New π-conjugated polymers as acceptors designed for all polymer solar cells based on imide/amide-derivatives†
Received
22nd August 2015
, Accepted 17th November 2015
First published on 17th November 2015
Abstract
In this paper we report two n-type polymeric acceptors P1 and P2 based on imide/amide-derivatives (PDI, NDI and DPP). P1 and P2 exhibit very different optical properties which may be related to the different molecular configuration and polymeric crystallinity. All polymer solar cells were fabricated with conventional configuration based on polymeric donor PBDTTT-C-T and P1/P2. Results reveal that devices based on P1 show the best performance with a PCE of 2.01%, a VOC of 0.68 V, a JSC of 7.06 mA cm−2 and a FF of 41.78%, which is attributed to the appropriate energy levels, relatively high electron mobility and preferable micro-phase separation. The poor performance for P2 (PCE = 0.30%) was mainly ascribed to its extremely low JSC which was probably caused by the small HOMO energy level difference between donor and P2.
Introduction
All polymer solar cells (all-PSCs), which consist of an n-type polymer semiconductor instead of a fullerene derivative as electron acceptor, have some potential advantages over polymer/fullerene BHJ solar cells, including high electron mobilities, fine-tuned molecular energy levels, broad and strong absorptions, facile control of solution viscosity and miscibility.1 To date, many narrow band-gap polymeric donors with high PCEs (>9%) have been reported in polymer/fullerene BHJ solar cells and many of them have also been applied in the construction of high performance all-PSCs.2 However, far less polymeric acceptors have been reported compared to the donor counterpart, which is one of the key factors towards high performance all-PSCs. Therefore, it is necessary to design and synthesize new conjugated polymeric acceptors with expected properties, including proper molecular energy levels, broad absorption range, high electron mobilities and good miscibility with polymeric donors.
Conjugated polymeric acceptors based on imide/amide derivatives have drawn much attention in all-PSCs and other optoelectronic devices for the following reasons: (1) the electron-withdrawing effect of imide/amide groups can be utilized to tune the frontier molecular orbital energies (HOMO/LUMO); (2) alkyls substituted at the N-positions can adjust the solubility and intermolecular interactions of polymers; (3) they usually exhibit planar structures and show excellent chemical, thermal and photochemical stability which is favourable to the fabrication of devices.3 Perylenediimide (PDI) and naphthalenediimide (NDI) are among the most efficient imide-derivatives and have been widely explored as building blocks in n-type polymers for PSCs and other optoelectronic materials.4 Zhan and coworkers first reported an n-type polymeric acceptor based on PDI in all-PSCs with PCE up to 1.5% and 3.45% after optimization by binary additives.5 Thereafter, many polymeric acceptors based on PDI and NDI were designed and synthesized for all-PSCs with high performance.6 For example, n-type polymer P(NDI2OD-T2) has been intensely investigated by several research groups with PCE>4.5% after utilizing suitable electron donors and device configurations.7 Recently, polymer P(NDI2OD-FT2) based on NDI and F-substituted bithiophene was reported, and the derived all-polymer BHJ solar cell showed a PCE up to 6.71%, which indicates that there are no fundamental reasons as to why all-PSCs cannot achieve the efficiency of fullerene–polymer devices.8 In addition, as a classical representation of π-conjugated amide derivatives, diketopyrrolopyrrole (DPP) and its analogues, such as dithienyl-DPP, have been widely incorporated into conjugated polymers as electron accepting moieties for organic optoelectronic materials.9 However, compared to PDI and NDI moieties, DPP exhibits both high-lying HOMO and LUMO energy levels, which suggests that the DPP moiety may serve as an electron-donating unit when combined with the PDI or NDI moiety.10,11 The optoelectronic properties of polymeric acceptors based on all imide/amide-derivatives and the potential applications in all-PSCs attract us to explore them.
In this work, we report two n-type polymers P1 and P2, based on PDI/DPP and NDI/DPP, respectively. It was found that they exhibit very different solubilities with the same alkyl chains in PDI and NDI moieties, which may be ascribed to the different molecular configuration and crystallinity. The thin film of P2 shows a broad and strong absorption in the near-infrared region (∼925 nm), which is quite different from that of P1 (weak absorption at 737 nm). All-PSCs were fabricated with conventional configuration based on narrow band-gap polymers PBDTTT-C-T and PTB7-Th as electron donors and P1/P2 as the acceptor, respectively. Results demonstrate that all-PSCs based on P1 show the best performance with a PCE of 2.01%, a VOC of 0.68 V, a JSC of 7.06 mA cm−2 and a FF of 41.78%. The better performance of devices based on P1 than P2 is owing to several factors including suitable energy levels, relatively high electron mobility and preferable micro-phase separation. The poor performance of devices based on P2 may be mainly caused by the small HOMO energy level difference between donor and P2. Therefore, to obtain high efficiency of all-PSCs, the HOMO/LUMO energy levels of the polymers should be carefully regulated, especially for narrow band-gap materials with broad absorptions in the near-infrared range.
Results and discussion
Synthesis and characterization
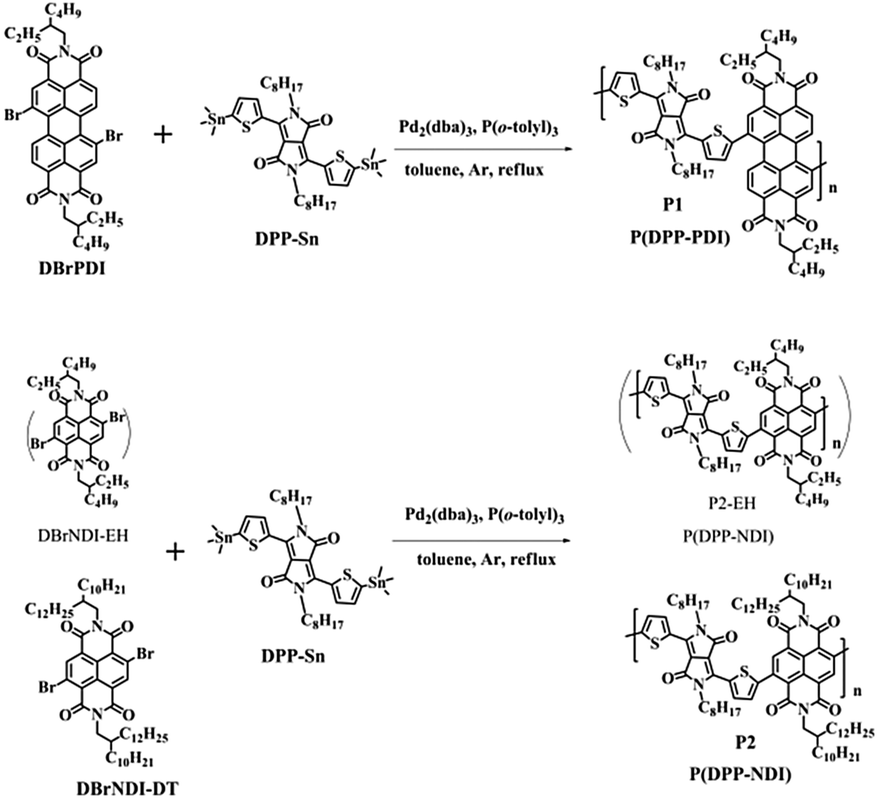
As depicted in Scheme 1, P1 and P2 (including P2-EH) were prepared by the Stille coupling reaction between dibromo-substituted PDI/NDI and DPP-Sn, in the presence of Pd2(dba)3 and P(o-toly)3 as catalyst and ligand, respectively, and purified by precipitation followed by Soxhlet extraction. P1 with ethyl-hexyl (EH) as the branched alkyl chains in the PDI monomer is highly soluble (>20 mg mL−1) in common organic solvents such as chloroform, tetrahydrofuran, toluene and 1,2-dichlorobenzene (o-DCB) at room temperature. However, P2-EH (see Scheme 1) with the same alkyl chains in the NDI monomer which is a smaller π-conjugated system than PDI shows extremely poor solubility in common solvents mentioned above and even in hot o-DCB (150 °C, <0.5 mg mL−1), making it difficult to be further purified and characterized. To investigate the optoelectronic properties of polymer P(DPP-NDI), P2 with decyl-tetradecanyl (DT) as the long branched alkyl chains in the NDI monomer was synthesized. P2 shows good solubility in common solvents mentioned above.
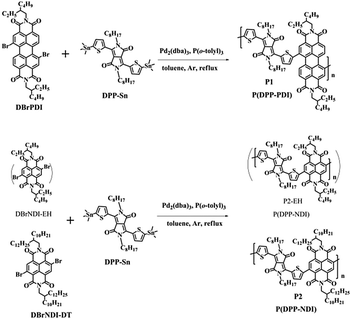 |
| | Scheme 1 Chemical structures and synthetic routes to P1 and P2. | |
The number average molecular weight (Mn) and weight average molecular weight (Mw) as well as polydispersity index (PDI = Mw/Mn) of P1 and P2 were determined by gel permeation chromatography (GPC) at 40 °C using polystyrene as standard with tetrahydrofuran as the eluent. P1 shows a Mn of 22.6 kDa and a PDI of 1.89. For P2, Mn is 37.7 kDa and PDI is 1.31.
Thermal properties
The thermal property of the polymer was measured by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) in a nitrogen atmosphere at a heating rate of 10 °C min−1. TGA plots (see Fig. S1a, ESI†) reveal that P1 and P2 both exhibit good thermal stability with decomposition temperatures (Td) above 425 °C at 5% weight loss under nitrogen. Obviously, the thermal stability of these two polymers is adequate for their application in all-PSCs and other optoelectronic devices. Moreover, P1 and P2 show no detectable thermal transitions up to 300 °C as observed from DSC plots (Fig. S1b, ESI†).
Optical properties
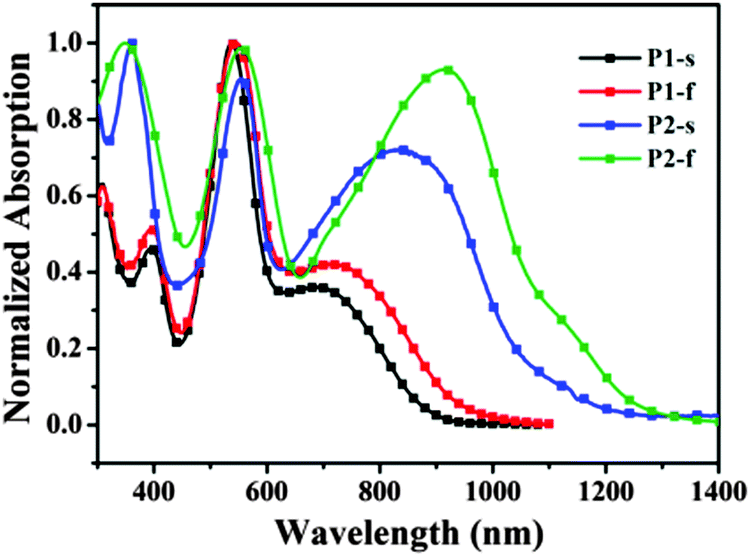
The UV-Vis-NIR (near infrared region) absorption properties of P1 and P2 were measured in chloroform solutions (1.0 × 10−5 M) and as thin films on quartz slides (Fig. 1), and the data are summarized in Table 1. In dilute solutions, P1 mainly exhibits three absorption peaks at around 395, 539 and 700 nm, and P2 shows three absorption peaks at around 360, 553 and 841 nm. Compared to those in solutions, the absorption spectra of thin films of P1 and P2 are largely red-shifted. For instance, as to the thin film of P2, the absorption band at 841 nm in solution is shifted to 925 nm. Such red-shifts are likely owing to the intermolecular interactions within thin films which could facilitate charge transportation for photovoltaic and other optoelectronic applications.12 Notably as to these red-shifts at long wavelength between solutions and thin films, P2 exhibits a larger red-shift (84 nm) than P1 (37 nm), which could be explained from that the NDI moiety has a more intense tendency to crystallize than PDI, inducing a stronger interaction in thin films.6h Also the better crystallization of P2 will partly reduce its solubility (for example, P2-EH shows an extremely poor solubility than P1, as discussed above). It is surprising that absorptions of P2 (both in solution and in thin film) are largely red-shifted compared to those of P1; however, the NDI moiety within P2 has a much smaller π-conjugated system than the PDI moiety. The reason for this is probably that the PDI moiety has a larger torsion configuration with the DPP moiety within P1 (see Fig. 3), which will reduce the efficient conjugation length. Furthermore, the optical band-gap of P2 estimated from the absorption onset in thin films is 1.00 eV, much smaller than that of P1 (1.28 eV), and the broad absorption of P2 may contribute to a NIR photovoltaic response.
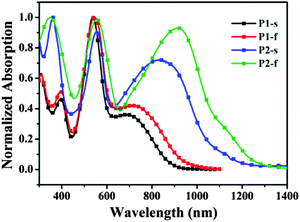 |
| | Fig. 1 Normalized absorption UV-Vis-NIR spectra for P1 and P2 in CHCl3 solutions (P1-s, P2-s) and thin films (P1-f, P2-f). | |
Table 1 Absorption data, electrochemical properties, HOMO/LUMO energy levels and band gaps of P1 and P2
| Polymer |
λ
max
/nm |
E
oxonset/V |
E
redonset/V |
HOMO/eV |
LUMO/eV |
E
cvg/eV |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b/eV b/eV |
| Solution |
Film |
Δλ |
|
Measured in CHCl3 with a concentration of 1.0 × 10−5 M of the respective repeating units.
Calculated based on the onset of thin film absorption spectra.
|
|
P1
|
539 |
548 |
9 |
1.01 |
0.34 |
−5.42 |
−4.07 |
1.35 |
1.28 |
| 700 |
737 |
37 |
|
P2
|
553 |
558 |
5 |
0.89 |
0.42 |
−5.30 |
−3.99 |
1.31 |
1.00 |
| 841 |
925 |
84 |
Electrochemical properties
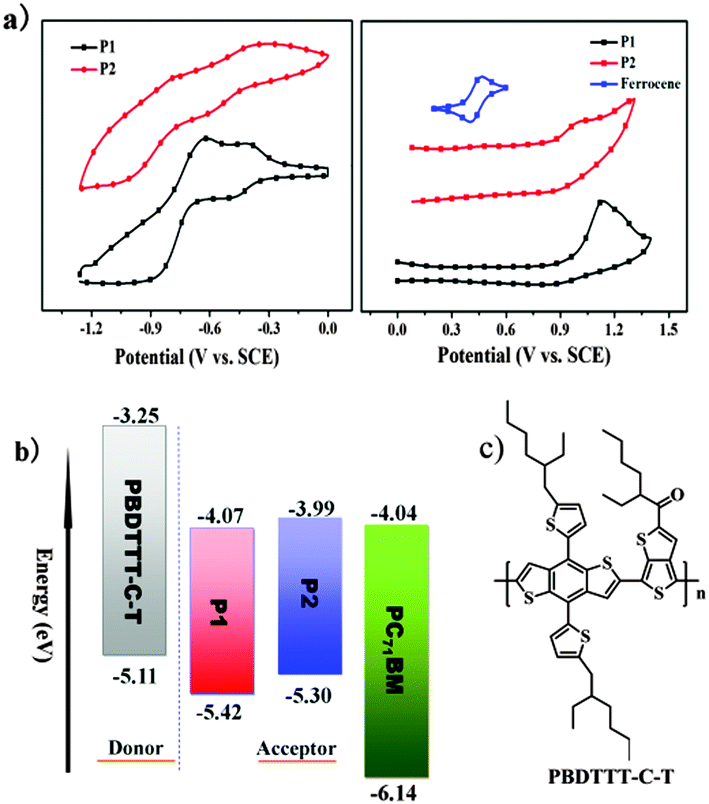
To investigate the differences in electrochemical properties, the redox potentials of P1 and P2 films deposited onto a platinum electrode were studied by cyclic voltammetry (CV) measurements. The HOMO and LUMO levels of polymers were calculated from their onset oxidation potential (φox) and onset reduction potential (φred) according to the empirical equations EHOMO = −e(φox + 4.8 − φ1/2, FeCp2) (eV) and ELUMO = −e(φred + 4.8 − φ1/2, FeCp2) (eV), where the unit of potential is V (vs. SCE).13 As shown in Fig. 2a, the onset oxidation potentials of P1 and P2 were estimated to be 1.01 V and 0.89 V, corresponding to the HOMO energy levels of −5.42 eV and −5.30 eV, respectively. The onset reduction potentials of P1 and P2 were estimated to be 0.34 V and 0.42 V, corresponding to the LUMO energy levels of −4.07 eV and −3.99 eV, respectively. From the onset potentials of the oxidation and reduction processes, the band gaps (Ecvg) of P1 and P2 were calculated to be about 1.35 and 1.31 eV. The values are higher than those obtained by the above described optical method, which is mainly due to the energy barriers of the charge transfer at the electrodes during the CV measurement.14 The HOMO/LUMO energy levels of the polymers and PC71BM are shown in Fig. 2b. As P1 and P2 have relatively low-lying LUMO levels (around −4.00 eV, in line with that of PC71BM), they are generally n-type organic semiconductors and have potential application in all-PSCs as non-fullerene acceptor materials.
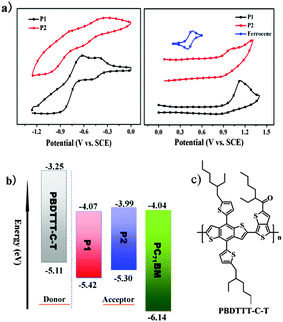 |
| | Fig. 2 (a) Cyclic voltammograms of thin films of P1 and P2 on Pt electrodes in 0.1 M Bu4NPF6–CH3CN at a scan rate of 100 mV s−1; (b) energy level diagrams of the materials in the photovoltaic device; (c) chemical structure of PBDTTT-C-T. | |
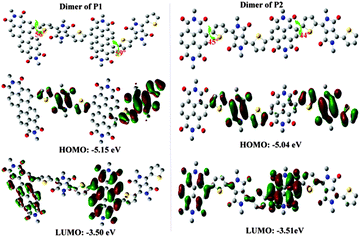 |
| | Fig. 3 The calculated structures, dihedral angles and HOMO/LUMO orbitals and energies of P1 and P2 obtained with DFT-calculations (B3LYP/6-31G); the alkyl chains were replaced by methyl groups in the calculations and all the hydrogen atoms were hidden for simplification. | |
DFT-calculated absorptions, molecular geometries and orbitals
To further understand the connection between molecular structures and optical/electrochemical properties of P1 and P2, quantum-chemical calculations (based on density functional theory, B3LYP/6-31G) were performed for the dimers of both polymers. Long alkyl chains were replaced by methyl groups in order to reduce the calculation time. This simplification does not affect the results because the influence of alkyl chains on the electronic structures of the polymers can be ignored.
As shown in Fig. S2 (ESI†), the absorption profiles calculated in the Vis-NIR range are almost consistent with our experiment results, and the absorptions in the NIR region are mainly contributed by HOMO → LUMO energy level transitions. Fig. 3 reveals that the dihedral angle between the NDI core and the neighbouring thiophene units is about 44–45° in the dimer of P2, which is smaller than that of P1 (56–59°). A more intuitive diagram of molecular conformations is shown in Fig. S3 (ESI†). The smaller backbone torsion of P2 is more favourable for electron cloud delocalization along polymer chains, which may be one reason for the large red-shift in absorption spectra compared to that of P1. Moreover, better planarity of the polymer backbone of P2 would afford a stronger intermolecular interaction and also partly injure the solubility of polymers (see the solubility of P2-EH in “Synthesis and characterization” section). Meanwhile, the band gap of the dimer of P2 (1.53 eV) is smaller than that of P1 (1.65 eV), which is also consistent with the results obtained from CV measurements and optical absorption. Notably, the HOMO orbitals of both polymers are mainly delocalized over the DPP moieties, and the LUMO orbitals are mainly located in the PDI and NDI cores for P1 and P2, respectively. Therefore in this work, DPP moieties should serve as weak donors, though DPP building blocks are generally considered as electron-deficient parts in most D–A polymers.
Electron mobilities
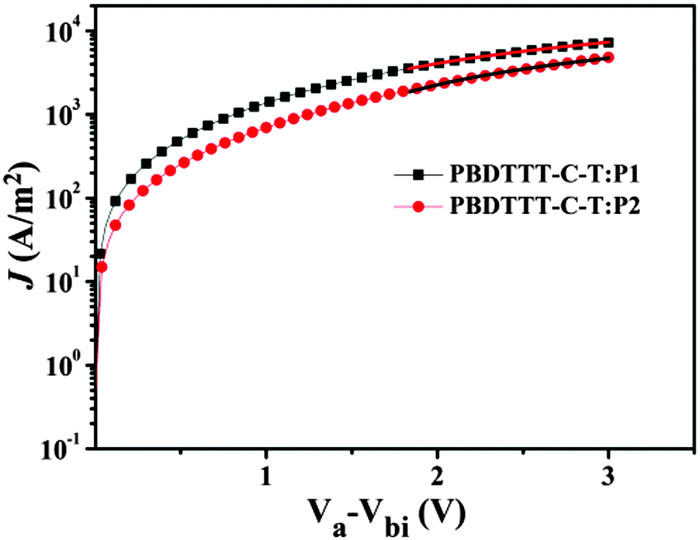
For n-type non-fullerene acceptor materials in all-PSCs, one of the important properties is electron mobility. In this work, we used the space-charge-limited current (SCLC) method with a device configuration of ITO/ZnO/blend film/Ca/Al to measure electron mobility. The SCLC is described as| | 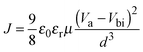 | (1) |
where J is the current density, ε0 is the permittivity of vacuum, εr is the relative permittivity of the transport medium, μ is the mobility, Va is the applied voltage, Vbi is the built-in voltage, and d is the thickness of the active film. According to eqn (1) and Fig. 4, the electron mobility of P1 is calculated to be 3.51 × 10−4 cm2 V−1 s−1, which is 2.5 times that of P2 (1.38 × 10−4 cm2 V−1 s−1). A higher electron mobility would facilitate charge transportation, affording a higher short-circuit current density (JSC) and consequently higher PCE of PSCs.15
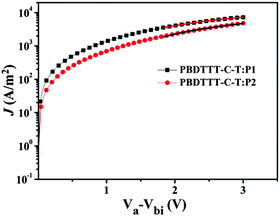 |
| | Fig. 4 Current–voltage characteristics of the SCLC devices with blend films of PBDTTT-C-T:P1 (w/w = 1/1) and PBDTTT-C-T:P2 (w/w = 1/1). | |
Photovoltaic properties
Judging from their LUMO/HOMO levels, relatively high electron mobilities and broad absorption spectra, P1 and P2 may function as electron acceptors for all-PSCs as discussed above. In this work, considering the relatively low LUMO energy levels of P1 and P2, to achieve a high VOC and further a high PCE, narrow bandgap polymer PBDTTT-C-T (see Fig. 2c) with a low HOMO energy level (−5.11 eV, lower than P3HT) was chosen as the donor material to couple with polymer acceptors P1 and P2. As reported before, PBDTTT-C-T exhibited high mobility (>0.01 cm2 V−1 s−1, measured using a field-effect transistor), broad absorption (300–800 nm), and good film-forming ability, and has been intensely investigated in organic solar cells.5a,b,6a,16 Moreover, as shown in Fig. S4a (ESI†), thin film absorptions of donor PBDTTT-C-T and acceptors P1 and P2 are exactly complementary, especially in the range of 500–800 nm, which is beneficial for a higher JSC and a broad photo-response. In fact, the blend thin films of PBDTTT-C-T:P1 and PBDTTT-C-T:P2 exhibited broad absorptions in the region of 300–900 nm and 300–1100 nm, respectively, as shown in Fig. S4b (ESI†). The HOMO/LUMO energy levels of donor PBDTTT-C-T are higher (by more than 0.3 eV) than those of P1, indicating that the energy level positions of the donor and acceptor are suitable for efficient exciton migration and separation at the interface between the two polymers. However, the HOMO energy level difference between PBDTTT-C-T and P2 is only 0.19 eV, much smaller than 0.3 eV which is considered to be an essential empirical threshold for efficient exciton separation.17 Thus the smaller HOMO energy level gap might affect the efficient exciton separation and result in a lower short-circuit current density for the polymer/P2 system.
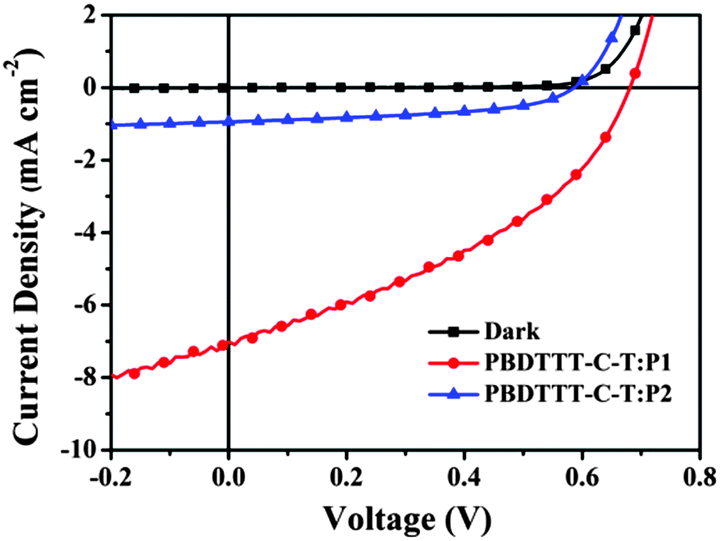
All-PSCs were fabricated with the configuration of ITO/PEDOT:PSS/PBDTTT-C-T:P1 or P2/Ca/Al (PEDOT:PSS = poly-(3,4-ethylenedioxythiophene):poly(styrenesulfonate)) under the illumination of AM 1.5 G (100 mW cm−2). The weight ratios of PBDTTT-C-T:P1 and PBDTTT-C-T:P2 varied from 2:1 to 1:2 for device optimization. Current density versus voltage (J–V) curves of all-PSCs are shown in Fig. 5 and the corresponding photovoltaic parameters including open circuit voltage (VOC), short-circuit current density (JSC) and fill factor (FF) are summarized in Table 2.17 One can observe that both PBDTTT-C-T:P1 and PBDTTT-C-T:P2 systems exhibit the best photovoltaic performance at 1:1 weight ratio. All-PSCs based on the blend films of PBDTTT-C-T/P1 give the best results with a maximum PCE of 2.01%, a VOC of 0.68 V, a JSC of 7.06 mA cm−2 and a FF of 41.78%. However, all-PSCs based on the blend films of PBDTTT-C-T/P2 show extremely poor performance with a maximum PCE of 0.30%, which is mainly ascribed to the much lower JSC (1.16 mA cm−2). As we have discussed above, the HOMO energy level gap between PBDTTT-C-T and P2 is much smaller than 0.30 eV, which may cause an insufficient driving force for efficient exciton separation.18 In addition, we have chosen another p-type conjugated polymer PTB7-Th in our available donor materials to couple P1 and P2 in all-PSCs. Results show that devices based on PTB7-Th/P1 and PTB7-Th/P2 exhibit slightly lower photovoltaic performance than those based on PBDTTT-C-T/P1 and PBDTTT-C-T/P2, respectively, with no obvious change in VOC and FF, but an obvious reduction in JSC (from 7.06 mA cm−2 to 6.24 mA cm−2 for the P1 device, reduced by 12%). The decrease in JSC probably resulted from the further reduced HOMO energy level differences between PTB7-Th and P1/P2 (see Table S1 and Fig. S6, ESI†). Therefore, the HOMO/LUMO energy levels should be carefully regulated, especially for narrow band-gap materials with broad absorptions in the near-infrared range. Furthermore, thermal annealing or introduction of additives could not obviously improve the photovoltaic performance for the blend thin films of PBDTTT-C-T/P1 (or P2) under the current device configuration.
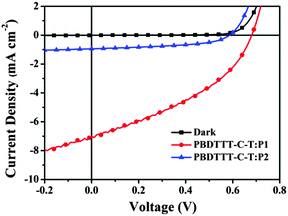 |
| | Fig. 5
J–V curves of the photovoltaic devices with the blend thin films of PBDTTT-C-T:P1 (1:1, w/w) and PBDTTT-C-T:P2 (1:1, w/w) under the illumination of AM 1.5 G, 100 mW cm−2. | |
Table 2 Characteristics of all-PSCs based on blend films of PBDTTT-C-T/P1 and PBDTTT-C-T/P2a
| Donor:acceptor |
w/w |
V
OC (V) |
J
SC (mA cm−2) |
FF (%) |
PCEb (%) |
|
The data were based on more than 10 devices.
Data were provided in the “highest (average)” format.
|
| PBDTTT-C-T:P1 |
2:1 |
0.68 |
5.65 |
40.89 |
1.57 (1.49) |
| 1:1 |
0.68 |
7.06 |
41.78 |
2.01 (1.96) |
| 1:2 |
0.67 |
4.78 |
38.66 |
1.24 (1.16) |
|
|
| PBDTTT-C-T:P2 |
2:1 |
0.57 |
0.85 |
43.53 |
0.21 (0.16) |
| 1:1 |
0.58 |
1.16 |
44.32 |
0.30 (0.27) |
| 1:2 |
0.58 |
0.68 |
40.12 |
0.16 (0.14) |
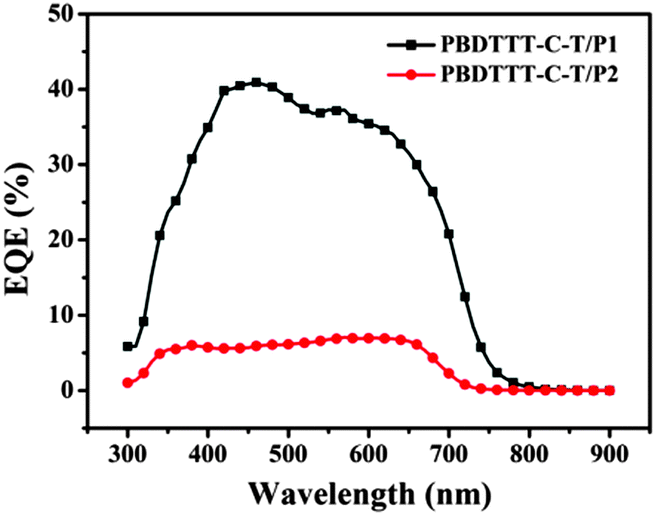
To verify the accuracy of the J–V measurements, the corresponding external quantum efficiencies (EQEs) of the devices were measured. As depicted in Fig. 6, the PBDTTT-C-T:P1 based device (1:1, w/w) displays a broad EQE spectrum from 300 to 750 nm. However, the PBDTTT-C-T:P2 based device exhibits extremely weak EQE response, corresponding to its low JSC. Note that although P2 can offer light absorption up to 1300 nm, photocurrent response of all-PSCs in the long wavelength region is still very low, suggesting a very small contribution to JSC. Furthermore, the calculated JSC by integrating the spectral response of the cells were 7.02 and 1.08 mA cm−2 for PBDTTT-C-T:P1 and PBDTTT-C-T:P2, respectively, which agree well with the measured ones (Table 2).
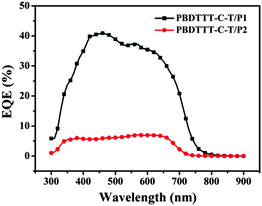 |
| | Fig. 6 EQE curves of the photovoltaic cells with the blend thin films of PBDTTT-C-T:P1 (1:1, w/w) and PBDTTT-C-T:P2 (1:1, w/w) illuminated by monochromatic light. | |
Morphology
In order to deeply understand the different photovoltaic performance of the devices based on PBDTTT-C-T:P1 and PBDTTT-C-T:P2, the morphologies of both blend films were investigated by atomic force microscopy (AFM) with tapping mode height images (4 μm × 4 μm) and phase images (4 μm × 4 μm). As shown in Fig. 7, blend films of both PBDTTT-C-T:P1 and PBDTTT-C-T:P2 exhibit relatively smooth surface with a root-mean square (RMS) roughness of 0.98 nm and 1.23 nm, respectively. However, compared to the blend film of PBDTTT-C-T:P1, that of PBDTTT-C-T:P2 obviously shows larger crystalline domains and phase separation, which are unfavourable for the charge transfer from donor to P2, thus limiting the efficiency of the resulting device.19 The AFM images partially support the results that the device based on PBDTTT-C-T:P1 shows better photovoltaic performance than that based on PBDTTT-C-T:P2 (Table 2).
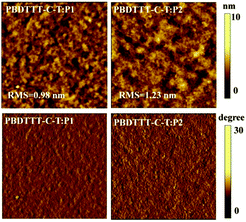 |
| | Fig. 7 AFM height (top) and phase (bottom) images (4.0 μm × 4.0 μm) of blend films of PBDTTT-C-T with P1 and P2 (1:1, w/w), respectively. | |
Conclusions
Two new conjugated polymers P1 and P2 based on diketopyrrolopyrrole (DPP), perylenediimide (PDI) and naphthalenediimide (NDI) were synthesized and characterized. The two conjugated polymers show good thermal stability up to 425 °C. Compared to P1 (PPDI-DPP), polymer P2 (PNDI-DPP) exhibits a more red-shifted and broad absorption from 300 to 1300 nm. The optical bandgaps of P1 and P2 were estimated to be 1.28 eV and 1.00 eV, respectively, based on the onset of thin film absorption spectra. Quantum-chemical calculations were performed to further understand the relationship between molecular structures and optical/electrochemical properties. Results reveal that polymer P2 shows a smaller torsion angle along the backbone of polymer chains, resulting in a narrower band gap and stronger molecular interactions. All polymer solar cells (All-PSCs) were fabricated using conventional configuration with PBDTTT-C-T as donor and P1 or P2 as acceptor materials. It was demonstrated that all-PSCs based on P1 show the best performance with a maximum PCE of 2.01%, a VOC of 0.68 V, a JSC of 7.06 mA cm−2 and a FF of 41.78%, which is ascribed to the appropriate energy levels, relatively high electron mobility and preferable micro-phase separation. The poor performance of the device based on P2 was attributed to its extremely low JSC which probably resulted from the small HOMO energy level difference between donor and P2, causing insufficient driving force for exciton separation. Therefore, the HOMO/LUMO energy levels should be carefully regulated, especially for narrow band-gap polymers with broad absorptions in the near-infrared range.
Experimental section
Materials and characterization techniques
DBrPDI, DBrNDI-EH and DBrNDI-DT were synthesized according to previous reports.20,21 Monomer DPP-Sn was purchased from Beijing Allmers Chemical S&T Co., Ltd, and recrystallized from acetone before polymerization. The donor materials PBDTTT-C-T and PTB7-Th were purchased from Solarmer Materials Inc. 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE III 600 MHz spectrometer. Mass spectra were measured in the MALDI-TOF mode. TGA and DSC measurements were performed using a SDT Q600 V20.9 Build 20 at a heating rate of 10 °C min−1. The absorption spectra were recorded using a Hitachi U-4100 UV-Vis-NIR scanning spectrophotometer. The molecular weight of the polymer was measured by gel permeation chromatography (GPC) performed using an ELEOS system, and polystyrene was used as the standard (40 °C, tetrahydrofuran (THF) as the eluent). Cyclic voltammetry (CV) measurements were performed on a CHI660D electrochemical workstation, which is equipped with a three-electrode cell consisting of a platinum working electrode, a saturated calomel electrode (SCE) as reference electrode and a platinum wire as counter electrode. The measurements were carried out in anhydrous acetonitrile containing 0.1 M n-Bu4NPF6 as a supporting electrolyte under an argon atmosphere at a scan rate of 100 mV s−1. Thin films were deposited from chloroform solution onto the working electrodes.
Characterization of the compound DPP-Sn.
1H NMR (600 MHz, CDCl3): δ 8.98 (d, J = 3.6 Hz, 2H), 7.34 (d, J = 3.7 Hz, 2H), 4.11–4.07 (m, 4H), 1.75 (m, 4H), 1.42–1.22 (m, 20H), 0.87 (t, J = 6.9 Hz, 6H), 0.56–0.33 (m, 18H). 13C NMR (150 MHz, CDCl3): δ 161.60, 146.36, 139.60, 136.50, 135.94, 135.42, 107.19, 42.32, 31.92, 30.03, 29.35, 29.32, 27.01, 22.77, 14.23. MS (MALDI-TOF): m/z 850.3 (M + 1)+.
General synthetic procedures of P1 and P2.
DBrPDI or DBrNDI (1.0 eq.), DPP-Sn (1.0 eq.) and P(o-tolyl)3 (0.08 eq.) were dissolved in toluene (10 mL). The solution was purged with Ar for 30 min and then Pd2(dba)3 (0.04 eq.) was added. The reaction mixture was stirred at 110 °C for 2 days. After cooling to room temperature, the resulting mixture was poured into methanol and stirred for 1.0 h. The dark precipitates were filtered off and subjected to Soxhlet extraction for 2 days successively with methanol, acetone and hexane for the removal of remaining monomers, oligomers and catalytic impurities. The residue was re-dissolved in chloroform and precipitated against methanol; the precipitates were filtered, washed with methanol, and dried under vacuum at 50 °C.
Synthesis of P1.
DBrPDI (115.9 mg, 0.15 mmol), DPP-Sn (127.6 mg, 0.15 mmol), P(o-tolyl)3 (3.6 mg, 0.012 mmol) and Pd2(dba)3 (5.5 mg, 0.006 mmol), and 10 mL of dried toluene were used. P1 was obtained as a purple solid (150 mg) in 88% yield. 1H NMR (600 MHz, CDCl3): δ 9.10 (br, 2H), 8.76 (br, 2H), 8.37 (br, 2H), 8.22 (br, 2H), 4.45 (br), 1.98 (br), 1.77 (br), 1.59–1.14 (m), 0.98–0.83 (m). Mn (GPC, THF, 40 °C) = 22611, PDI = 1.89.
Synthesis of P2.
DBrNDI-DT (164.6 mg, 0.15 mmol), DPP-Sn (127.6 mg, 0.15 mmol), P(o-tolyl)3 (3.6 mg, 0.012 mmol) and Pd2(dba)3 (5.5 mg, 0.006 mmol), and 10 mL of dried toluene were used. P2 was obtained as a dark-purple solid (177 mg) in 81% yield. 1H NMR (600 MHz, CDCl3): δ 9.15 (br, 2H), 8.84 (br, 2H), 7.49 (br, 2H), 4.43 (br), 2.00–1.11 (m), 0.88–1.86 (m). Mn (GPC, THF, 40 °C) = 37717, PDI = 1.31.
Polymer P2-EH shows extremely poor solubility in common solvents (hexane, chloroform, THF, toluene, chlorobenzene) and even in hot o-DCB (150 °C, <0.5 mg mL−1). Therefore, it is difficult to characterize it with conventional methods.
Fabrication of all polymer solar cells
The all-PSCs were fabricated with conventional device architecture, ITO/PEDOT:PSS/PBDTTT-C-T:P1 or P2/Ca/Al. The patterned ITO glass (sheet resistance = 15 Ω square−1) was pre-cleaned in an ultrasonic bath of acetone and isopropyl alcohol and treated in an ultraviolet-ozone chamber (PREEN II-862) for 30 min. Then a thin layer (∼30 nm) of PEDOT:PSS was spin-coated onto the ITO glass and baked at 150 °C for 15 min. Solutions of PBDTTT-C-T:P1 or P2 in o-DCB were spin-coated on the PEDOT:PSS layer to form the active layer of about 90–110 nm. The thickness of the active layer was measured using a Veeco Dektak 150 profilometer. Finally, a Ca (ca. 20 nm) and Al (ca. 70 nm) layer was evaporated onto the surface of the active layer under vacuum.
The photo and dark current density–voltage (J–V) characteristics were recorded with a Keithley 2400 source measurement unit under simulated 100 mW cm−2 irradiation from a Newport solar simulator. The external quantum efficiencies (EQEs) of solar cells were analysed using a certified Newport incident photon conversion efficiency (IPCE) measurement system.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21502205, 21204097, 51173199, 51573205 and 61107090), the Ministry of Science and Technology of China (2010DFA52310), the Outstanding Young Scientists Research Award Fund of Shandong Province (BS2014CL013) and China Postdoctoral Science Foundation (2015M570615).
Notes and references
-
(a) A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS;
(b) Y. Li and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC;
(c) Y.-J. Hwang, T. Earmme, S. Subramaniyan and S. A. Jenekhe, Chem. Commun., 2014, 50, 10801–10804 RSC.
-
(a) J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446–1455 CrossRef PubMed;
(b) J. Subbiah, B. Purushothaman, M. Chen, T. Qin, M. Gao, D. Vak, F. H. Schole, X. Chen, S. E. Watkins, G. J. Wilson, A. B. Holmes, W. W. H. Wong and D. J. Jones, Adv. Mater., 2015, 27, 702–705 CrossRef CAS PubMed;
(c) K. Yao, X.-K. Xin, C.-C. Chueh, K.-S. Chen, Y.-X. Xu and A. K.-Y. Jen, Adv. Funct. Mater., 2015, 25, 567–574 CrossRef CAS;
(d) Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293–5300 CrossRef CAS PubMed;
(e) Z. Zheng, S. Zhang, M. Zhang, K. Zhao, L. Ye, Y. Chen, B. Yang and J. Hou, Adv. Mater., 2015, 27, 1189–1194 CrossRef CAS PubMed;
(f) L. Huo, T. Liu, X. Sun, Y. Cai, A. J. Heeger and Y. Sun, Adv. Mater., 2015, 27, 2938–2944 CrossRef CAS PubMed;
(g) T. L. Nguyen, H. Choi, S. J. Ko, M. A. Uddin, B. Walker, S. Yum, J. E. Jeong, M. H. Yun, T. J. Shin, S. Hwang, J. Y. Kim and H. Y. Woo, Energy Environ. Sci., 2014, 7, 3040–3051 RSC.
- Z. Liu, G. Zhang, Z. Cai, X. Chen, H. Luo, Y. Li, J. Wang and D. Zhang, Adv. Mater., 2014, 26, 6965–6977 CrossRef CAS PubMed.
-
(a) L. Tan, Y. Guo, Y. Yang, G. Zhang, D. Zhang, G. Yu, W. Xu and Y. Liu, Chem. Sci., 2012, 3, 2530–2541 RSC;
(b) X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed;
(c) W. Zhou, Y. Wen, L. Ma, Y. Liu and X. Zhan, Macromolecules, 2012, 45, 4115–4121 CrossRef CAS;
(d) X. Gao, C. Di, Y. Hu, X. Yang, H. Fan, F. Zhang, Y. Liu, H. Li and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3697–3699 CrossRef CAS PubMed;
(e) Y. Li, G. Zhang, G. Yang, Y. Guo, C. Di, X. Chen, Z. Liu, H. Liu, Z. Xu, W. Xu, H. Fu and D. Zhang, J. Org. Chem., 2013, 78, 2926–2934 CrossRef CAS PubMed;
(f) P. Wang, H. Li, C. Gu, H. Dong, Z. Xu and H. Fu, RSC Adv., 2015, 5, 19520–19527 RSC;
(g) Z. Zhao, J. He, J. Wang, W. Chen, N. Wang, Y. Zhang and R. Yang, J. Mater. Chem. C, 2015, 3, 4515–4521 RSC;
(h) Z.-G. Zhang, B. Qi, Z. Jin, D. Chi, Z. Qi, Y. Li and J. Wang, Energy Environ. Sci., 2014, 7, 1966–1973 RSC.
-
(a) X. Zhan, Z. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS PubMed;
(b) P. Cheng, L. Ye, X. Zhao, J. Hou, Y. Li and X. Zhan, Energy Environ. Sci., 2014, 7, 1351–1356 RSC.
-
(a) E. Zhou, J. Cong, K. Hashimoto and K. Tajima, Adv. Mater., 2013, 25, 6991–6996 CrossRef CAS PubMed;
(b) T. Earmme, Y.-J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960–14963 CrossRef CAS PubMed;
(c) Y.-J. Hwang, T. Earmme, S. Subramaniyan and S. A. Jenekhe, Chem. Commun., 2014, 50, 10801–10804 RSC;
(d) H. Huang, N. Zhou, R. P. Ortiz, Z. Chen, S. Loser, S. Zhang, X. Guo, J. Casado, J. T. L. Navarrete, X. Yu, A. Facchetti and T. J. Marks, Adv. Funct. Mater., 2014, 24, 2782–2793 CrossRef CAS;
(e) Y. Jiang, L. Lu, M. Yang, C. Zhan, Z. Xie, F. Verpoortacd and S. Xiao, Polym. Chem., 2013, 4, 5612–5620 RSC;
(f) E. Zhou, J. Cong, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 2799–2803 CrossRef CAS PubMed;
(g) Y. Zhou, T. Kurosawa, W. Ma, Y. Guo, L. Fang, K. Vandewal, Y. Diao, C. Wang, Q. Yan, J. Reinspach, J. Mei, A. L. Appleton, G. I. Koleilat, Y. Gao, S. C. B. Mannsfeld, A. Salleo, H. Ade, D. Zhao and Z. Bao, Adv. Mater., 2014, 26, 3767–3772 CrossRef CAS PubMed;
(h) Y.-J. Hwang, T. Earmme, B. A. E. Courtright, F. N. Eberle and S. A. Jenekhe, J. Am. Chem. Soc., 2015, 137, 4424–4434 CrossRef CAS PubMed;
(i) C. Wu, C. Chue, Y. Xi, H. Zhong, G. Gao, Z. Wang, L. D. Pozzo, T. Wen and A. K.-Y. Jen, Adv. Funct. Mater., 2015, 25, 5326–5332 CrossRef CAS.
-
(a) H. Kang, K.-H. Kim, J. Choi, C. Lee and B. J. Kim, ACS Macro Lett., 2014, 3, 1009–1014 CrossRef CAS;
(b) H. Kang, M. A. Uddin, C. Lee, K.-H. Kim, T. L. Nguyen, W. Lee, Y. Li, C. Wang, H. Y. Woo and B. J. Kim, J. Am. Chem. Soc., 2015, 137, 2359–2365 CrossRef CAS PubMed;
(c) C. Mu, P. Liu, W. Ma, K. Jiang, J. Zhao, K. Zhang, Z. Chen, Z. Wei, Y. Yi, J. Wang, S. Yang, F. Huang, A. Facchetti, H. Ade and H. Yan, Adv. Mater., 2014, 26, 7224–7230 CrossRef CAS PubMed.
- J. W. Jung, J. W. Jo, C.-C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K.-Y. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef CAS PubMed.
-
(a) J. Huang, X. Wang, X. Zhang, Z. Niu, Z. Lu, B. Jiang, Y. Sun, C. Zhan and J. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 3853–3862 CrossRef CAS PubMed;
(b) A. Tang, L. Li, Z. Lu, J. Huang, H. Jia, C. Zhan, Z. Tan, Y. Li and J. Yao, J. Mater. Chem. A, 2013, 1, 5747–5757 RSC;
(c) H. Bürckstümmer, A. Weissenstein, D. Bialas and F. Würthner, J. Org. Chem., 2011, 76, 2426–2432 CrossRef PubMed;
(d) W. Li, W. S. C. Roelofs, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2012, 134, 13787–13795 CrossRef CAS PubMed.
-
(a) X. Xia, T. Lei, J. Pei and C. Liu, Chin. J. Org. Chem., 2014, 34, 1905–1915 CrossRef CAS;
(b) W. Yue, A. Lv, J. Gao, W. Jiang, L. Hao, C. Li, Y. Li, L. E. Polander, S. Barlow, W. Hu, S. D. Motta, F. Negri, S. R. Marder and Z. Wang, J. Am. Chem. Soc., 2012, 134, 5770–5773 CrossRef CAS PubMed.
- The HOMO/LUMO energy levels were calculated by DFT (density functional theory) of DPP, NDI and PDI moieties. Results show that HOMO/LUMO energy levels were −4.98/−2.47 eV, −7.03/−3.41 eV and −6.00/−3.46 eV for DPP, NDI and PDI moieties, respectively (see Fig. S5 in ESI†).
- Y. Li, G. Zhang, Z. Liu, X. Chen, J. Wang, C. Di and D. Zhang, Macromolecules, 2013, 46, 5504–5511 CrossRef CAS.
- J. L. Bredas, R. Silbey, D. S. Boudreaux and R. R. Chance, J. Am. Chem. Soc., 1983, 105, 6555–6559 CrossRef CAS.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS PubMed.
- D. Ouyang, M. Xiao, D. Zhu, W. Zhu, Z. Du, N. Wang, Y. Zhou, X. Bao and R. Yang, Polym. Chem., 2015, 6, 55–63 RSC.
- L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697–9702 CrossRef CAS PubMed.
- As for the characteristics of all-PSCs, it is surprising that the VOC value of the PBDTTT-C-T/P2 device (0.58 V) is lower than that of PBDTTT-C-T/P1 (0.68 V), considering that the LUMO energy level of P2 is a little higher than that of P1. In fact, there are many factors that will influence VOC of polymer solar cells. According to the previous report (Flexible Conjugated Polymer-Based Plastic Solar Cells: From Basics to Applications, Proceedings of the IEEE, 2005, 93, 1429–1439), the maximum value of VOC appears to be limited by the energy difference between the HOMO of the donor and the LUMO of the acceptor. However, apart from this, different donor–acceptor distance, morphologies of active layers, device
structures and many other factors can also affect VOC (Origin of the Open Circuit Voltage of Plastic Solar Cells, Adv. Funct. Mater., 2001, 11, 374–380). As for this work, we think that the unfavorable morphology of the blend film of PBDTTT-C-T:P2 (see Fig. 7) will do harm to the open circuit voltage.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- X. Liu, W. Shen, R. He, Y. Luo and M. Li, J. Phys. Chem. C, 2014, 118, 17266–17278 CAS.
- B. C. Popere, A. M. D. Pell and S. Thayumanavan, Macromolecules, 2011, 44, 4767–4776 CrossRef CAS.
- X. Guo and M. D. Watson, Org. Lett., 2008, 10, 5333–5336 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: TGA and DSC curves, calculated results by quantum-chemical calculations, NMR spectra. See DOI: 10.1039/c5tc02615f |
| ‡ These two authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.