
Red wine polyphenol extract efficiently protects intestinal epithelial cells from inflammation via opposite modulation of JAK/STAT and Nrf2 pathways
Carla
Nunes
a,
Natércia
Teixeira
b,
Diana
Serra
a,
Víctor
Freitas
b,
Leonor
Almeida
a and
João
Laranjinha
*a
aCenter for Neurosciences and Cell Biology and Faculty of Pharmacy, University of Coimbra, Health Sciences Campus, Azinhaga de Santa Comba, 3000-548 Coimbra, Portugal. E-mail: laranjin@ci.uc.pt
bDepartment of Chemistry, Faculty of Sciences, University of Porto, Portugal
First published on 5th October 2015
Abstract
The development of therapeutic approaches combining efficacy and safety represents an important goal in intestinal inflammation research. Recently, evidence has supported dietary polyphenols as useful tools in the treatment and prevention of chronic inflammatory diseases, but the mechanisms of action are still poorly understood. We here reveal molecular mechanisms underlying the anti-inflammatory action of a non-alcoholic polyphenol red wine extract (RWE), operating at complementary levels via the Janus kinase/signal transducer and activator of transcription (JAK/STAT) and Nuclear factor-erythroid 2-related factor-2 (Nrf2) pathways. RWE significantly reduced the nuclear levels of phosphorylated STAT1 and also the cellular levels of phosphorylated JAK1 induced by cytokines, suppressing the JAK/STAT inflammatory signalling cascade. In turn, RWE increased the Nrf2 nuclear level, activating the Nrf2 pathway, leading not only to an up-regulation of the heme oxygenase-1 (HO-1) expression but also to an increase of the glutamate–cysteine ligase subunit catalytic (GCLc) gene expression, enhancing the GSH synthesis, thereby counteracting GSH depletion that occurs under inflammatory conditions. Overall, data indicate that the anti-inflammatory action of RWE is exerted at complementary levels, via suppression of the JAK/STAT inflammatory pathway and positive modulation of the activity of Nrf2. These results point to the potential use of the RWE as an efficient, readily available and inexpensive therapeutic strategy in the context of gastrointestinal inflammation.
1. Introduction
Inflammatory Bowel Diseases (IBD), which include Crohn's disease and ulcerative colitis, are idiopathic chronic inflammatory pathologies of the gastrointestinal tract that affect millions of people worldwide. The etiology of IBD remains unclear but it is thought to involve a combination of environmental, genetic, microbial and immunological factors that leads to a deregulated synthesis and release of a variety of pro-inflammatory mediators, including cytokines, reactive oxygen species (ROS) and nitric oxide (˙NO), resulting in a disruption of the epithelial barrier, excessive tissue injury and a persistent inflammatory state.1–4A specific treatment of IBD is still not available and the most current drugs used in its treatment such as 5-amino salicylic acid (5-ASA), antibiotics, steroids, immunosuppressive agents, have problems related to lower efficacy and serious side effects that limit their use.5–7 Therefore, the development of new therapeutic approaches combining efficacy and safety has emerged as an important goal in intestinal inflammation and IBD research.
Recently, red wine has attracted significant interest because its high polyphenol content is associated with positive health effects, including improvements in cardiovascular and endothelial function and also in the prevention and treatment of inflammatory-mediated diseases by modulating key signaling cascades.8–10 Thus, considering that the gastrointestinal tract is a compartment where the concentration of the dietary polyphenols might achieve its higher concentration in the body and also considering the potential anti-inflammatory properties of polyphenols, we hypothesized that a non-alcoholic red wine extract (RWE) rich in polyphenols can be useful in the prevention and/or treatment of intestinal inflammation, namely of IBD, as an adjunct nutritional therapy. In this regard, it is important to note that polyphenols interact with each other in a way that might interfere with their individual effects (e.g. inhibitory or synergistic effects). Therefore, the use of an extract, conversely to the use of pure compounds, permitted us to study the combined effects of the several polyphenols present in red wine in a manner that can be more readily translated to an in vivo condition. Therefore, this strategy is of relevance if one foresees the use of wine components as therapeutic agents in the context of gastrointestinal inflammation, not only because of the stability of the several compounds in the whole matrix (as compared with isolated compounds), but also because it would be less expensive and readily available in terms of drug development.
In a previous study, using a cellular model of intestinal inflammation, consisting of cytokine-stimulated HT-29 colon epithelial cells, we showed that a non-alcoholic Portuguese RWE had a significant anti-inflammatory effect, protecting the intestinal epithelial cells (IECs) against inflammation via the modulation of cascades orchestrated by NF-κB.11
Inflammatory cascades are highly complex and, in addition to NF-κB activation, the JAK/STAT signalling pathway has been implicated in the pathogenesis of inflammatory diseases, including IBD.12 The binding of several cytokines, including interferons (INFs), to their corresponding transmembrane receptors induces receptor dimerization, triggering an intracellular cascade of events that include autophosphorylation of receptor-associated JAKs that, in turn, phosphorylate specific receptor tyrosine residues, which then serve as docking sites for STATs. Once recruited to the receptor, STATs are phosphorylated on a single tyrosine residue by JAKs. Then, phosphorylated (activated) STATs dissociate from the receptor, dimerize, and translocate into the nucleus to regulate the transcription of several genes that codify pro-inflammatory mediators as well as mediators of cell death.13,14 Therefore, it sounds pertinent that inhibitors of the JAK/STAT inflammatory pathway might be useful in the treatment/prevention of inflammatory diseases such as IBD.
Nrf2 is a redox-sensitive transcription factor that, under the basal resting conditions, is sequestered in the cytoplasm as an inactive complex with its cytosolic repressor Keap1. The oxidation of critical cysteine sulfhydryl groups of Keap1 or the phosphorylation of serine/threonine residues in Nrf2 leads to dissociation of the complex and subsequent nuclear translocation of Nrf2 followed by its binding to ARE sequences located in the promoter region of genes that codify many phase II detoxifying or antioxidant enzymes and related stress-responsive proteins, including heme oxygenase-1 (HO-1) and glutamate–cysteine ligase (GCL), among many others.15–17 So, the Nrf2 pathway plays a key role in the protection of cells against the adverse effects of nitroxidative stress. In addition, the notion that Nrf2 pathway activation might confer protection against inflammation might support the development of therapeutic and preventive strategies for the management of inflammation-associated disorders,18 such as IBD.
In summary, both JAK/STAT and Nrf2 orchestrate major signalling pathways involved in the regulation of cell response to inflammatory stress. Thus, a major goal of the present work was to analyse the potential modulation of both, the JAK/STAT and Nrf2 pathways, by RWE under inflammatory conditions.
2. Materials and methods
2.1 Reagents
Dulbecco's Modified Eagle Medium (DMEM) with Glutamax, foetal bovine serum (FBS) and trypsin were purchased from Gibco-Invitrogen (Grand Island, NY, USA). Primary antibody anti-phospho-JAK1 (Tyr 1022/Tyr 1023), primary antibody anti-phospho-STAT1 (Tyr 701) and primary antibody anti-Nrf2 were purchased from Santa Cruz (Santa Cruz, CA, USA). Primary antibody anti-actin was purchased from Sigma-Aldrich (St Louis, MO, USA). Primary antibody anti-Heme Oxygenase-1 (HO-1), primary antibody anti-Lamin B1 and the alkaline phosphatase-conjugated secondary antibodies (anti-mouse, anti-rabbit, anti-goat) were purchased from Abcam (Cambridge, CB, UK). Polyvinylidene difluoride (PVDF) membranes and Enhanced Chemifluorescence (ECF) substrate were purchased from Amersham/GE Healthcare (Buckinghamshire, BKM, UK). All other chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA) with the highest purity available.2.2. Preparation of red wine extract and determination of polyphenolic content
We used a non-alcoholic Red Wine Extract (RWE) obtained from Portuguese red wine from the Douro Portuguese region. This wine is composed of a blend of “touriga nacional”, “touriga francesa”, “touriga franca” and “tinta roriz”. To obtain a free alcohol Red Wine Extract powder (RWE), the wine was concentrated through a nanofiltration system, which also allows eliminating the ethanol present in the wine. Next, the sample was applied on a silica gel C18-reversed phase in order to remove inorganic salts, sugars and other impurities by elution with water. Further elution with methanol allows recovering polyphenols present initially in the wine. Methanol was evaporated in a rotary evaporator at 38 °C, and the RWE sample was freeze-dried until use.Polyphenols of the RWE were identified and quantified by several methods previously described.19–22 The composition of the RWE is indicated in Table 1. The total phenolic content of the extract was determined according to the Folin-Ciocalteu assay adjusted to a microscale23 and expressed as mg catechin equivalents per gram of RWE (144 mg g−1).
| Chemical class | Compounds | Concentration (mg g−1 of extract) |
|---|---|---|
| a Expressed in equivalents of gallic acid. | ||
| Catechins and oligomeric procyanidins | B1 | 6.50 |
| B3 | 0.72 | |
| B4 | 1.08 | |
| B2 | 1.94 | |
| C1 | 0.45 | |
| B2-Gallate | 0.36 | |
| (+)-Catechin | 0.07 | |
| (−)-Epicatechin | 0.11 | |
| Flavonolsa | Myricetin glucoside | 3.27 |
| Myricetin arabinoside | 8.80 | |
| Myricetin rhamnoside | 3.06 | |
| Anthocyanin-3-monoglucosides | Delphinidin-3-glucoside | 2.33 |
| Petunidin-3-glucoside | 2.94 | |
| Peonidin-3-glucoside | 0.62 | |
| Malvidin-3-glucoside | 11.67 | |
| Delphinidin-3-acetylglucoside | 0.38 | |
| Cyanidin-3-acetylglucoside | 0.19 | |
| Petunidin-3-acetylglucoside | 0.47 | |
| Peonidin-3-acetylglucoside | 0.49 | |
| Delfinidin-3-coumaroylglucoside | 3.60 | |
| Malvidin-3-acetylglucoside | 0.45 | |
| Malvidin-3-caffeoylglucoside | 0.46 | |
| Peonidin-3-coumaroylglucoside | 0.23 | |
| Malvidin-3-coumaroylglucoside | 2.08 | |
| Phenolic acidsa | Gallic acid | 6.52 |
| Syringic acid | 7.93 | |
| Diethyl fertaric acid | 6.40 | |
| Ethyl cinnamate | 9.37 | |
| Condensed tannins | 230 | |
2.3. Cell culture
HT-29 cell line was purchased from Sigma-Aldrich (St Louis, MO, USA). The HT-29 cells are a well characterised epithelial cell line derived from a primary colon tumour, which exhibit characteristics of normal intestinal epithelium.24 Cell cultures were routinely grown in DMEM medium with FBS (10% vol/vol) and no antibiotic supplements in 75 cm2 flasks and maintained at 37 °C, under a humidified atmosphere of 5% CO2. For the experiments, twenty-four hours before stimulation, cells were washed and cultured in fresh medium without FBS.HT-29 cells were stimulated with a cocktail of cytokines relevant in the context of the intestinal inflammation consisting of 20 ng ml−1 TNF-α, 10 ng ml−1 IL-1 and 50 ng ml−1 INF-γ.
Cells were pre-treated with different concentrations of the RWE (200, 400 and 600 μg ml−1) for 30 minutes before exposure to the cytokines for different time intervals, depending on the assay.
2.4. Analysis of cell viability
The cell viability was assessed by using the MTT assay, which is based on the reduction of the dye MTT to formazan, an insoluble intracellular blue product, by cellular dehydrogenases.25 Briefly, after incubation of the cells for 24 hours with several concentrations of the RWE (200, 400 and 600 μg ml−1), the culture medium was removed, and the cells were washed twice with PBS. Then MTT was added to each well at a final concentration of 0.5 mg ml−1. Following incubation for 1 h at 37 °C, MTT was removed and 1 ml of DMSO was added and mixed thoroughly until formazan crystals were dissolved. The mixture was then collected from each well and the extent of MTT reduction was measured spectrophotometrically at 540 nm using a Synergy HT plate reader. Cell viability was expressed as a percentage of the control cultures.2.5. Measurement of the intracellular content of phospho-JAK1, phospho-STAT1, Nrf2 and HO-1
The intracellular contents of phospho-JAK1 and HO-1 were assessed by western blot in whole cell lysates. Briefly, to prepare whole cell lysates, following incubation under the specified conditions, the cells were washed twice with PBS and resuspended in an ice-cold lysis buffer [50 mM Hepes pH 7.4, 150 mM NaCl, 2 mM EDTA, 10% (w/v) glycerol, 0.5% (w/v) sodium deoxycholate, 1% (v/v) Triton X-100] supplemented with 1 mM NaVO4, 5 mM NaF, 1 mM PMSF, and 1/100 (v/v) protease cocktail inhibitor and left in ice for 20 min. Lysates were subsequently centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 4 °C and supernatants (whole cell lysates) were then collected and stored at −80 °C. The phospho-STAT1 and Nrf2 nuclear contents were analysed by western blot in nuclear fractions. First it was necessary to prepare cytoplasmic lysates. For this propose, cells were washed twice with PBS, resuspended in ice cold buffer [10 mM Tris-HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.5%(v/v) Igepal] supplemented with 1 mM NaVO4, 5 mM NaF, 1 mM PMSF, and 1/100 (v/v) protease cocktail inhibitor, maintained in ice for 5 minutes and centrifuged at 5000 rpm for 5 minutes at 4 °C. The supernatants were cytoplasmic lysates. For nuclear cellular protein extracts, the pellets were resuspended in an ice cold buffer [20 mM Hepes pH 7.4, 5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 300 mM NaCl, 20% (w/v) glycerol] supplemented with 1 mM NaVO4, 5 mM NaF, 1 mM PMSF, and 1/100 (v/v) protease cocktail inhibitor, left on ice for 30 min and then subjected to three rapid cycles of freezing and thawing (N2 liquid/37 °C). After that, the lysates were centrifuged at 14000 rpm for 20 minutes at 4 °C. The supernatants (nuclear extracts) were stored at −80 °C until used.
000 rpm for 10 min at 4 °C and supernatants (whole cell lysates) were then collected and stored at −80 °C. The phospho-STAT1 and Nrf2 nuclear contents were analysed by western blot in nuclear fractions. First it was necessary to prepare cytoplasmic lysates. For this propose, cells were washed twice with PBS, resuspended in ice cold buffer [10 mM Tris-HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.5%(v/v) Igepal] supplemented with 1 mM NaVO4, 5 mM NaF, 1 mM PMSF, and 1/100 (v/v) protease cocktail inhibitor, maintained in ice for 5 minutes and centrifuged at 5000 rpm for 5 minutes at 4 °C. The supernatants were cytoplasmic lysates. For nuclear cellular protein extracts, the pellets were resuspended in an ice cold buffer [20 mM Hepes pH 7.4, 5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 300 mM NaCl, 20% (w/v) glycerol] supplemented with 1 mM NaVO4, 5 mM NaF, 1 mM PMSF, and 1/100 (v/v) protease cocktail inhibitor, left on ice for 30 min and then subjected to three rapid cycles of freezing and thawing (N2 liquid/37 °C). After that, the lysates were centrifuged at 14000 rpm for 20 minutes at 4 °C. The supernatants (nuclear extracts) were stored at −80 °C until used.
The cellular protein content was quantified by the Bio-Rad protein assay dye (Bio-Rad, USA), using bovine serum albumin as the standard.
Equal amounts of reduced and denatured proteins were separated by electrophoresis on a 7.5%–12% SDS-polyacrylamide gel and transferred onto PVDF membranes using the Trans-Blot Turbo Transfer System of Bio-Rad (Bio-Rad, USA). To avoid non-specific binding, membranes were blocked for 1 h at room temperature with 5% (w/v) non-fat dried milk in TBS-T buffer [25 mM Tris-HCl pH 7.6, 150 mM NaCl, 0.1% (v/v) Tween 20]. The membranes were then incubated overnight at 4 °C with a primary antibody (anti-phospho-JAK1, anti-phospho-STAT1, anti-Nrf2 and anti-HO-1). After 3 washes of 10 minutes with TBS-T, the membranes were incubated with a phosphatase alkaline-labelled secondary antibody for 2 h at room temperature. Then, the membranes were washed again 3 times with TBS-T. Immunoreactive complexes were detected by fluorescence after exposition of blots to ECF substrate using a Typhoon 9000 scanner (Amersham Biosciences). Bands were analyzed using ImageQuant™ software from Amersham Biosciences. Actin or Lamin B1 was used as the internal standard to monitor protein loading per lane.
2.6. RNA extraction and quantitative real-time PCR analysis
Following incubation under the specified conditions, total RNA was extracted from HT-29 cells using the RNA extraction kit Aurum Total RNA Mini (Bio-Rad, Hercules, CA, USA), according to the procedure described by the manufacturer. Extracted RNA was quantified using a NanoDrop ND-1000 spectrophotometer. RNA (1 μg per sample) was transcribed to first-strand cDNA using the NZY First-Stand cDNA Synthesis Kit (NZYtech, Portugal), according to the manufacturer's protocol.RT-PCR was performed using a CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Briefly, first strand cDNA (2 μL) was amplified in 20 μL PCR reaction volume containing, 2 μl of each primer (250 nM), 10 μl of the IQ SYBR Green Supermix (Bio-Rad) and 4 μl RNase/DNase-free distilled water. The primers for HO-1, the Glutamate Cysteine Ligase catalytic subunit (GCLc) and hypoxanthine phosphoribosyltransferase-1 (HPRT-1) were designed using Beacon Designer software (PREMIER Biosoft International, Palo Alto, CA). The sequences of the primers used were as follows: HO-1 (forward) 5′-TCACTGTGTCCCTCTCTC-3′ and (reverse) 5′-ATTGCCTGGATGTGCTTT-3′; GCLc (forward) 5′-ATTCTGAACTCTTACCTTGA-3′ and (reverse) 5′-ATCTGGCAACTGTCATTA-3′; HPRT-1 (forward) 5′-TGACACTGGCAAAACAATG-3′ and (reverse) 5′-GGCTTATATCCAACACTT-3′.
PCR conditions were as follows: 1 cycle at 95 °C for 3 min followed by 45 cycles, each consisting of a denaturation step (95 °C, 10 seconds), an annealing step (55 °C, 30 seconds) and an elongation step (72 °C, 30 seconds). The specificity of the amplification products was verified through the analysis of the melting curve. The efficiency of the amplification reaction for each gene was calculated by running a standard curve of serially diluted cDNA sample. Gene expression was analyzed using Bio-Rad CFX Manager 3.0 software (Bio-Rad, Hercules, CA, USA). The results for each gene of interest were normalized against the housekeeping gene HPRT-1 and expressed as fold increase above control cells (cells without any treatment).
2.7. Quantification of intracellular levels of reduced glutathione (GSH)
Intracellular levels of reduced glutathione (GSH) were determined using a fluorimetric assay as described previously.26 After the period of incubation, the medium was removed and cells were scrapped with 500 μl of 100 mM phosphate buffer containing 5 mM EDTA, pH 8.0 at 4 °C. Then 500 μl of 0.6 M perchloric acid was added. After a vigorous vortex, the suspension was centrifuged at 14000 rpm for 5 min at 4 °C. For the GSH determination, a volume of 100 μl of the supernatant was incubated with 100 μl of ortho-phtaldehyde (0.1% w/v in methanol) and 1.8 ml of 100 mM Na2HPO4, pH 8.0 for 15 min at room temperature. After that, fluorescence intensity was read using a Synergy HT plate reader (Bio-Tek Instruments) using an excitation wavelength of 350 nm and an emission wavelength of 420 nm. The intracellular GSH content was calculated using the respective standard curves, containing known concentrations of GSH (0–2 μg) and expressed as a percentage of control cells. The cellular protein content was quantified by the Bio-Rad protein assay dye, using bovine serum albumin as the standard.
2.8. Statistical analysis
Statistical analyses were performed using GraphPad Prism software. All data were expressed as mean ± SEM of at least three independent assays, each one in duplicate or triplicate. Differences between groups were assessed by one-way analysis of variance (ANOVA). Student's t-test was used to determine differences between two groups. A value of p < 0.05 was considered as statistically significant.3. Results
3.1. RWE did not affect the viability of the HT-29 cells
In order to assess the cytotoxic profile of the RWE, the viability of HT-29 cells incubated with several concentrations of the RWE (200, 400 and 600 μg ml−1) for twenty-four hours was evaluated by the MTT assay as described in the Materials and methods section. As shown in Fig. 1, RWE did not affect the viability of HT-29 cells at the concentrations tested.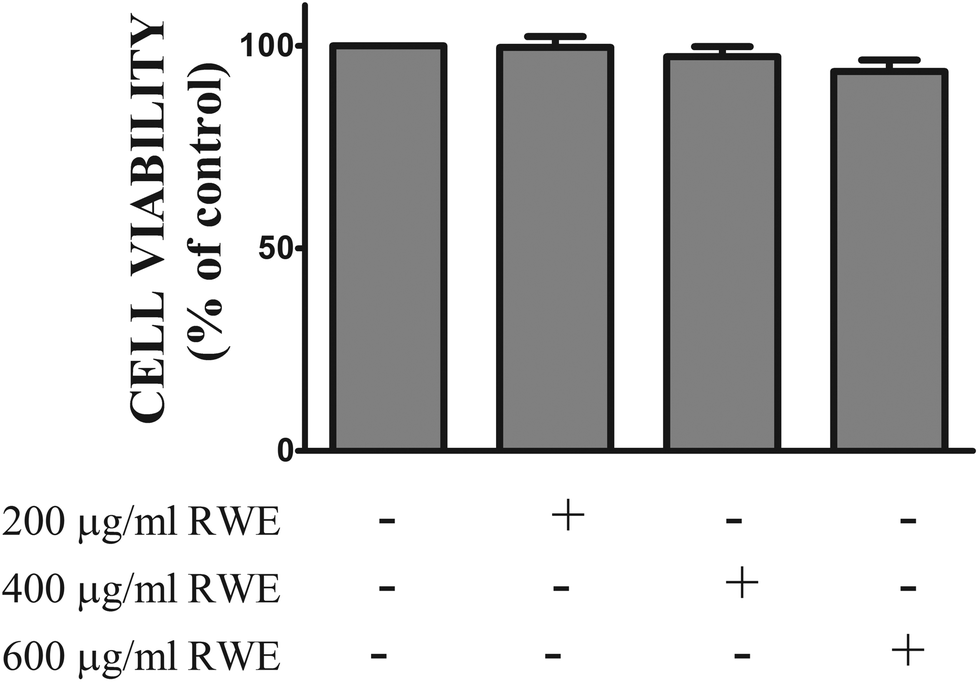 | ||
| Fig. 1 Effect of Red Wine Extract (RWE) on cell viability. Cells were pre-treated with three different concentrations of RWE (200, 400 and 600 μg ml−1). After 24 hours, cell viability was measured by following the extent of MTT reduction. Data represent the mean ± SEM of at least four independent experiments run in duplicate and are expressed as a percentage of control cells (100%). | ||
3.2. RWE decreases the levels of cytokine-induced phosphorylated STAT1 in the nucleus of HT-29 cells
INF-γ, one of the cytokines used in this study as an inflammatory stimulus, is a known activator of the JAK/STAT pathway.27 Following INF-γ stimulation, STAT1 is phosphorylated (Tyr701) by tyrosine kinases of the JAK family, namely by JAK1. This phosphorylation induces STAT1 dimerization and subsequent nuclear translocation, regulating the transcription of genes that codify pro-inflammatory mediators, including iNOS among others.Therefore, the effect of RWE on the nuclear levels of phosphorylated (activated) STAT1 was examined by western blot using a phosphotyrosine STAT1 (Tyr701) antibody. For this purpose, the activation time course of STAT1 induced by cytokines in HT-29 cells was firstly evaluated. Phospho-STAT1 was not detectable in the nucleus of unstimulated cells (Fig. 2) but the phosphorylation and nuclear translocation of STAT1 induced by cytokines was visible after 15 minutes of incubation and became stronger after 30 minutes (data not shown). Pre-treatment with RWE showed a significant concentration-dependent decrease of the nuclear content of this activated transcription factor in the cells stimulated with cytokines for 30 minutes (200 μg RWE per ml – 55.3 ± 5.6%; 400 μg RWE per ml – 33.1 ± 6.9%; 600 μg RWE per ml – 19.9 ± 5.3% of cells incubated with cytokines) as shown in Fig. 1.
 | ||
| Fig. 2 Effect of Red Wine Extract (RWE) on the nuclear levels of phosphorylated-STAT-1. Cells were pre-treated with three different concentrations of RWE (200, 400 and 600 μg ml−1) for 30 min and then stimulated with a mix of cytokines (20 ng ml−1 TNF-α; 10 ng ml−1 IL-1; 50 ng ml−1 INF-γ). After 30 min of incubation, nuclear protein extracts were obtained and analysed by western blot using an anti-phospho-STAT-1 antibody. In the bar graph, the relative expression of phospho-STAT-1 normalized to the lamin B1 level represents the mean ± SEM from at least three independent experiments and is expressed as a percentage of cells incubated with cytokines (100%). Statistical significance: ***p < 0.001 and *p < 0.05 as compared to control cells; &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
3.3. RWE decreases the levels of cytokine-induced phosphorylated JAK1 in the HT-29 cells
As phosphorylation of STATs is dependent of the JAK activation, we have examined whether the inhibitory effect of the RWE on the STAT1 activation is due to the suppression of JAK1 activity. For this propose, cells were stimulated with cytokines in the presence and absence of RWE and the levels of phospho-JAK1 were evaluated by western blot. As shown in Fig. 3 phospho-JAK1 was not detectable in unstimulated cells. However, cytokines induced JAK1 phosphorylation after 15 minutes of incubation, this effect being stronger after 30 minutes (data not shown). Considering the cells treated only with cytokines for 30 minutes as 100%, we can observe that the pre-treatment with 200, 400 and 600 μg RWE per ml significantly decreased the phospho-JAK1 cellular content to 83.3, 32 and 8.7% respectively (Fig. 3).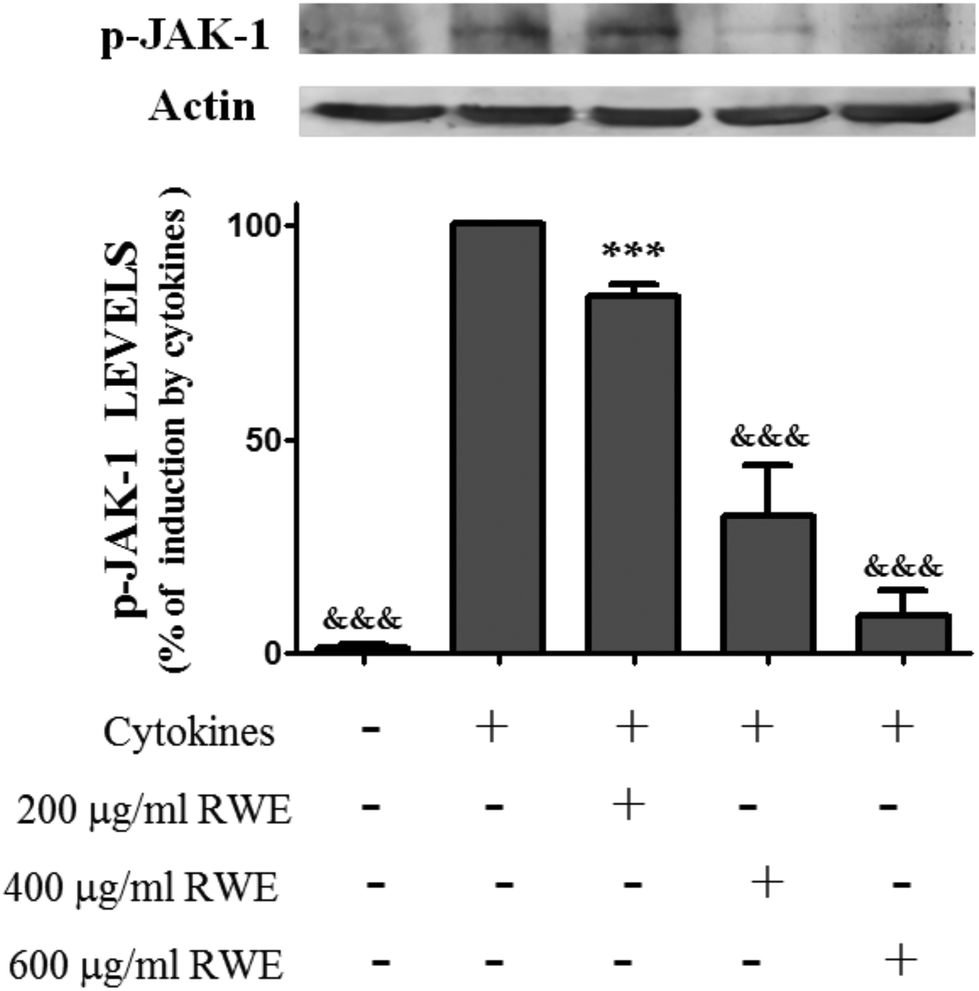 | ||
| Fig. 3 Effect of Red Wine Extract (RWE) on the total levels of phosphorylated-JAK-1. Cells were incubated as described in Fig. 1. After 30 min of incubation, total protein extracts were obtained and analysed by western blot using an anti-phospho-JAK-1 antibody. In the bar graph, the relative expression of phospho-JAK-1 normalized to the actin level represents the mean ± SEM from at least three independent experiments and is expressed as a percentage of cells incubated with cytokines (100%). Statistical significance: ***p < 0.001 as compared to control cells; &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
3.4. RWE induces nuclear Nrf2 translocation
Recent studies have demonstrated that Nrf2 pathway activation confers protection against inflammation-associated pathogenesis in several diseases, including IBD.18Thus, compounds capable of activating this signaling pathway can be used as therapeutic and preventive agents for the management of inflammatory diseases,18 such as IBD. Therefore, the effect of RWE on the Nrf2 pathway activation was analyzed. For this propose, the nuclear level of Nrf2, indicative of Nrf2 activation, was evaluated by western blot in control cells, cells incubated with RWE, cells incubated with cytokines and cells pre-treated with RWE and then challenged with cytokines. At 8 h, the nuclear Nrf2 level was slightly lower in cells challenged with cytokines than that of the control cells (66 ± 12% of control levels) (Fig. 4). However, the pre-treatment with RWE, in a concentration-dependent manner, significantly increased the Nrf2 nuclear level in cytokine-treated cells (200 μg RWE per ml – 188.4 ± 38%; 400 μg RWE per ml – 225.6 ± 21.3%; 600 μg RWE per ml – 295.3 ± 49% of control levels) (Fig. 4), suggesting that RWE could trigger a cellular defence mechanism under inflammatory conditions via Nrf2. RWE alone also induced a slight increase of the nuclear Nrf2 levels compared to control cells (Fig. 4).
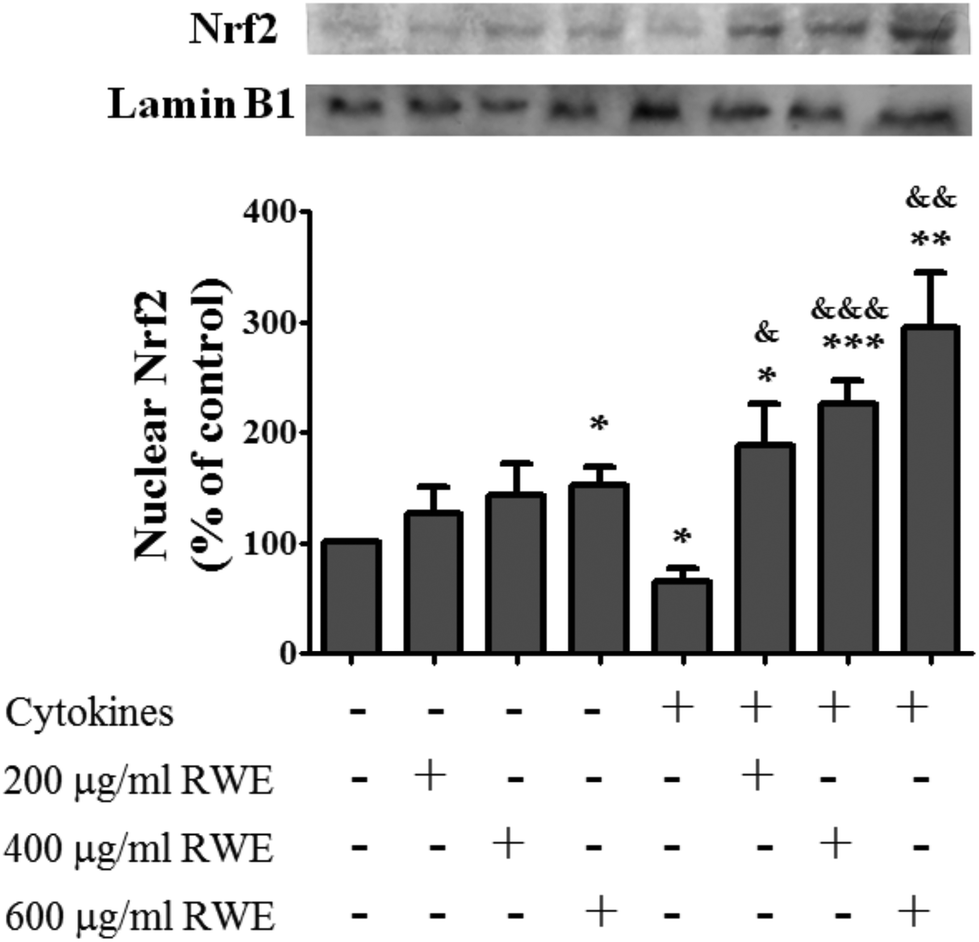 | ||
| Fig. 4 Effect of Red Wine Extract (RWE) on the Nrf2 nuclear translocation. Cells were incubated as described in Fig. 1. After 8 hours of incubation, nuclear protein extracts were obtained and then analysed by western blot using an anti-human Nrf2 antibody. In the bar graph, the Nrf2 content normalised to the lamin B1 level represents the mean ± SEM from at least six independent experiments and is expressed as a percentage of control cells (100%). Statistical significance: *p < 0.05, **p < 0.005 and ***p < 0.001 as compared to control cells; &p < 0.05, &&p < 0.005 and &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
3.5. RWE up-regulates the HO-1 expression
Among the enzymes up-regulated by Nrf2, HO-1 has pronounced anti-inflammatory as well as anti-oxidative properties, modulating the innate immunity and inflammation28 Moreover, up-regulation of HO-1 seems to prevent an inflammatory response in the colon.29,30 In this context, and because RWE activated the Nrf2 pathway, next we examined the effect of the RWE on the cellular HO-1 content by western blot. As shown in Fig. 5, the pre-treatment with RWE significantly enhanced the protein levels of HO-1 in cytokine-incubated cells as a function of concentration (200 μg RWE per ml – 145.3 ± 1.9%; 400 μg RWE per ml – 169.9 ± 18%; 600 μg RWE per ml – 186.5 ± 23.8% of control levels). Interestingly, the highest concentration of RWE (600 μg ml−1) used in this study also significantly increased the HO-1 intracellular levels from a background in control cells (181.5 ± 16.8% of control levels) (Fig. 5A). | ||
| Fig. 5 Effect of Red Wine Extract (RWE) on the HO-1 protein (A) and gene expression (B). (A) Cells were pre-treated with several concentrations of RWE (200, 400 and 600 μg ml−1) for 30 min and then stimulated with cytokines (20 ng ml−1 TNF-α; 10 ng ml−1 IL-1; 50 ng ml−1 INF-γ). After 24 hours of incubation, total protein extracts were obtained and analysed by western blot using an anti-human HO-1 antibody. In the bar graph, the HO-1 content normalised to the actin level represents the mean ± SEM from at least three independent experiments and is expressed as a percentage of control cells (100%). (B) Cells were pre-treated with RWE (600 μg ml−1) for 30 min and then stimulated with the mix of cytokines. After 18 hours of incubation, mRNA production was evaluated by qRT-PCR, as described in the “Materials and methods” section, and expressed as fold increase above control cells. Statistical significance: *p < 0.05, **p < 0.005 as compared to control cells; &p < 0.05, &&p < 0.005 and &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
Moreover, HO-1 mRNA concentrations were determined by real-time quantitative PCR after 18 h of incubation. In this assay, we only tested the highest concentration of RWE (600 μg ml−1). As expected, pre-treatment with RWE significantly increased the HO-1 mRNA levels in both control cells (10 ± 0.2 fold increase above control) and cytokine-incubated cells (a 7.6 ± 0.6 fold increase above the control) (Fig. 5B).
These results are in agreement with the results concerning to the activation of Nrf2 presented above and show that the Nrf2 pathway activation by RWE can effectively induce HO-1 gene expression and consequently increase the protein levels of HO-1.
3.6. RWE increases the intracellular GSH and the GCLc gene expression
Besides the HO-1 gene, Nrf2 also regulates genes that codify glutathione related enzymes which confers on Nrf2 an important role in the regulation of cellular GSH levels.31–35 Several studies showed that GSH, a key antioxidant and also an important modulator of inflammation and cell death, decreases in the course of inflammatory disorders,36–40 namely in IBD.Firstly we evaluated the effect of the RWE on the GSH intracellular content. At 24 h of incubation, cytokines induced a significant depletion of intracellular GSH (40.1 ± 7.3% of control levels) but, as shown in Fig. 6, RWE significantly prevented this depletion in a dose dependent manner (200 μg RWE per ml – 107.4 ± 7.9%; 400 μg RWE per ml – 110.5 ± 6.4%; 600 μg RWE per ml – 123.3 ± 8.8% of control levels). Also, the highest concentration of RWE (600 μg ml−1) used in this study significantly increased the intracellular GSH levels from a background in control cells (139.8 ± 18% of control levels) (Fig. 6).
 | ||
| Fig. 6 Effect of Red Wine Extract (RWE) on the GSH intracellular content. Cells were treated as described in Fig. 1. After 24 hours of incubation, GSH was quantified using a fluorimetric assay as described in the “Materials and methods” section. The results represent the mean ± SEM of at least 7 independent experiments, run in duplicate, and are expressed as a percentage of control cells (100%). Statistical significance: *p < 0.05, **p < 0.005 and ***p < 0.001, as compared to control cells; &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
The enzyme glutamate–cysteine ligase (GCL), composed by a catalytic (GCLc) and a modifier (GCLm) subunit, is the rate-limiting enzyme in the GSH synthesis. So, in this context and considering that the Nrf2 pathway activation leads to the transcription of the genes that codify the subunits of the GCL, next we examined the effect of the highest RWE used (600 μg ml−1) in the mRNA levels of the GCLc by real-time quantitative PCR. RWE significantly increased the GCLc mRNA levels in both control cells (a 8.6 ± 1.3 fold increase above the control) and cytokine-incubated cells (a 9.8 ± 1.2 fold increase above the control) (Fig. 7). Cytokines had no effect on the GCLc mRNA levels (Fig. 7).
 | ||
| Fig. 7 Effect of Red Wine Extract (RWE) on the GCLc gene expression. Cells were pre-treated with RWE (600 μg ml−1) for 30 min and then stimulated with the mix of cytokines. After 18 hours of incubation, GCLc mRNA production was evaluated by qRT-PCR, as described in the “Materials and methods” section, and expressed as fold increase above control cells. Statistical significance: ***p < 0.001, as compared to control cells; &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
These results show that the Nrf2 pathway activation by RWE induces GCLc gene expression, leading to the GSH synthesis and consequently counteracts the GSH depletion observed in cells incubated with cytokines.
4. Discussion
Despite intensive research, a specific treatment of IBD is still not available and the most current drugs used in its treatment have problems of low efficacy and serious side effects that limit their use.5–7 Consequently, a widespread increase in the use of complementary alternative therapeutic strategies, including natural products, has been recognized.41,42 However, the less clear effects of many of the compounds used highlight the need for a study of their inherent molecular mechanisms.This is the case with dietary polyphenols (or dietary products containing polyphenols) which are natural compounds with recognized health benefits and a growing body of evidence suggests their beneficial role for use in IBD.43–45 The main problem is that the molecular mechanisms underlying the anti-inflammatory effect of these compounds are not completely understood. Initially, the health benefits of polyphenols were exclusively assigned to their well-known in vitro antioxidant properties but now it is becoming evident that many of the effects of polyphenols rely on the modulation of key signalling cascades in connection with physiological and pathophysiological conditions such as inflammatory processes.46–48
Although, most of the research has been focused on the antioxidant and anti-inflammatory activity of pure polyphenols, several studies in the literature also reported the anti-inflammatory effect of plant polyphenol extracts in several cellular models, including models of intestinal inflammation, with the final concentrations of polyphenolic extracts used being widely variable (2–2000 μg ml−1).49–55 Furthermore, it is noteworthy that strong evidence indicates that the protective effect of the polyphenolic extracts may not be due to any one component, but rather due to a synergic action of various phenolic compounds including catechin, phenolic acids, anthocyanins, etc.,54–56 suggesting that the use of combinations of several polyphenols, instead pure polyphenols, might be an interesting therapeutic approach. Rossetto and colleagues also reported, in a model of lipid peroxidation, a synergistic antioxidant effect of catechin and malvidin-3-glucoside by a mechanism that involves malvidin-3-glucoside recycling by catechin.57
Previously, in a cellular model of intestinal inflammation using cytokine-stimulated HT-29 colon epithelial cells, we presented strong evidence that the non-alcoholic RWE rich in several different polyphenols (Table 1) (100–600 μg ml−1) used in the present study protects against intestinal inflammation by attenuating cytokine-dependent expression of interleukin-8 (IL-8), COX-2 and iNOS, in addition to reducing protein nitration and inhibiting NF-κB pathway activation,11 the anti-inflammatory effect of the RWE being likely due to a combined activity of these polyphenols. Therefore, and given the complexity of cellular inflammatory pathways, in the present work, we further explore the molecular mechanisms underlying the anti-inflammatory action of the RWE with special attention to the JAK/STAT pathway and also to the Nrf2 pathway.
The JAK/STAT pathway plays a crucial role in the pathogenesis of several inflammatory diseases, including IBD.12 In fact, an inhibitor of JAKs, tofacitinib, has been already approved for the treatment of rheumatoid arthritis58 and has also been shown to be effective in the treatment of ulcerative colitis,59,60 reinforcing the importance of this cellular pathway in the molecular mechanisms underlying IBD. In this context and also considering that INF-γ, one of the cytokines used as an inflammatory stimulus in this study, is a well known activator of the JAK/STAT pathway,27 we explored the potential effects of the RWE on this cellular pathway. The binding of INF-γ to its receptor induces receptor dimerization and the consequent activation of associated JAKs (JAK1 and JAK2) by mutual phosphorylation. JAKs, in turn, phosphorylate specific tyrosine residues in the cytoplasmic domain of the receptor, providing STAT1 as a docking site.61,62 Once recruited to the receptor, STAT1 is phosphorylated on conserved tyrosine 701 by JAKs, and it homo-dimerizes and translocates into the nucleus, where it regulates the transcription of several genes that encode pro-inflammatory cytokines, chemokines and enzymes, such as cyclooxygenase-2 (COX-2) and iNOS as well as mediators of cell death.13,14,63
Therefore, we first determined the effect of RWE on the nuclear levels of phosphorylated (activated) STAT1. The observed RWE-dependent reduction in the nuclear levels of this activated transcription factor (Fig. 2) may contribute to the anti-inflammatory activity of the RWE against intestinal inflammation that we observed previously.11 In this context, Tedeschi and colleagues also showed that both a green tea polyphenol extract51 and an extract from Hypericum perforatum64 efficiently inhibited STAT1 phosphorylation in colon DLD-1 cells. A cocoa extract enriched in polyphenols showed an anti-inflammatory effect against ulcerative colitis in mice by the inhibition of STAT1 and STAT3 phosphorylation.65
In addition to the important role of STAT1 in inflammation, this transcription factor is also central in promoting various forms of cell death (apoptosis, necrosis and autophagic cell death).14,66–69 Moreover, the activation of STAT1 induced by INF-γ seems to render cells more susceptible to death induced by other stimuli, such as TNF-α.66,70 The possibility that a synergism between INF-γ and TNF-α in promoting cell death via activation (phosphorylation) of STAT1 could occur turns the STAT1 inhibitory effect of RWE more relevant.
With the aim of knowing how RWE reduced the nuclear levels of phospho-STAT1 in cells stimulated with cytokines, we further investigated the effect of the RWE upstream of STAT1 phosphorylation on the signaling pathway, more precisely on the cellular levels of phosphorylated JAK1. The observation that phosphorylation of JAK1 was significantly suppressed by RWE in cytokine-challenged HT-29 cells (Fig. 3) suggests that the RWE effect on the inactivation of STAT1 could, at least partially, probably be via inhibition of JAK1 phosphorylation.
It was previously reported that certain polyphenols suppressed the JAK/STAT pathway via activation of protein tyrosine phosphatases (PTPs).71,72 Thus, the activation of PTPs by RWE could be one possible explanation for the decrease of JAK1 phosphorylated levels. The activation of these phosphatases involves their phosphorylation and consequent attachment to residues of phosphotyrosine in the JAKs, leading to their dephosphorylation. PTPs also dephosphorylate activated receptors and STATs.73–75 Moreover, we cannot exclude a potential effect of the RWE on the Suppressor of Cytokine Signaling (SOCS) proteins or on the protein inhibitors of activated STAT (PIAS) proteins, other key proteins that also negatively regulate the JAK/STAT pathway.73,74
Although many targets might be found in future, it is clear that RWE exerts an inhibitory effect on the JAK/SAT signaling pathway in cells under inflammatory stimuli, operating at early steps of the pathway, notably by preventing JAK phosphorylation. It is also of note that such inhibition is a function of RWE concentration.
In IBD, as well as in the case of other chronic inflammatory diseases, occurs an intense immune response that consisted of nitroxidative stress as well as an overproduction of a myriad of pro-inflammatory mediators, such as cytokines, chemokines, cell adhesion molecules and inflammatory enzymes, notably iNOS and COX-2. Therefore, nitroxidative stress and inflammation are closely related processes that play a key role in the pathogenesis of inflammatory diseases and IBD.1,76–79
Nrf2 is a transcription factor responsible for the expression of many phase II detoxifying or antioxidant enzymes and related stress-responsive proteins32,80,81 relieving nitroxidative stress. Also, Nrf2 protects against inflammation by reducing the production of several pro-inflammatory mediators and by enhancing the expression of genes that codify several cytoprotective agents that are able to fight inflammation.18 These notions suggest that activators of the Nrf2 pathway may trigger the development of new therapeutic and preventive strategies for the management of inflammatory diseases such as IBD.
Therefore, considering that RWE was able to inhibit the JAK/STAT pro-inflammatory pathway it would be of relevance to examine whether the anti-inflammatory action of RWE includes the activation of a detoxifying (Nrf2) pathway. Moreover, studies in the literature show that some plant extracts are inducers of the Nrf2 pathway.81–83
In accordance with several studies in the literature, the pro-inflammatory cytokines suppressed the Nrf2 activation in colonic epithelial cells by decreasing the translocation of Nrf2 into the nucleus (Fig. 4). In fact, although consistent data about the Nrf2 expression in the mucosa of IBD patients are not available, studies using animal models of IBD show that the Nrf2 pathway activation is impaired in this disease,84,85 which can justify a decreased expression of antioxidant proteins in the inflamed areas of IBD patients.86 Actually, the Nrf2 pathway is also impaired in other inflammatory disorders.87–89
The cells pre-treated with RWE rich in polyphenols increased the Nrf2 nuclear level in the presence and absence of cytokines (Fig. 4), activating the Nrf2 pathway. However, this effect was stronger in the cells stimulated with cytokines (Fig. 4), suggesting that the pre-treatment with RWE is able to put the cells in a state of alert, preparing cells to efficiently fight a pro-inflammatory stimulus. This result is in accordance with the concept of phytohormesis, in which polyphenols may be beneficial for human health because they can act as “low-dose stressors”, increasing the expression of cellular defenses via the Nrf2 pathway and rendering cells tolerant or adaptive to a subsequent cytotoxic stimulus.90,91
In this context, a study performed by Erlank and colleagues suggests that polyphenols such as curcumin, resveratrol and tert-butylhydroquinone activate Nrf2 signaling through the generation of H2O2 and polyphenol-oxidized species, inducing cell adaptation to oxidative stress.92 Moreover, Lee-Hilz and colleagues also showed that the pro-oxidant chemistry of flavonoids plays a key role in the Nrf2 pathway activation,93 probably via ROS and/or flavonoid quinone production. In fact, ROS can both oxidize critical cysteine residues present in Keap1 and activate some specific protein kinases that phosphorylate Nrf2, mechanisms that lead to the dissociation of the Keap1–Nrf2 complex and subsequent nuclear translocation of Nrf2 followed by its binding to ARE sequences.15,16 Flavonoid quinones have electrophilic properties and therefore rapidly react with cysteine residues of Keap1 to form adducts, lowering Keap1 affinity for Nrf2 and consequently leading to Nrf2 release and nuclear translocation. The direct alkylation of Keap1 by polyphenols with electrophilic properties such as polyphenols with an α,β-unsaturated carbonyl moiety also activates the Nrf2 pathway.94,95 Consequently, considering that RWE is rich in polyphenols with a high structural diversity, it is likely that a combination of the pro-oxidant and electrophilic properties of these polyphenols is responsible for the RWE-induced Nrf2 activation.
It is well known that the Nrf2 pathway activation is a major trigger for the up-regulation of HO-1 expression, an inducible enzyme with recognized anti-inflammatory, anti-oxidative and anti-apoptotic properties. Several studies showed that the up-regulation of HO-1 expression inhibits the production of pro-inflammatory cytokines and chemokines and also inhibits the induction of pro-inflammatory enzymes such as COX-2 and iNOS.96–98 For instance, in the colon, the up-regulation of HO-1 seems to prevent an inflammatory response.29,30
HO-1 catalyzes the rate-limiting step of the conversion of intracellular heme into biliverdin, carbon monoxide and free iron.99 HO-1 products act as antioxidant, anti-inflammatory and cytoprotective molecules,100–102 mainly in the gastrointestinal tract, having a key role in the protection that HO-1 offers against nitroxidative stress and inflammation.
Therefore, as expected from Nrf2 activation, RWE increased the HO-1 mRNA levels and HO-1 protein expression, either in the presence or absence of cytokines, indicating that the Nrf2 pathway activation induced by RWE resulted in HO-1 up-regulation.
Besides the HO-1 gene, Nrf2 also regulates genes that codify glutathione related enzymes, including GCL, which confers on Nrf2 an important role in maintaining cellular GSH homeostasis.31–35 GSH is not only a vital protective antioxidant that plays a key role against nitroxidative stress103 but is also an important modulator of inflammation104,105 and cell death.106–108 In fact, several studies show that GSH decreases in the course of inflammatory disorders and that GSH supplementation results in beneficial effects on the treatment of these diseases.36–40 In this context, several studies have reported GSH depletion in human and experimental colitis.105,109–111 It seems that the reduced activity of key enzymes involved in GSH synthesis, namely of GCL and also the decreased availability of cysteine/cystine, can be mainly responsible for the mucosal GSH depletion observed in IBD patients.105
Expectedly, pro-inflammatory cytokines induced a significant depletion of intracellular GSH content in HT-29 cells but had no effect on the GCLc mRNA levels, suggesting that GSH depletion induced by cytokines is not related with an impairment of the GSH synthesis. Given this observation, GSH extrusion through specific carriers reported in several studies as occurring in cells undergoing apoptosis112–114 can explain the GSH depletion induced by the mix of cytokines. Although this mechanism was not explored (not a major goal of the work), RWE counteracts GSH depletion via increased GCL mRNA in both control and cytokine-stimulated cells.
This result indicates that RWE operates in the prevention of the inflammatory response by positively modulating the activity of the Nrf2, leading not only to an increase in HO-1 expression but also to the prevention of the GSH depletion that occurs under inflammatory conditions.
There is considerable evidence supporting a complex and functional interplay between Nrf2 and NF-κB pathways on inflammatory signaling. For instance, the NF-κB pathway activation, by mechanisms not completely understood, could be attenuated by several Nrf2-activating compounds.115–117 Also, several studies showed that the activation of the NF-κB and the consequent overproduction of pro-inflammatory mediators is more intense in Nrf2 knockout mice compared to wild-type mice.118 In this context, Nrf-2 knockout mice had an increased susceptibility to DSS-induced colitis,119 suggesting a protective role of the Nrf2 pathway in this inflammatory disease. Recently, it was reported that NF-κB can repress the Nrf2 pathway.120,121 So, the inhibition of the NF-κB pathway by RWE that we have observed before11 and the activation of the Nrf2 pathway shown here suggests a potential cross-talk between NF-κB and Nrf2 in the presence of RWE.
In conclusion, the data presented here indicate that the intestinal anti-inflammatory effect of the RWE is complex, involving several molecular mechanisms. A point of particular relevance is the complementary action of RWE in JAK/STAT and Nrf2 pathways. The opposite effects of RWE in the inhibition of a pro-inflammatory pathway (JAK/STAT) and activation of a detoxifying pathway (Nrf2) are of great relevance for their potential efficacy against inflammation. Thus, RWE not only inhibited key inflammatory pathways such as the JAK/STAT signalling cascade but also activated the Nrf2 pathway, increasing the expression of cellular defenses.
Considering the increased awareness of alternative approaches for mitigating intestinal inflammatory disorders, this study may contribute to the development of an emerging therapeutic approach in gastrointestinal inflammation, namely nutritional red wine supplements with well characterized anti-inflammatory actions for disease prevention and treatment.
Abbreviations
| COX-2 | Cyclooxygenase-2 |
| DMEM | Dulbecco's modified Eagle medium |
| ECF | Enhanced chemifluorescence |
| EDTA | Ethylenediaminetetraacetic acid |
| FBS | Foetal bovine serum |
| GCL | Glutamate–cysteine ligase |
| GCLc | Glutamate–cysteine ligase catalytic subunit |
| GCLm | Glutamate–cysteine ligase modifier subunit |
| Hepes | 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid |
| HO-1 | Heme oxygenase-1 |
| IBD | Inflammatory bowel disease |
| HPRT-1 | Hypoxanthine phosphoribosyltransferase-1 |
| IECs | Intestinal epithelial cells |
| IL | Interleukin |
| INF-γ | Interferon-γ |
| iNOS | Inducible nitric oxide synthase |
| JAK | Janus kinase |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide |
| LPS | Lipopolysaccharide |
| Nrf2 | Nuclear factor-erythroid 2-related factor-2, phosphate buffer |
| PIAS | Protein inhibitors of activated STATs |
| PMSF | Phenylmethanesulfonyl fluoride |
| PTPs | Protein tyrosine phosphatases |
| PVDF | Polyvinylidene difluoride |
| ROS | Reactive oxygen species |
| RWE | Red wine extract |
| SOCs | Suppressor of cytokine signaling |
| STAT | Signal transducer and activator of transcription |
| SDS | Sodium dodecyl sulfate |
| TBS-T | Tris-buffered saline with Tween |
| TNF-α | Tumor necrosis factor-α |
| Tris | Tris(hydroxymethyl)aminomethane |
Acknowledgements
This work is funded by FEDER funds through the Operational Programme Competitiveness Factors – COMPETE and national funds by FCT – Foundation for Science and Technology under the project PTDC/BBB-BQB/3217/2012 and strategic project UID/NEU/04539/2013. Carla Nunes acknowledges FCT fellowship SFRH/BPD/46149/2008 and Natércia Teixeira acknowledges FCT fellowship SFRH/BD/70053/2010.References
- R. K. Cross and K. T. Wilson, Inflammatory Bowel Dis., 2003, 9, 179–189 CrossRef.
- R. J. Xavier and D. K. Podolsky, Nature, 2007, 448, 427–434 CrossRef CAS PubMed.
- B. Khor, A. Gardet and R. J. Xavier, Nature, 2011, 474, 307–317 CrossRef CAS PubMed.
- M. Scharl and G. Rogler, Curr. Opin. Gastroenterol., 2012, 28, 301–309 CrossRef PubMed.
- R. B. Stein and S. B. Hanauer, Drug Saf., 2000, 23, 429–448 CrossRef CAS PubMed.
- L. N. Rosenberg and M. A. Peppercorn, Expert Opin. Drug Saf., 2010, 9, 573–592 CrossRef CAS PubMed.
- C. T. Xu, S. Y. Meng and B. R. Pan, World J. Gastroenterol., 2004, 10, 2311–2317 CrossRef CAS PubMed.
- P. Dolara, C. Luceri, C. De Filippo, A. P. Femia, L. Giovannelli, G. Caderni, C. Cecchini, S. Silvi, C. Orpianesi and A. Cresci, Mutat. Res., 2005, 591, 237–246 CrossRef CAS PubMed.
- B. Gago, J. O. Lundberg, R. M. Barbosa and J. Laranjinha, Free Radical Biol. Med., 2007, 43, 1233–1242 CrossRef CAS PubMed.
- P. Janega, J. Klimentova, A. Barta, M. Kovacsova, S. Vrankova, M. Cebova, Z. Cierna, Z. Matuskova, V. Jakovljevic and O. Pechanova, Food Funct., 2014, 5, 2202–2207 CAS.
- C. Nunes, E. Ferreira, V. Freitas, L. Almeida, R. M. Barbosa and J. Laranjinha, Food Funct., 2013, 4, 373–383 CAS.
- M. Coskun, M. Salem, J. Pedersen and O. H. Nielsen, Pharmacol. Res., 2013, 76, 1–8 CrossRef CAS PubMed.
- Y. Q. Huang, J. J. Li and S. Karpatkin, J. Biol. Chem., 2000, 275, 6462–6468 CrossRef CAS PubMed.
- H. S. Kim and M. S. Lee, Cell. Signalling, 2007, 19, 454–465 CrossRef CAS PubMed.
- H. K. Bryan, A. Olayanju, C. E. Goldring and B. K. Park, Biochem. Pharmacol., 2013, 85, 705–717 CrossRef CAS PubMed.
- Y. S. Keum and B. Y. Choi, Molecules, 2014, 19, 10074–10089 CrossRef PubMed.
- W. Li and A. N. Kong, Mol. Carcinog., 2009, 48, 91–104 CrossRef CAS PubMed.
- J. Kim, Y. N. Cha and Y. J. Surh, Mutat. Res., 2010, 690, 12–23 CrossRef CAS PubMed.
- A. Fernandes, A. Sousa, N. Mateus, M. Cabral and V. de Freitas, Food Chem., 2011, 125, 1398–1405 CrossRef CAS.
- N. Mateus, S. Proença, P. Ribeiro, J. M. Machado and V. De Freitas, Cienc. Tecnol. Aliment., 2001, 3, 102–110 CrossRef.
- A. Peña-Neira, T. Hernández, M. C. García-Vallejo, E. Cadahía, B. Fernández de Simón and J. A. Suarez, Am. J. Enol. Vitic., 1999, 50, 285–290 Search PubMed.
- P. Ribéreau-Gayon and E. Stonestreet, Chim. Anal., 1966, 627–631 Search PubMed.
- A. Arnous, D. P. Makris and P. Kefalas, J. Agric. Food Chem., 2001, 49, 5736–5742 CrossRef CAS PubMed.
- I. Chantret, A. Barbat, E. Dussaulx, M. G. Brattain and A. Zweibaum, Cancer Res., 1988, 48, 1936–1942 CAS.
- F. Denizot and R. Lang, J. Immunol. Methods, 1986, 89, 271–277 CrossRef CAS PubMed.
- P. J. Hissin and R. Hilf, Anal. Biochem., 1976, 74, 214–226 CrossRef CAS PubMed.
- C. Schindler and C. Plumlee, Semin. Cell Dev. Biol., 2008, 19, 311–318 CrossRef CAS PubMed.
- D. Willis, A. R. Moore, R. Frederick and D. A. Willoughby, Nat. Med., 1996, 2, 87–90 CrossRef CAS PubMed.
- Y. Naito, T. Takagi and T. Yoshikawa, Aliment. Pharmacol. Ther., 2004, 20(Suppl 1), 177–184 CrossRef CAS PubMed.
- M. Yalniz, U. Demirel, C. Orhan, I. H. Bahcecioglu, I. H. Ozercan, C. Aygun, M. Tuzcu and K. Sahin, Inflammation, 2012, 35, 1213–1221 CrossRef CAS PubMed.
- L. M. Aleksunes and J. E. Manautou, Toxicol. Pathol., 2007, 35, 459–473 CrossRef CAS PubMed.
- T. Nguyen, P. J. Sherratt and C. B. Pickett, Annu. Rev. Pharmacol. Toxicol., 2003, 43, 233–260 CrossRef CAS PubMed.
- J. Y. Chan and M. Kwong, Biochim. Biophys. Acta, 2000, 1517, 19–26 CrossRef CAS.
- S. A. Chanas, Q. Jiang, M. McMahon, G. K. McWalter, L. I. McLellan, C. R. Elcombe, C. J. Henderson, C. R. Wolf, G. J. Moffat, K. Itoh, M. Yamamoto and J. D. Hayes, Biochem. J., 2002, 365, 405–416 CrossRef CAS PubMed.
- J. Zheng, M. J. Piao, K. C. Kim, C. W. Yao, J. W. Cha and J. W. Hyun, Mar. Drugs, 2014, 12, 4214–4230 CrossRef CAS PubMed.
- P. Santus, A. Corsico, P. Solidoro, F. Braido, F. Di Marco and N. Scichilone, COPD, 2014, 11, 705–717 CrossRef PubMed.
- R. Tirouvanziam, C. K. Conrad, T. Bottiglieri, L. A. Herzenberg and R. B. Moss, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4628–4633 CrossRef CAS PubMed.
- J. De Backer, W. Vos, C. Van Holsbeke, S. Vinchurkar, R. Claes, P. M. Parizel and W. De Backer, Int. J. Chronic Obstruct. Pulm. Dis., 2013, 8, 569–579 CrossRef PubMed.
- S. Uraz, G. Tahan, H. Aytekin and V. Tahan, Scand. J. Clin. Lab. Invest., 2013, 73, 61–66 CrossRef CAS PubMed.
- W. Jeong, S. H. Yoon, D. J. An, S. H. Cho, K. K. Lee and J. Y. Kim, Parasitology, 2010, 137, 241–249 CrossRef CAS PubMed.
- J. O. Clarke and G. E. Mullin, Nutr. Clin. Pract., 2008, 23, 49–62 CrossRef PubMed.
- S. C. Kong, D. P. Hurlstone, C. Y. Pocock, L. A. Walkington, N. R. Farquharson, M. G. Bramble, M. E. McAlindon and D. S. Sanders, J. Clin. Gastroenterol., 2005, 39, 138–141 CrossRef PubMed.
- H. Shapiro, P. Singer, Z. Halpern and R. Bruck, Gut, 2007, 56, 426–435 CrossRef CAS PubMed.
- S. J. Hur, S. H. Kang, H. S. Jung, S. C. Kim, H. S. Jeon, I. H. Kim and J. D. Lee, Nutr. Res., 2012, 32, 801–816 CrossRef CAS PubMed.
- F. Biasi, M. Astegiano, M. Maina, G. Leonarduzzi and G. Poli, Curr. Med. Chem., 2011, 18, 4851–4865 CrossRef CAS PubMed.
- B. S. Rocha, C. Nunes, C. Pereira, R. M. Barbosa and J. Laranjinha, Food Funct., 2014, 5, 1646–1652 CAS.
- C. G. Fraga, M. Galleano, S. V. Verstraeten and P. I. Oteiza, Mol. Aspects Med., 2010, 31, 435–445 CrossRef CAS PubMed.
- I. Rahman, S. K. Biswas and P. A. Kirkham, Biochem. Pharmacol., 2006, 72, 1439–1452 CrossRef CAS PubMed.
- J. S. Kim, A. S. Narula and C. Jobin, Clin. Exp. Immunol., 2005, 141, 288–297 CrossRef CAS PubMed.
- M. I. Netsch, H. Gutmann, C. Aydogan and J. Drewe, Planta Med., 2006, 72, 697–702 CrossRef CAS PubMed.
- E. Tedeschi, M. Menegazzi, Y. Yao, H. Suzuki, U. Forstermann and H. Kleinert, Mol. Pharmacol., 2004, 65, 111–120 CrossRef CAS PubMed.
- B. Romier-Crouzet, J. Van De Walle, A. During, A. Joly, C. Rousseau, O. Henry, Y. Larondelle and Y. J. Schneider, Food Chem. Toxicol., 2009, 47, 1221–1230 CrossRef CAS PubMed.
- S. J. Jeong, H. S. Lim, C. S. Seo, J. H. Kim, S. E. Jin, S. R. Yoo and H. K. Shin, Phytomedicine, 2015, 22, 326–332 CrossRef PubMed.
- A. N. Carey, D. R. Fisher, A. M. Rimando, S. M. Gomes, D. F. Bielinski and B. Shukitt-Hale, J. Agric. Food Chem., 2013, 61, 5979–5986 CrossRef CAS PubMed.
- A. S. Hole, S. Grimmer, M. R. Jensen and S. Sahlstrøm, Food Chem., 2012, 133, 969–977 CrossRef CAS.
- M. Herranz-Lopez, S. Fernandez-Arroyo, A. Perez-Sanchez, E. Barrajon-Catalan, R. Beltran-Debon, J. A. Menendez, C. Alonso-Villaverde, A. Segura-Carretero, J. Joven and V. Micol, Phytomedicine, 2012, 19, 253–261 CrossRef CAS PubMed.
- M. Rossetto, P. Vanzani, F. Mattivi, M. Lunelli, M. Scarpa and A. Rigo, Arch. Biochem. Biophys., 2002, 408, 239–245 CrossRef CAS PubMed.
- D. Vyas, K. M. O'Dell, J. L. Bandy and E. G. Boyce, Ann. Pharmacother., 2013, 47, 1524–1531 CrossRef CAS PubMed.
- J. Panes, C. Su, A. G. Bushmakin, J. C. Cappelleri, C. Mamolo and P. Healey, BMC Gastroenterol., 2015, 15, 14 CrossRef CAS PubMed.
- W. J. Sandborn, S. Ghosh, J. Panes, I. Vranic, C. Su, S. Rousell and W. Niezychowski, N. Engl. J. Med., 2012, 367, 616–624 CrossRef CAS PubMed.
- E. A. Bach, M. Aguet and R. D. Schreiber, Annu. Rev. Immunol., 1997, 15, 563–591 CrossRef CAS PubMed.
- C. Schindler, K. Shuai, V. R. Prezioso and J. E. Darnell Jr., Science, 1992, 257, 809–813 CAS.
- R. W. Ganster, B. S. Taylor, L. Shao and D. A. Geller, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8638–8643 CrossRef CAS PubMed.
- E. Tedeschi, M. Menegazzi, D. Margotto, H. Suzuki, U. Forstermann and H. Kleinert, J. Pharmacol. Exp. Ther., 2003, 307, 254–261 CrossRef CAS PubMed.
- I. Andujar, M. C. Recio, R. M. Giner, E. Cienfuegos-Jovellanos, S. Laghi, B. Muguerza and J. L. Rios, J. Agric. Food Chem., 2011, 59, 6474–6483 CrossRef CAS PubMed.
- K. Suk, S. Kim, Y. H. Kim, K. A. Kim, I. Chang, H. Yagita, M. Shong and M. S. Lee, J. Immunol., 2001, 166, 4481–4489 CrossRef CAS.
- J. J. Sironi and T. Ouchi, J. Biol. Chem., 2004, 279, 4066–4074 CrossRef CAS PubMed.
- P. A. Townsend, T. M. Scarabelli, S. M. Davidson, R. A. Knight, D. S. Latchman and A. Stephanou, J. Biol. Chem., 2004, 279, 5811–5820 CrossRef CAS PubMed.
- H. S. Kim and M. S. Lee, Mol. Cell. Biol., 2005, 25, 6821–6833 CrossRef CAS PubMed.
- K. Suk, I. Chang, Y. H. Kim, S. Kim, J. Y. Kim, H. Kim and M. S. Lee, J. Biol. Chem., 2001, 276, 13153–13159 CrossRef CAS PubMed.
- T. K. Kao, Y. C. Ou, S. Y. Lin, H. C. Pan, P. J. Song, S. L. Raung, C. Y. Lai, S. L. Liao, H. C. Lu and C. J. Chen, J. Nutr. Biochem., 2011, 22, 612–624 CrossRef CAS PubMed.
- H. Y. Kim, E. J. Park, E. H. Joe and I. Jou, J. Immunol., 2003, 171, 6072–6079 CrossRef CAS.
- W. Chen, M. O. Daines and G. K. Khurana Hershey, J. Allergy Clin. Immunol., 2004, 114, 476–489 CrossRef CAS PubMed ; quiz 490.
- S. Wormald and D. J. Hilton, J. Biol. Chem., 2004, 279, 821–824 CrossRef CAS PubMed.
- D. Xu and C. K. Qu, Front. Biosci., 2008, 13, 4925–4932 CrossRef CAS.
- K. P. Pavlick, F. S. Laroux, J. Fuseler, R. E. Wolf, L. Gray, J. Hoffman and M. B. Grisham, Free Radical Biol. Med., 2002, 33, 311–322 CrossRef CAS PubMed.
- S. M. Karp and T. R. Koch, DM, Dis. – Mon., 2006, 52, 199–207 CrossRef PubMed.
- S. Tanida, T. Mizoshita, T. Mizushima, M. Sasaki, T. Shimura, T. Kamiya, H. Kataoka and T. Joh, J. Clin. Biochem. Nutr., 2011, 48, 112–116 CrossRef CAS PubMed.
- H. H. Arab, S. A. Salama, A. H. Eid, H. A. Omar, S. A. Arafa el and I. A. Maghrabi, Food Chem. Toxicol., 2014, 69, 294–302 CrossRef CAS PubMed.
- V. O. Tkachev, E. B. Menshchikova and N. K. Zenkov, Biochemistry, 2011, 76, 407–422 CAS.
- R. Patel and G. Maru, Free Radical Biol. Med., 2008, 44, 1897–1911 CrossRef CAS PubMed.
- Y. P. Hwang, J. H. Choi, H. J. Yun, E. H. Han, H. G. Kim, J. Y. Kim, B. H. Park, T. Khanal, J. M. Choi, Y. C. Chung and H. G. Jeong, Food Chem. Toxicol., 2011, 49, 93–99 CrossRef CAS PubMed.
- S. Chen, Y. Zhu, Z. Liu, Z. Gao, B. Li, D. Zhang, Z. Zhang, X. Jiang, L. Meng, Y. Yang and B. Shi, PLoS One, 2015, 10, e0126457 Search PubMed.
- A. L. Theiss, M. Vijay-Kumar, T. S. Obertone, D. P. Jones, J. M. Hansen, A. T. Gewirtz, D. Merlin and S. V. Sitaraman, Gastroenterology, 2009, 137, 199–208 CrossRef CAS PubMed.
- K. Choi, J. Chen, S. Mitra and S. K. Sarna, Gastroenterology, 2011, 141, 1293–1301 CrossRef CAS PubMed.
- L. Kruidenier, I. Kuiper, W. Van Duijn, M. A. Mieremet-Ooms, R. A. van Hogezand, C. B. Lamers and H. W. Verspaget, J. Pathol., 2003, 201, 17–27 CrossRef CAS PubMed.
- F. Khodagholi, B. Eftekharzadeh, N. Maghsoudi and P. F. Rezaei, Mol. Cell. Biochem., 2010, 337, 39–51 CrossRef CAS PubMed.
- K. Sahin, M. Tuzcu, H. Gencoglu, A. Dogukan, M. Timurkan, N. Sahin, A. Aslan and O. Kucuk, Life Sci., 2010, 87, 240–245 CrossRef CAS PubMed.
- M. A. Aminzadeh, S. A. Reisman, N. D. Vaziri, S. Shelkovnikov, S. H. Farzaneh, M. Khazaeli and C. J. Meyer, Redox Biol., 2013, 1, 527–531 CrossRef CAS PubMed.
- A. Speciale, J. Chirafisi, A. Saija and F. Cimino, Curr. Mol. Med., 2011, 11, 770–789 CrossRef CAS PubMed.
- M. P. Mattson, Ageing Res. Rev., 2008, 7, 43–48 CrossRef PubMed.
- H. Erlank, A. Elmann, R. Kohen and J. Kanner, Free Radical Biol. Med., 2011, 51, 2319–2327 CrossRef CAS PubMed.
- Y. Y. Lee-Hilz, A. M. Boerboom, A. H. Westphal, W. J. Berkel, J. M. Aarts and I. M. Rietjens, Chem. Res. Toxicol., 2006, 19, 1499–1505 CrossRef CAS PubMed.
- R. Sirota, D. Gibson and R. Kohen, Redox Biol., 2015, 4, 48–59 CrossRef CAS PubMed.
- H. O. Pae, G. S. Jeong, S. O. Jeong, H. S. Kim, S. A. Kim, Y. C. Kim, S. J. Yoo, H. D. Kim and H. T. Chung, Exp. Mol. Med., 2007, 39, 267–277 CrossRef CAS PubMed.
- H. Y. Lin, S. H. Juan, S. C. Shen, F. L. Hsu and Y. C. Chen, Biochem. Pharmacol., 2003, 66, 1821–1832 CrossRef CAS PubMed.
- A. Haider, R. Olszanecki, R. Gryglewski, M. L. Schwartzman, E. Lianos, A. Kappas, A. Nasjletti and N. G. Abraham, J. Pharmacol. Exp. Ther., 2002, 300, 188–194 CrossRef CAS PubMed.
- H. G. Chen, K. L. Xie, H. Z. Han, W. N. Wang, D. Q. Liu, G. L. Wang and Y. H. Yu, Int. J. Surg., 2013, 11, 1060–1066 CrossRef PubMed.
- R. Tenhunen, H. S. Marver and R. Schmid, Proc. Natl. Acad. Sci. U. S. A., 1968, 61, 748–755 CrossRef CAS.
- T. Takagi, Y. Naito, K. Uchiyama, T. Suzuki, I. Hirata, K. Mizushima, H. Tsuboi, N. Hayashi, O. Handa, T. Ishikawa, N. Yagi, S. Kokura, H. Ichikawa and T. Yoshikawa, Dig. Dis. Sci., 2011, 56, 1663–1671 CrossRef CAS PubMed.
- M. Lenicek, D. Duricova, O. Hradsky, P. Dusatkova, A. Jiraskova, M. Lukas, P. Nachtigal and L. Vitek, Inflammatory Bowel Dis., 2014, 20, 481–487 CrossRef PubMed.
- K. A. Kirkby and C. A. Adin, Am. J. Physiol.: Renal, Fluid Electrolyte Physiol., 2006, 290, F563–F571 CrossRef CAS PubMed.
- K. Aquilano, S. Baldelli and M. R. Ciriolo, Front. Pharmacol., 2014, 5, 196 Search PubMed.
- P. Ghezzi, Int. J. Gen. Med., 2011, 4, 105–113 CrossRef CAS PubMed.
- B. Sido, V. Hack, A. Hochlehnert, H. Lipps, C. Herfarth and W. Droge, Gut, 1998, 42, 485–492 CrossRef CAS PubMed.
- C. Nunes, R. M. Barbosa, L. Almeida and J. Laranjinha, Mol. Cell. Neurosci., 2011, 48, 94–103 CrossRef CAS PubMed.
- A. G. Hall, Adv. Exp. Med. Biol., 1999, 457, 199–203 CrossRef CAS PubMed.
- M. L. Circu and T. Y. Aw, Biochim. Biophys. Acta, 2012, 1823, 1767–1777 CrossRef CAS PubMed.
- R. O. Ek, M. Serter, K. Ergin, S. Cecen, C. Unsal, Y. Yildiz and M. D. Bilgin, Int. J. Clin. Exp. Med., 2014, 7, 989–997 Search PubMed.
- N. Nieto, M. I. Torres, M. I. Fernandez, M. D. Giron, A. Rios, M. D. Suarez and A. Gil, Dig. Dis. Sci., 2000, 45, 1820–1827 CrossRef CAS PubMed.
- H. H. Arab, M. Y. Al-Shorbagy, D. M. Abdallah and N. N. Nassar, PLoS One, 2014, 9, e97193 Search PubMed.
- M. D'Alessio, C. Cerella, M. De Nicola, A. Bergamaschi, A. Magrini, G. Gualandi, A. M. Alfonsi and L. Ghibelli, Ann. N. Y. Acad. Sci., 2003, 1010, 449–452 CrossRef.
- R. Franco and J. A. Cidlowski, Antioxid. Redox Signaling, 2012, 17, 1694–1713 CrossRef CAS PubMed.
- L. Ghibelli, C. Fanelli, G. Rotilio, E. Lafavia, S. Coppola, C. Colussi, P. Civitareale and M. R. Ciriolo, FASEB J., 1998, 12, 479–486 CAS.
- J. H. Lyu, K. H. Kim, H. W. Kim, S. I. Cho, K. T. Ha, J. Y. Choi, C. W. Han, H. S. Jeong, H. K. Lee, K. S. Ahn, S. R. Oh, R. T. Sadikot, J. W. Christman and M. Joo, J. Ethnopharmacol., 2012, 140, 107–116 CrossRef PubMed.
- N. Wakabayashi, S. L. Slocum, J. J. Skoko, S. Shin and T. W. Kensler, Antioxid. Redox Signalling, 2010, 13, 1649–1663 CrossRef CAS PubMed.
- W. Li, T. O. Khor, C. Xu, G. Shen, W. S. Jeong, S. Yu and A. N. Kong, Biochem. Pharmacol., 2008, 76, 1485–1489 CrossRef CAS PubMed.
- R. K. Thimmulappa, H. Lee, T. Rangasamy, S. P. Reddy, M. Yamamoto, T. W. Kensler and S. Biswal, J. Clin. Invest., 2006, 116, 984–995 CAS.
- T. O. Khor, M. T. Huang, K. H. Kwon, J. Y. Chan, B. S. Reddy and A. N. Kong, Cancer Res., 2006, 66, 11580–11584 CrossRef CAS PubMed.
- G. H. Liu, J. Qu and X. Shen, Biochim. Biophys. Acta, 2008, 1783, 713–727 CrossRef CAS PubMed.
- M. Yu, H. Li, Q. Liu, F. Liu, L. Tang, C. Li, Y. Yuan, Y. Zhan, W. Xu, W. Li, H. Chen, C. Ge, J. Wang and X. Yang, Cell. Signalling, 2011, 23, 883–892 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |