef CAS.
- E. Ahmed, G. Ren, F. S. Kim, E. C. Hollenbeck and S. A. Jenekhe, Chem. Mater., 2011, 23, 4563–4577 CrossRef CAS.
- A. Sharenko, C. M. Proctor, T. S. van der Poll, Z. B. Henson, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2013, 25, 4403–4406 CrossRef CAS PubMed.
- Z. Lu, B. Jiang, X. Zhang, A. Tang, L. Chen, C. Zhan and J. Yao, Chem. Mater., 2014, 26, 2907–2914 CrossRef CAS.
- X. Zhang, Z. Lu, L. Ye, C. Zhan, J. Hou, S. Zhang, B. Jiang, Y. Zhao, J. Huang, S. Zhang, Y. Liu, Q. Shi, Y. Liu and J. Yao, Adv. Mater., 2013, 25, 5791–5797 CrossRef CAS PubMed.
- Y. Zhong, B. Kumar, S. Oh, M. T. Trinh, Y. Wu, K. Elbert, P. Li, X. Zhu, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 8122–8130 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- Y. Lin, Z.-G. Zhang, H. Bai, J. Wang, Y. Yao, Y. Li, D. Zhu and X. Zhan, Energy Environ. Sci., 2015, 8, 610–616 CAS.
- K. Cnops, B. P. Rand, D. Cheyns, B. Verreet, M. A. Empl and P. Heremans, Nat. Commun., 2014, 5, 3406 Search PubMed.
- Y. Zhong, M. T. Trinh, R. Chen, W. Wang, P. P. Khlyabich, B. Kumar, Q. Xu, C.-Y. Nam, M. Y. Sfeir, C. Black, M. L. Steigerwald, Y.-L. Loo, S. Xiao, F. Ng, X. Y. Zhu and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 15215–15221 CrossRef CAS.
- Y. Zang, C.-Z. Li, C.-C. Chueh, S. T. Williams, W. Jiang, Z.-H. Wang, J.-S. Yu and A. K. Y. Jen, Adv. Mater., 2014, 26, 5708–5714 CrossRef CAS PubMed.
- J. Wang, Y. Yao, S. Dai, X. Zhang, W. Wang, Q. He, L. Han, Y. Lin and X. Zhan, J. Mater. Chem. A, 2015, 3, 13000–13010 CAS.
- P. E. Hartnett, A. Timalsina, H. S. S. R. Matte, N. Zhou, X. Guo, W. Zhao, A. Facchetti, R. P. H. Chang, M. C. Hersam, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 16345–16356 CrossRef CAS PubMed.
- Y. Liu, J. Y. L. Lai, S. Chen, Y. Li, K. Jiang, J. Zhao, Z. Li, H. Hu, T. Ma, H. Lin, J. Liu, J. Zhang, F. Huang, D. Yu and H. Yan, J. Mater. Chem. A, 2015, 3, 13632–13636 CAS.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C.-Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y.-L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242 CrossRef CAS PubMed.
- S. Holliday, R. S. Ashraf, C. B. Nielsen, M. Kirkus, J. A. Röhr, C.-H. Tan, E. Collado-Fregoso, A.-C. Knall, J. R. Durrant, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 898–904 CrossRef CAS PubMed.
- J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821–3838 CrossRef CAS.
- Y. Jiang, C. Cabanetos, M. Allain, P. Liu and J. Roncali, J. Mater. Chem. C, 2015, 3, 5145–5151 RSC.
- A. Leliege, R. C.-H. Le, M. Allain, P. Blanchard and J. Roncali, Chem. Commun., 2012, 48, 8907–8909 RSC.
- V. Jeux, D. Demeter, P. Leriche and J. Roncali, RSC Adv., 2013, 3, 5811–5814 RSC.
- O. Vybornyi, Y. Jiang, F. Baert, D. Demeter, J. Roncali, P. Blanchard and C. Cabanetos, Dyes Pigm., 2015, 115, 17–22 CrossRef CAS.
- J.-W. Mun, I. Cho, D. Lee, W. S. Yoon, O. K. Kwon, C. Lee and S. Y. Park, Org. Electron., 2013, 14, 2341–2347 CrossRef CAS.
- P. G. W. P. Van, F. Gholamrezaie, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2012, 22, 20387–20393 RSC.
- C. H. Woo, P. M. Beaujuge, T. W. Holcombe, O. P. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 15547–15549 CrossRef CAS PubMed.
- T. Kono, T. Sakaguchi, Y. Hu, M. Shiotsuki, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5943–5953 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS.
- M. T. Dang, L. Hirsch and G. Wantz, Adv. Mater., 2011, 23, 3597–3602 CrossRef CAS PubMed.
- J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619–3623 CrossRef CAS PubMed.
- U. Vongsaysy, B. Pavageau, G. Wantz, D. M. Bassani, L. Servant and H. Aziz, Adv. Energy Mater., 2014, 4, 1300752 Search PubMed.
- A. Sharenko, D. Gehrig, F. Laquai and T.-Q. Nguyen, Chem. Mater., 2014, 26, 4109–4118 CrossRef CAS.
- J. Warnan, A. El Labban, C. Cabanetos, E. T. Hoke, P. K. Shukla, C. Risko, J.-L. Brédas, M. D. McGehee and P. M. Beaujuge, Chem. Mater., 2014, 26, 2299–2306 CrossRef CAS.
- M. A. Khan, W. Xu, H. P. Lin Khizar-ul-Haq, Y. Bai, X. Y. Jiang, Z. L. Zhang, W. Q. Zhu, Z. L. Zhang and W. Q. Zhu, J. Appl. Phys., 2008, 103, 014509 CrossRef.
- S. Berson, R. de Bettignies, S. Bailly and S. Guillerez, Adv. Funct. Mater., 2007, 17, 1377–1384 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta09171c |
| This journal is © The Royal Society of Chemistry 2016 |