
Mechanistic understanding of molecular initiating events (MIEs) using NMR spectroscopy
Paul N.
Sanderson
*,
Wendy
Simpson
,
Richard
Cubberley
,
Maja
Aleksic
,
Stephen
Gutsell
and
Paul J.
Russell
Unilever Safety & Environmental Assurance Centre, Colworth Science Park, Sharnbrook, Bedford, MK44 1LQ, UK. E-mail: paul.sanderson@unilever.com
First published on 15th September 2015
Abstract
Toxicological risk assessments in the 21st century are increasingly being driven by the Adverse Outcome Pathways (AOP) conceptual framework in which the Molecular Initiating Event (MIE) is of fundamental importance to pathway progression. For those MIEs that involve covalent chemical reactions, such as protein haptenation, determination of relative rates and mechanisms of reactions is a prerequisite for their understanding. The utility of NMR spectroscopy as an experimental technique for effectively providing reaction rate and mechanistic information for early assessment of likely MIE(s) has been demonstrated. To demonstrate the concept, model systems exemplifying common chemical reactions involved in the covalent modification of proteins were utilized; these involved chemical reactions of electrophilic species (representing different mechanistic classes) with simple amine and thiol nucleophiles acting as surrogates for the reactive groups of lysine and cysteine protein side chains respectively. Such molecular interactions are recognized as critical mechanisms in a variety of chemical and drug toxicities, including respiratory and skin sensitization and liver toxicity as well as being the key mechanism of action for a number of therapeutic agents.
Introduction
Consumer and environmental toxicology is currently undergoing a paradigm shift, following TT21C principles1 further underpinned by the drive to use species-relevant in vitro data as alternatives to surrogate animal models, particularly for humans.2 Increasingly, focus is directed towards risk assessment based upon the Adverse Outcome Pathways (AOP) conceptual framework;3–6 a logical sequence of events or processes within biological systems that can be used to understand adverse effects. This approach shifts the focus away from traditional toxicity endpoints (e.g. allergy, cancer) to the development of mechanistic understanding of a chemical's effect at a molecular and sub-cellular level. The overall aim is to reduce uncertainty and increase confidence in risk assessments through characterisation of the impact of a chemical on human health,7 or the environment, across different levels of biological organization; i.e. from initial exposure and molecular interactions through cellular effects and tissue effects, up to organ and individual effects. One of the strengths of the AOP approach is the potential for using inter-species synergies to perform integrated research towards producing risk assessments for both human and environmentally relevant species.8Central to an AOP is the ‘molecular initiating event’ (MIE), which has been defined as ‘the initial interaction between a molecule and a biomolecule or biosystem that can be causally linked to an outcome via a pathway’.9 Gaining detailed understanding of MIEs is seen as a key step to understanding AOPs and their utility in risk assessment approaches.10 In several AOPs, including liver necrosis (induced by a metabolite of paracetamol11), skin blistering (caused by exposure to warfare agents12) and skin sensitization (resulting from exposure to electrophilic molecules in the environment, occupational setting13 or household and cosmetic products14), the MIE involves covalent modification of proteins (haptenation) by electrophilic chemicals.15 A large number of xenobiotics or their metabolites have electrophilic characteristics and hence have the potential for covalent modification of proteins.16,17 In order to understand MIES of this nature, there is a need for novel robust methodologies to study chemical reactivity to proteins.
The extent of protein modification (and hence biological response) of MIEs that involve haptenation is a function of the rate of reaction between the hapten and the protein; consequently, a measurement of relative rates of chemical reactivity could be used predictively to inform toxicological risk assessments.14,18–22 Indeed, as the application of mathematical modelling in the use of MIEs and AOPs in risk assessments becomes more commonplace,23 the need for reliable relative reaction rate data will become increasingly important. A further important consideration for exposure and hazard based risk assessment is the rate of creation of a reactive product from an unreactive parent compound, such information is needed to understand the potential for overwhelming the adaptive mechanisms of a biological system.24
The study of haptenation reactions has largely been restricted to the use of nucleophilic protein-mimetic models as the protein target is not always identified.25–29 Indeed, several approaches for the assessment of chemical reactivity towards proteins have been developed,22,30–39 largely driven by the need to assess chemical reactivity in skin sensitization, although it should be noted that they are equally applicable wherever a covalent mechanism of action is involved in the MIE; they include spectrophotometric assays that monitor changes in UV absorbance and fluorescence32,33 as well as peptide reactivity assays that measure reactivity of nucleophiles to synthetic peptides using HPLC-UV, MS and MS/MS platforms.30,34–37 These assays do, however, have some limitations which include providing limited mechanistic information and indirect reactivity data that are not suitable for risk modelling (i.e. percentage depletion, rather than rate constants40), as well as the over- or under-estimation of relative reactivity due, for example, to secondary reactivity or adduct instability. These limitations could, potentially, be overcome by the use of NMR spectroscopy as a complementary technique that can provide detailed reactivity information.
The intrinsic quantitative nature of NMR spectroscopy data makes the technique particularly useful for the generation of reaction rates, as both reactant and product concentrations can be measured directly and concomitantly, in ‘real-time’, without need to determine response factors; also, as NMR measurements are non-destructive they do not affect reaction outcomes. Furthermore, the non-selective nature of NMR data makes this technique applicable to a wide range of chemical entities. NMR spectroscopy can, therefore, provide a rapid initial assessment of the reactivity of a new chemical entity; such information would allow straightforward comparisons of relative reactivity with known reactants in a given system. In addition to reaction rates, the ‘information rich’ nature of NMR data allows for NMR spectroscopy to simultaneously provide mechanistic information,41 as the technique can be used to determine (or confirm) the chemical structures of both the starting materials and the reaction product(s) at any stage of the reaction. NMR spectroscopy data can also be obtained under different solution conditions, including aqueous (physiological) and non-aqueous; and consequently can be of particular benefit for assessing relative reactivity of chemicals with low aqueous solubility which cannot be assessed using peptide depletion assays. For MIEs, in which the location of the reactive nucleophilic protein site remains unknown, it is not possible to infer whether any particular solvent system is more representative of the in vivo reactive environment than any other solvent; hence, for the purpose of determining comparative reactivity, the choice of solvent is not important.40 The only requirements for NMR analysis being that reactants and products must have NMR active nuclei and must remain in solution throughout, with adequate NMR signal dispersion and signal-to-noise. NMR spectroscopy is, however, less sensitive than mass spectrometry and so requires larger quantities of materials for analysis.
In this paper we demonstrate the effectiveness of NMR spectroscopy for the concomitant elucidation of reaction rates and mechanisms (knowledge of both being essential for gaining a mechanistic understanding of MIEs that involve a covalent reaction) using a set of electrophilic chemicals that represent common mechanistic domains42 and which are implicated in numerous adverse outcomes including skin sensitization.43–54 As the target protein(s) have not yet been established for most MIEs, it cannot be assumed that any one particular model nucleophile is more representative of the in vivo reactive sites than any other;40 consequently, for the sake of simplicity and to demonstrate the utility of the NMR technique, the simple amine and thiol nucleophiles, n-butylamine and 1-butanethiol, were used as surrogates for nucleophilic amine and thiol groups of lysine and cysteine, respectively. Reactions with the amine nucleophile were performed in an organic solvent (acetonitrile) to keep the reaction system as simple as possible for the generation of comparative reaction rate constants. For the thiol reactivity studies, an aqueous/acetonitrile co-solvent was used; a similar solvent system to that used in peptide reactivity30,34–37 and spectrophotometric assays.32,33 One of the aims of the reactivity studies was to generate reaction rate constants that provide an absolute measure of relative reactivity. Rate constants are independent of reactant concentration and so could be obtained using solution concentrations that allowed rapid acquisition of NMR data from the reactions; it should be noted that the reactant concentrations used were similar to those used in peptide reactivity assays30,34–37 and spectrophotometric assays.32,33 Data for each nucleophile were obtained under standardized conditions of reaction environment; this being a prerequisite for obtaining reaction rate constant data to be used comparatively and predictively.
Experimental
Materials and methods
All chemicals and solvents used in this work were obtained from Sigma-Aldrich.Validation of methodology: reaction of 4-nitrobenzyl halides with n-butylamine
In order to obtain measurable quantities of reaction products within an hour, conditions for the reaction of n-butylamine with 4-nitrobenzyl chloride were: 4-nitrobenzyl chloride (100 mM) with excess n-butylamine (concentrations of 0.45, 0.91 and 1.75 M). For the reaction of n-butylamine with 4-nitrobenzyl bromide, the concentration of 4-nitrobenzyl bromide was 10 mM with n-butylamine concentrations of 57, 104, 221 and 395 mM. 1H NMR spectra were obtained at 310 K in methanol-d4 (NS = 1) at T = 4 minutes after mixing, and then at 1 minute intervals until T = 10 minutes and then at further intervals until T = 60 minutes.General reaction conditions with n-butylamine
For determination of amine nucleophile reaction rates and mechanisms, reactions were undertaken in deuterated acetonitrile (CD3CN) at 295 K. Initial electrophile concentrations were: phthalic anhydride (1 mM), 3,4-dihydrocoumarin (10 mM), phenyl benzoate (100 mM), trans-2-decenal (10 mM), cinnamaldehyde (10 mM), ethylene glycol dimethacrylate (100 mM), phenylacetaldehyde (1 mM) and 2,4-dimethyl-3-cyclohexenecarboxaldehyde (1 mM); reactions were started by addition of an approximately ten-fold molar excess of n-butylamine. The first spectrum was recorded after approximately 3 minutes (allowing time for transfer of sample to the NMR magnet and equilibration) with subsequent spectra obtained as reactions progressed.General reaction conditions with 1-butanethiol
For determination of thiol nucleophile reaction rates and mechanisms, reactions were undertaken in CD3CN/phosphate buffer (50 mM potassium phosphate; pH9) (50/50) at 295 K. Initial electrophile concentrations were: phthalic anhydride (10 mM), 3,4-dihydrocoumarin (10 mM), phenyl benzoate (10 mM), trans-2-decenal (5 mM), cinnamaldehyde (5 mM), ethylene glycol dimethacrylate (20 mM), phenylacetaldehyde (1 mM) and 2,4-dimethyl-3-cyclohexenecarboxaldehyde (1 mM); reactions were started by addition of an approximately ten-fold molar excess of 1-butanethiol. The first spectrum was recorded generally after 3 minutes, with subsequent spectra obtained as reactions progressed.For rate constant calculations, the values used for the concentrations of electrophiles were the nominal solution concentrations as prepared, whereas the nucleophile concentrations were determined from the 1H NMR data as molar ratios relative to the electrophile concentration, to compensate for potential errors in handling small volumes of reactants.
NMR spectroscopy
Stock solutions of reactants were prepared in appropriate solvents; aliquots of these were mixed to a solution volume of 0.6 ml immediately prior to being transferred to 5 mm diameter NMR tubes and the time of mixing was noted (T = 0). 1H NMR spectra were recorded on a Bruker AV(II) 600 NMR spectrometer operating at 600.13 MHz, with a probe temperature of either 310 K or 295 K, using standard pulse sequences for data acquisition. The set-up time for the NMR experiments (instrument lock, tuning and shimming) was minimized as far as possible whilst allowing the sample to equilibrate in the NMR magnet. Multiple 1H NMR spectra were then acquired sequentially. For reactions with n-butylamine in deuterated solvents, NMR spectra were acquired at intervals of at least a minute, with a single ‘pulse-acquire’ sequence (i.e. number of scans (NS) = 1); this allowed measurement as “instantaneously” as possible at the given time point as well as maximizing sample relaxation in order to provide quantitative NMR data. For reactions with 1-butanethiol in 50% aqueous solution, presaturation of the water signal was necessary; for this the Bruker pulse sequence noesygppr1d was used, typically with NS = 8, two dummy scans and a pulse repetition rate of 6 s. All 1H NMR spectra, after Fourier transformation, were accurately phased and baseline corrected prior to measurement of integrated signal areas. Chemical shifts were referenced to an external TMS reference.For validation of the methodology for determining rate constants using NMR data from the reaction of 4-nitrobenzyl halides with n-butylamine, rate constants were calculated using signal integral values extracted individually at each time point from a unique, well-resolved signal of the reaction product in the NMR spectrum. The NMR data for one of the test chemicals (trans-2-decenal) was analysed by two methods: (i) using signal integral values for a unique, well-resolved signal from the reaction product extracted manually from each NMR spectrum, and (ii) by extracting integral values for the same signals in a semi-automated manner using Dynamics Centre (version 2.1.10) (Bruker), a commercial software package for the analysis of NMR data containing non-frequency dimensions, in which signal areas (I) and time values (t) are converted to reaction rate data by curve fitting to equations of the form: f(t) = Io × exp(−kt) + C. The reaction rates calculated by both methods were the same; so, for efficiency of time, reaction rate data for all other test chemicals were obtained using Dynamics Centre. All well-resolved signals in the NMR spectra (from both reactants and products) were analysed in this way; the mean of these values provided the reaction rate value for each reaction. Rate constants were calculated by dividing the average rate value with the nucleophile concentration.
Determination of rate constants
For the bimolecular reaction: A + B → C in which reaction rate is dependent on the concentrations of the two reactants, the reaction is 2nd order and the rate of the reaction (r) is given by:55| r = k[A][B] k = rate constant |
For experimental determination of the rate constant (k), procedures and calculations can be simplified by using pseudo first order reaction conditions; i.e. maintaining one of the reactants (e.g. [B]) in excess and therefore essentially constant. Under these conditions: k[B] in the above equation becomes the pseudo first order rate constant: kobs; i.e.:
| kobs = k[B] |
The rate of reaction is the rate of increase in [C] with time (d[C]/dt) or the rate of decrease of [A] with time ( = −d[A]/dt); thus:
| r = kobs[A] = −d[A]/dt |
Integrating gives: ln[A] = −kobst + ln[A0] (the “integrated rate equation”). One molecule of A becomes one molecule of C in the reaction; hence: [A] = [A0] − [C].
Experimentally, either (or both) the decrease in concentration of A or the increase in concentration of C can be measured directly at each time point using NMR spectroscopy. In the current work, the nucleophile [B] was in excess, with [A0] being the initial concentration of the electrophile.
Substituting ([A0] − [C]) for [A] in the integrated rate equation and plotting ln([A0] − [C]) against t (time in seconds) produces a line with a slope of −kobs.
If reaction rates are measured with several values of [B] (i.e. different concentrations of the nucleophile), the second order rate constant “k” is readily determined as the slope of a plot of kobsversus [B].
Results
Preliminary NMR experiments to obtain information about reaction mechanism (i.e. the nature and number of reaction products) and to establish appropriate reaction conditions are required prior to obtaining data for determining rate constants. Appropriate sample conditions are those under which a measurable amount of reaction product is formed over a suitable time period (generally at least 10% conversion of reactant within a period of less than one hour), ensuring that the reaction does not proceed so quickly that it is essentially complete before measurements can be made. Reaction conditions can be optimised by varying reactant concentrations and/or temperature. After initial experiments, NMR signals from both reactants and products can be assigned and then measured in subsequent reaction rate experiments, prior to calculation of reaction rate constants.The information required for calculation of rate constants is the concentration of reactants and/or products at given time points. In NMR spectra, the area of a NMR signal is directly proportional to the concentration of the molecular species that gives rise to that signal. Therefore, for a solution of reactants at T = 0, the NMR signal areas will represent the concentrations of reactants at T = 0. It follows then that at any given time during the reaction, the concentrations of reactants and products at time t can be obtained directly from their NMR signal areas. As NMR spectra contain signals from all of the species present at any given time, mechanistic information can be obtained concurrently with reaction rate information.
The methodology of using NMR spectroscopy techniques to generate reaction rate constants was validated in this work using the reactions of 4-nitrobenzyl halides with n-butylamine as a model system. Subsequently, this approach was extended to demonstrate the mechanistic and chemical insights that can be obtained from NMR spectroscopy, using exemplar reactions of electrophiles chosen to represent reactions via common mechanistic domains (summarised in Scheme 1), with simple amine and thiol nucleophiles.
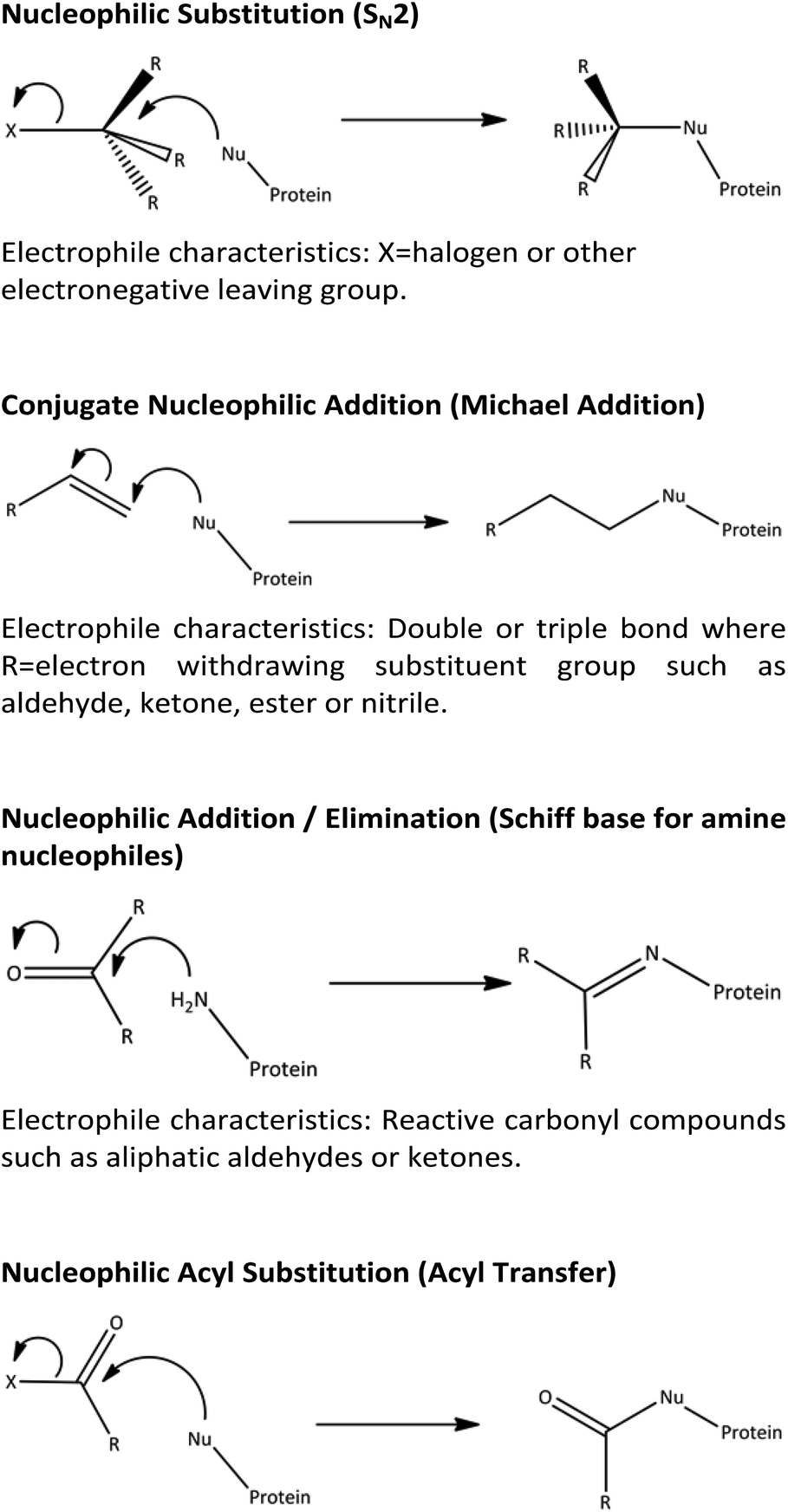 | ||
| Scheme 1 Mechanisms and nomenclature of nucleophile/electrophile reactions. | ||
Validation of the NMR approach: reactions of 4-nitrobenzyl halides with n-butylamine
1H NMR spectra from the reaction of 4-nitrobenzyl chloride with n-butylamine (Fig. 1), confirmed the mechanism (nucleophilic substitution; SN2) for this reaction and showed that there was just a single reaction product formed within the measurement time.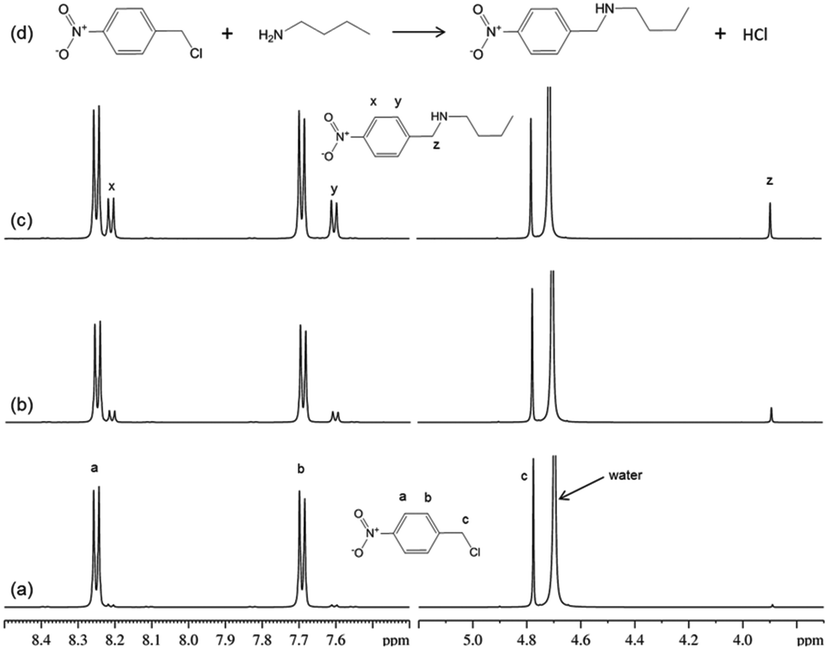 | ||
| Fig. 1 1H NMR signals and assignments from 4-nitrobenzyl chloride and its reaction product from the reaction between n-butylamine and 4-nitrobenzyl chloride in deuterated methanol; (a) T = 0.2 h, (b) T = 1 h, (c) T = 2.5 h, (d) the structures of reactants and products and the reaction scheme. Only the proton signals from 4-nitrobenzyl chloride (and its reaction product) are shown; the n-butylamine signals were all at <3.8 ppm. | ||
It was noted that reversing the relative concentrations of reactants (i.e. 4-nitrobenzyl chloride in excess) resulted in additional product signals in the NMR spectra; these were assigned to the tertiary amine product. Thus, when n-butylamine was in excess, the major reaction product was the secondary amine (as in Fig. 1); however, when the n-butylamine concentration was limiting (i.e. electrophile in excess), the secondary amine reacted further to produce the tertiary amine product. This simple reaction with potentially different outcomes therefore demonstrates the importance of verifying reaction mechanism prior to determining reaction rates.
1H NMR data were also obtained for the reaction of 4-nitrobenzyl bromide with n-butylamine. As the published rate constant for this reaction20 was reported to be approximately 160-fold greater than for the chloride, the concentrations of reactants for this reaction were reduced. The NMR data confirmed that this reaction also followed a SN2 mechanism.
NMR data for reaction rate constant determinations were obtained under reaction conditions in which n-butylamine was present in excess (pseudo first order reaction conditions). The concentrations of reaction products were obtained directly from the NMR data, using the NMR signal at ∼3.9 ppm which represented the phenyl methylene singlet from the reaction product. Data were collected from early time points (during the first ten minutes) of the reaction to ensure conditions were such that only the secondary amine product was formed. Several reactions were run, with different concentrations of n-butylamine, in order that second order rate constants could be obtained from plots of pseudo first order rate constants versus nucleophile concentrations.
For each reaction, the concentrations of product were measured directly from the NMR spectrum at each time point (from T = 4 to T = 10 minutes for each reaction) and the pseudo first order rate constant ‘kobs’ was then calculated for each reaction as the gradient of the slope of plots of −ln([Ao] − [C]) (M) vs. time (s) (Fig. 2a). The second order rate constant for each reaction was then obtained from the gradient of a plot of kobsvs. [Bo] as shown, for the reaction between n-butylamine and 4-nitrobenzyl bromide, in Fig. 2b.
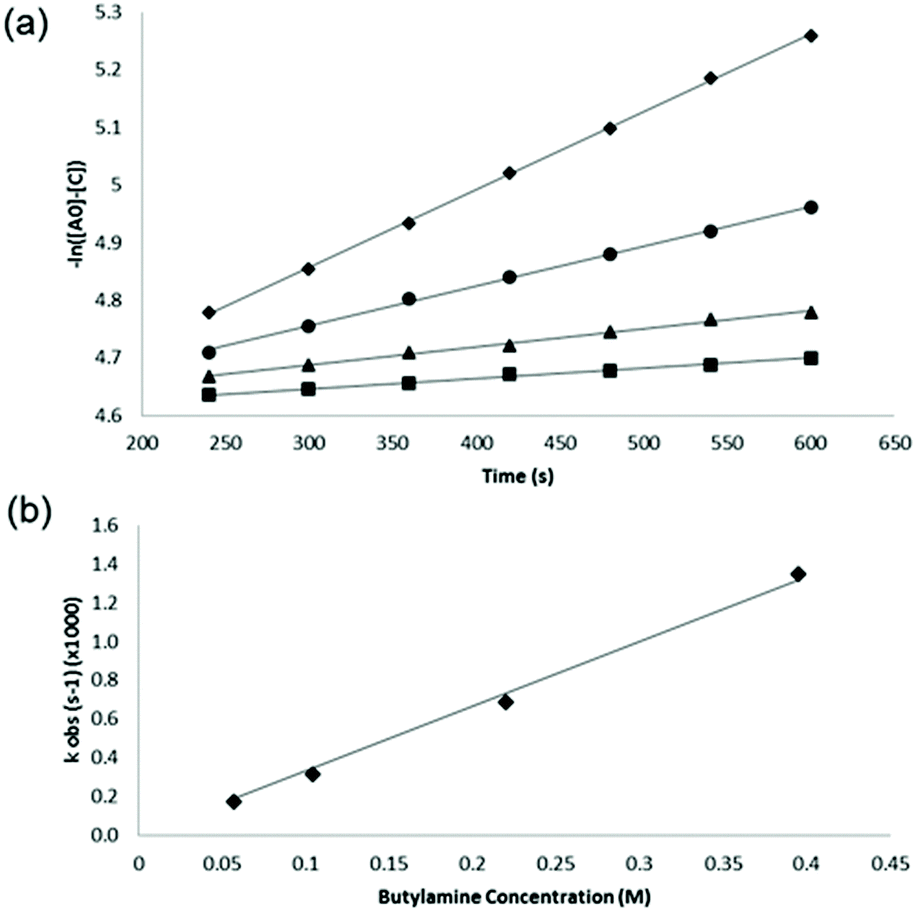 | ||
| Fig. 2 (a) Plots of −ln([Ao] − [C]) vs. time (s), for measurements made during the first 10 minutes of reaction, for the reaction between 4-nitrobenzyl bromide (10 mM) and various concentrations of n-butylamine (57 mM (squares), 104 mM (triangles), 221 mM (circles) & 395 mM (diamonds)); the pseudo first order rate constant ‘kobs’ was obtained as the gradient of the slope for each n-butylamine concentration. (b) Derivation of the second order rate constant (3.3 × 10−3 M−1 s−1) from the gradient of a plot of kobsvs. [Bo]. | ||
The second order rate constants determined from these data were: 8.1 × 10−5 M−1 s−1 for 4-nitrobenzyl chloride and 3.3 × 10−3 M−1 s−1 for 4-nitrobenzyl bromide; the magnitudes of these values were slightly higher than those reported previously,20 reflecting the higher reaction temperatures in the current work. The data clearly show the faster reactivity of the bromide relative to the chloride, the bromide ion being a larger, more effective leaving group.
Application of the NMR approach: characterisation of reactions across different mechanistic domains
Chemicals capable of reaction with both amine and thiol nucleophiles via a range of different mechanisms were investigated to confirm reaction mechanisms and to obtain reaction rate constants. Reactivity with amine nucleophiles was studied in an organic solvent (acetonitrile) using n-butylamine as the model nucleophile. For thiol nucleophile reactivity, 1-butanethiol was used as the model nucleophile and reactions were undertaken in a mixed organic/aqueous solvent system (acetonitrile/phosphate buffer). The reactivity data are summarised in Table 1.| Compound | Reaction with n-butylamine | Reaction with 1-butanethiol | |||
|---|---|---|---|---|---|
| Observed mechanism | Rate constant | Observed mechanism | Rate constant | ||
| a Reaction too fast to determine rate constant. b Three co-exisiting molecular species in aqueous solution before addition of thiol (see text). c Rate is depletion of electrophile, not generation of product as >1 product was formed. d This reaction mixture separated into two phases so derivation of rate constant was not possible. | |||||
| Phthalic anhydride |
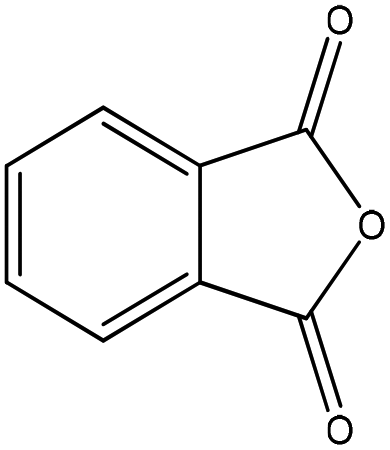
|
Nucleophilic acyl substitution | Nda | Nucleophilic acyl substitutionb | Nda |
| 3,4-Dihydrocoumarin |
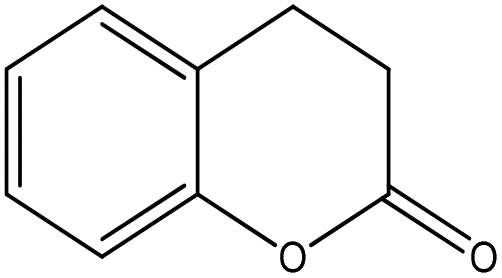
|
Nucleophilic acyl substitution | k = 7 × 10−3 M−1 s−1 | No reaction | Nd (no reaction) |
| Phenyl benzoate |
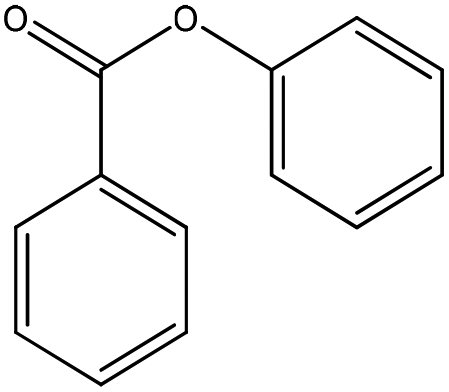
|
Nucleophilic acyl substitution | k = 3 × 10−5 M−1 s−1 | Nucleophilic acyl substitution | k ∼ 4 × 10−4 M−1 s−1 |
| Trans-2-decenal |
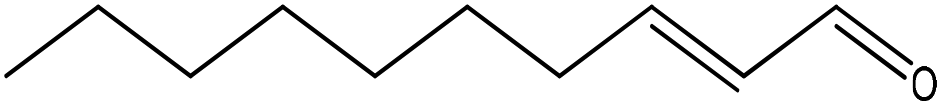
|
Nucleophilic addition/elimination & double adduct |
k![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c = 4.6 × 10−3 M−1 s−1 c = 4.6 × 10−3 M−1 s−1 |
Conjugate nucleophilic addition & double adduct |
kc = 3.6 × 10−2 M−1 s−1 |
| Trans-cinnamaldehyde |
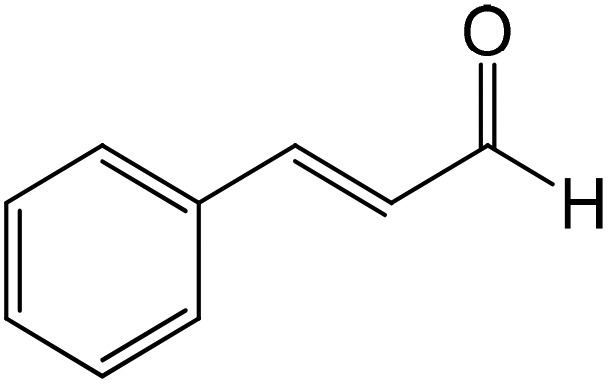
|
Nucleophilic addition/elimination | k = 4.7 × 10−3 M−1 s−1 | Conjugate nucleophilic addition & double adduct |
kc = 1 × 10−2 M−1 s−1 |
| Ethylene glycol dimethacrylate |
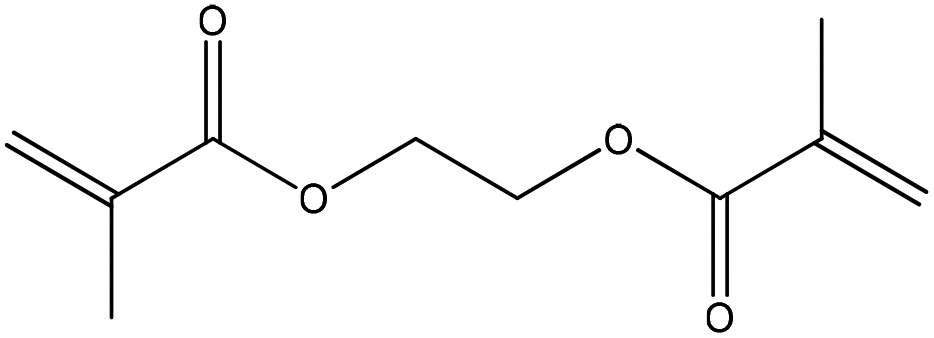
|
Conjugate nucleophilic addition | k ∼ 2 × 10−6 M−1 s−1 | Conjugate nucleophilic addition | Ndd |
| Phenylacetaldehyde |
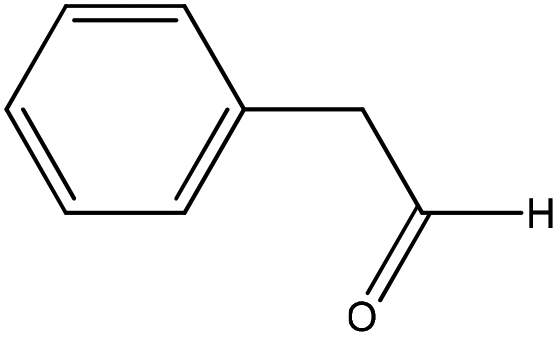
|
Nucleophilic addition/elimination | k = 0.04 M−1 s−1 | No reaction | Nd (no reaction) |
| 2,4-Dimethyl-3-cyclohexenecarboxaldehyde |
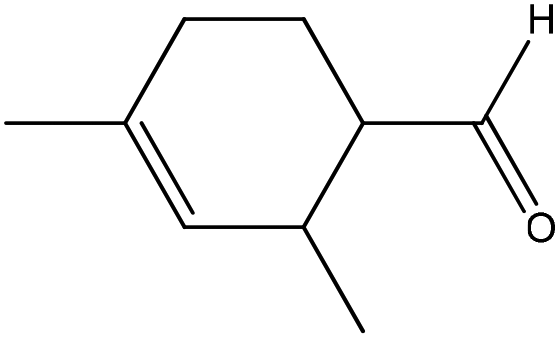
|
Nucleophilic addition/elimination | k = 0.03 M−1 s−1 | No reaction | Nd (no reaction) |
For phthalic anhydride, reaction with n-butylamine followed a nucleophilic acyl substitution mechanism; this reaction was fast and even with the lowest concentrations of reactants it had proceeded to completion before the first NMR spectrum had been recorded. Addition of 1-butanethiol to a semi-aqueous solution of phthalic anhydride also resulted in the fast disappearance of the phthalic anhydride. However, analysis of the data in this case was complicated by the behavior of phthalic anhydride in this solvent, in which three chemically-related species were identified in the 1H NMR spectrum prior to the addition of nucleophile. These species were relatively stable in solution as the sample composition in this solvent changed very little over a 30 minute period. The NMR data were consistent with the three species being: (i) the anhydride, (ii) a second symmetrical molecule: the dicarboxylate (i.e. phthalic acid) and (iii) a non-symmetrical molecule, assigned as phthaloyl monophosphate56 (Fig. 3). On addition of 1-butanethiol, the NMR signals from both the anhydride and the asymmetric species had disappeared before the first NMR measurement, demonstrating the fast reactivity. The NMR signals from the reaction product were consistent with a nucleophilic acyl substitution adduct; the formation of this being rationalized via a phthaloyl monophosphate intermediate (in equilibrium with phthalic anhydride) in the phosphate buffered reaction mixture (Fig. 3). The phthalic acid remained unchanged during this reaction.
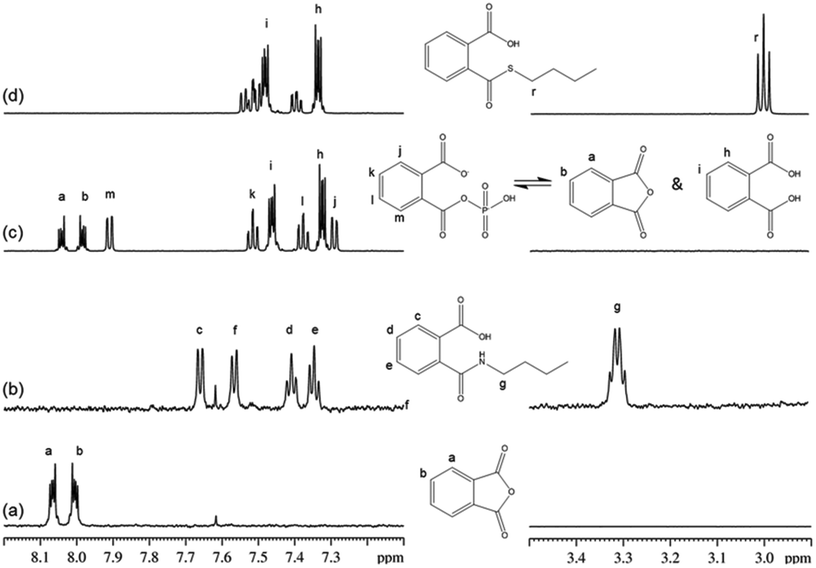 | ||
| Fig. 3 1H NMR spectra and assignments for phthalic anhydride and reaction products. (a) Phthalic anhydride in acetonitrile; (b) reaction product of phthalic anhydride and n-butylamine; (c) phthalic anhydride dissolved in 50% acetonitrile/50% phosphate buffer pH9; (d) reaction product of phthalic anhydride and 1-butanethiol, note phthalic acid did not react. | ||
The NMR results confirmed that both 3,4-dihydrocoumarin and phenyl benzoate also reacted with n-butylamine via a nucleophilic acyl substitution mechanism; the rate constant for 3,4-dihydrocoumarin was approximately two orders of magnitude greater than for phenyl benzoate. The reactivity of these two compounds towards the thiol differed. Phenyl benzoate reacted slowly under the standard conditions with the NMR data confirming that this reaction followed a nucleophilic acyl substitution mechanism. Conversely, 3,4-dihydrocoumarin demonstrated no significant reactivity (i.e. no adduct formation) in the presence of 1-butanethiol. The 1H NMR spectrum of 3,4-dihydrocoumarin in the semi aqueous solvent of the thiol reaction medium contained additional aromatic proton NMR signals; indicative of a small amount of ester hydrolysis prior to the addition of thiol. These putative hydrolysis product signals continued to increase with time in the presence of 1-butanethiol.
The reaction of trans-2-decenal with n-butylamine resulted in the concurrent formation of two distinct products; the NMR data for these two products were consistent with them being a nucleophilic addition/elimination adduct and a ‘nucleophilic addition/elimination plus conjugate nucleophilic addition’ double adduct (Fig. 4). The reaction of this compound with 1-butanethiol also resulted in the concurrent production of at least two distinct products. The NMR data (Fig. 4) contained evidence for one of these being a conjugate nucleophilic addition product (identified via a distinct aldehyde proton signal at approximately 9.6 ppm), and the second being a double adduct (NMR signals consistent with a proton on a carbon also bonded to sulphur and oxygen atoms). The appearance of an olefinic proton signal in the first few minutes of reaction (which decreased in intensity from 4 minutes onwards) indicated a potential short-lived intermediate in the formation of this double adduct. The NMR data show that the reactions of trans-2-decenal with nucleophiles clearly did not follow a single mechanistic pathway and consequently these reactions cannot be represented by a single rate constant value. A value for the rate of trans-2-decenal consumption was, however, readily obtained from the NMR data.
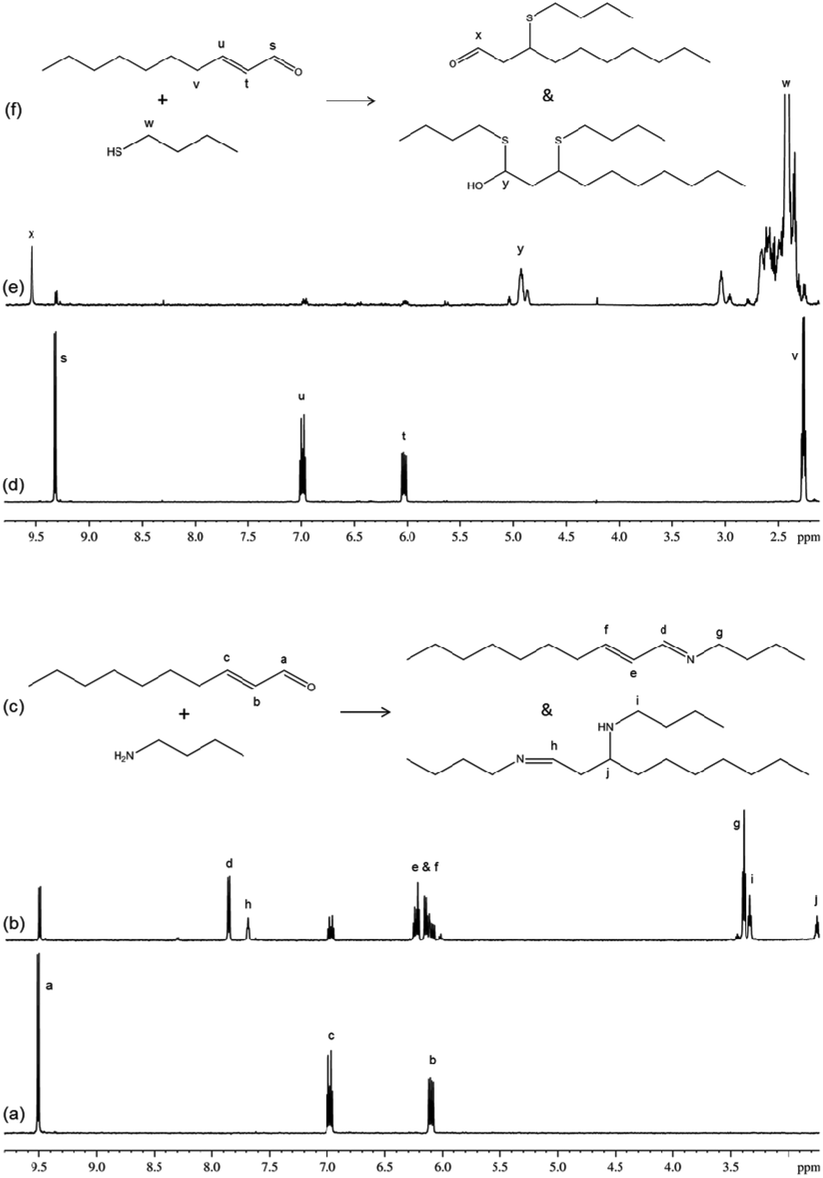 | ||
| Fig. 4 1H NMR spectra of indicative groups from trans-2-decenal and its reaction products (a) 1H NMR spectra of trans-2-decenal in acetonitrile; (b) 1H NMR spectra of trans-2-decenal plus n-butylamine, showing signals from reaction products; (c) structures of reactants and products; (d) 1H NMR spectra of trans-2-decenal in 50% acetonitrile/50% phosphate buffer pH9; (e) 1H NMR spectra of trans-2-decenal plus 1-butanethiol, highlighting indicative signals from reaction products; (f) structures of reactants and products. | ||
Cinnamaldehyde followed the same mechanistic pathways as trans-2-decenal, reacting by nucleophilic addition/elimination with n-butylamine (in this case with just a single reaction product) and reacting with 1-butanethiol to produce a product tentatively identified as a conjugate nucleophilic addition adduct. In the latter reaction the phenyl group proton NMR signals were readily observed for the major product, however, corresponding non-aromatic proton signals for this species were not unequivocally identified due to signal overlap.
Consequently this structure remained only tentatively assigned as a conjugate nucleophilic adduct. NMR signals from an additional reaction product were also present which had only 20% of a unit proton area compared to the major component and must therefore have represented a different reaction product, most likely a double adduct, as observed previously.30 As there was more than one reaction product, the rate constant derived from the NMR data was again for the depletion of the starting material.
Ethylene glycol dimethacrylate reacted very slowly with n-butylamine; new signals appearing in the NMR spectrum were consistent with a conjugate nucleophilic addition product. A correspondingly low rate constant was calculated from the data. The reaction of ethylene glycol dimethacrylate with 1-butanethiol was also very slow under the standard reaction conditions. However, in this reaction mixture an emulsion immediately formed which slowly separated into two clear phases and hence did not allow a rate constant for this reaction to be derived.
Phenylacetaldehyde reacted with n-butylamine to produce a nucleophilic addition/elimination reaction product. Reactions of this form are relatively straightforward to follow by NMR spectroscopy as the distinct aldehyde proton signal (at 9.7 ppm in this instance) decreases with the concomitant increase of an olefin signal approximately 2 ppm upfield (∼7.7 ppm) representing the nucleophilic addition/elimination product.
One of the benefits of using NMR spectroscopy for reaction monitoring is that the purity of reactants can be assessed immediately prior to, and under the same solution conditions of, the reaction. For 2,4-dimethyl-3-cyclohexenecarboxaldehyde this proved necessary as the commercial sample of 2,4-dimethyl-3-cyclohexenecarboxaldehyde was shown to be impure, being comprised of 3 species: (in approximate proportions of 82:15:3, each containing both aldehyde & olefin groups); plus a further component (present at approximately 10% of the total) with an olefin but no aldehyde group. For this sample the reaction of the major component (presumed to be 2,4-dimethyl-3-cyclohexenecarboxaldehyde) with n-butylamine, to produce a nucleophilic addition/elimination product, was monitored.
Both phenylacetaldehyde and 2,4-dimethyl-3-cyclohexenecarboxaldehyde were shown to react by a nucleophilic addition/elimination mechanism with n-butylamine. Neither of these compounds reacted with 1-butanethiol; this was not a surprising observation as thiol nucleophiles do not readily undergo nucleophilic addition/elimination reactions.
Discussion
It has been demonstrated that chemical reactions representative of haptenation MIEs can be characterized and measured using NMR spectroscopy. For conditions that give clearly resolved NMR signals from reactants and products, reaction mechanisms, rates and rate constants can be determined in a straightforward manner. The chemical information content of NMR spectroscopy data can provide direct confirmation of reaction mechanism, knowledge of which is a prerequisite for the determination of reaction rate constants and for developing understanding of MIEs.The mechanistic and reactivity data presented here are essentially consistent with previously published data from peptide reactivity assays30,33–37,39 and hence demonstrate that the technique can be used to support the study of skin sensitization AOP chemicals within TT21C1 approaches. Peptide reactivity assays are generally run over 24 h with the electrophile in excess, hence they differ from the NMR methodology described here in which reactions were generally followed for less than one hour and with excess nucleophile. Although there was a general consistency in terms of relative reactivity and putative reaction mechanisms, some differences were observed between the different approaches. These could, in most part, be ascribed to differences in reaction conditions, most notably solution pH, which will have a potentially large impact on, for example, rates of reactions involving thiol groups where the thiolate ion (S−) is the reactive species.
In the case of phthalic anhydride, the strong reactivity observed with the amine nucleophile was consistent with that observed previously for the skin sensitization AOP.34,35,37 Strong reactivity was also observed with the thiol nucleophile in the current study, in contrast to the apparent variable reactivity of phthalic anhydride with thiol nucleophiles previously reported.34,35,37 The differences are most likely explained by differences in reaction conditions; for example, lack of reactivity with thiol nucleophiles in carbonate buffer could be due to the absence of phosphate necessary to form a reactive phthaloyl monophosphate intermediate. It is also likely that the rapid conversion of phthalic anhydride to the un-reactive phthalic acid occurs under lower pH conditions. The previously observed reactivity of phthalic anhydride with glutathione but not with a cysteine nucleophile peptide34,35 could have been a result of disulphide formation or the formation of covalent adducts via the amine rather than the thiol moiety of the glutathione. Similarly, the single and double acyl adducts previously observed in the reaction of phthalic anhydride with a multiple nucleophile peptide AcNKKCDLF37 were most likely formed via the ε-amino groups of the lysine side chains. The results presented here provide a chemical explanation for the previously observed reactivity of phthalic anhydride34,35,37 and demonstrate the value of using NMR spectroscopy for gaining mechanistic insights into chemical reactivity, in this instance for the skin sensitization AOP.
The usefulness of NMR spectroscopy for shedding light on reaction mechanisms was demonstrated for 3,4-dihydrocoumarin which was clearly shown to react with n-butylamine via a nucleophilic acyl substitution mechanism whereas, in a previous study,30 it had not been possible to identify the putative mechanism definitively from mass spectrometry data.
Trans-2-decenal provided an example of an electrophile that reacted with nucleophiles via more than one mechanism. The reaction with n-butylamine produced a nucleophilic addition/elimination adduct and a ‘nucleophilic addition/elimination plus conjugate nucleophilic addition’ double adduct concurrently; it was not possible to establish whether the single adduct was a precursor of the double adduct or whether the double adduct arose via a different mechanism. In a previous study30 only a double adduct was observed; this could have been a consequence of the longer timescale of reactivity (24 h) in the earlier work. A single adduct and a double adduct was also formed with 1-butanethiol; in this case the single adduct was formed by conjugate nucleophilic addition. Previously30 the formation of a sulphone was reported for this reaction; however, there was no evidence for such a product in the NMR data.
Ethylene glycol dimethacrylate reacted very slowly with both n-butylamine and 1-butanethiol. This was consistent with previous reported reactivity with glutathione and a lysine containing peptide.34 The apparently higher reactivity of ethylene glycol dimethacrylate with a cysteine peptide in the previous work could possibly be a consequence of an increased proportion of reactive thiolate ion under the lower pH conditions.
In previous studies30,36,37,39 some dimerisation of thiol substrates through disulphide formation was observed after incubation with certain test chemicals. In the current work, disulphide dimers of 1-butanethiol were observed at low levels (<0.5% of the thiol) in the pH 9 buffer (with or without compounds) and this level increased with time but only to a maximum of less than 5% of the thiol concentration after 18 h. Therefore, for all of the test compounds in this study, for the one hour reaction period, the oxidation of thiols to form disulphide dimers was not a significant contributor to the overall reactivity.
The generation of rate constants under standard reaction conditions provides for accurate reaction rate data, such as required in TT21C approches for mathematical toxicokinetic modelling of MIEs for AOP-based risk assessments,23 in which a key task for a haptenation MIE is that of predicting how the free concentration of a hapten varies with time at the site of (protein) reaction. Accurate, standardised reaction rate data from NMR experiments could also be used comparatively to provide a ‘ranking’ of chemicals in order of reactivity for risk assessments.40 For comparative purposes, reaction conditions must be constant; this can readily be achieved for NMR measurements applied to chemicals and reactions in which both reactants and products are soluble (in either aqueous or organic solvents) and for which reaction is via a single mechanism. For more complex reactions, which perhaps more closely resemble in vivo reactivity, it may still be possible to obtain a full set of rate constants provided the individual reaction mechanisms can be determined; although it should be noted that, in such instances, NMR spectroscopy can readily provide relative reaction rate information through the monitoring of reactant depletion in a manner analogous to peptide depletion assays used for the skin sensitization AOP.
A further fundamental advantage of the NMR technique is that reactivity measurements can be recorded over a range of solution conditions, ranging from aqueous, or semi-aqueous (to mimic in vivo conditions), through to non-aqueous solvents; providing a greater range of solute solubility options than other techniques and bioassays. Consequently, the use of NMR can extend the scope of materials to be assessed for reactivity by providing opportunities for obtaining chemical understanding of organic compounds with low aqueous solubility. NMR spectroscopy is therefore complementary to other techniques and can provide additional information to answer questions that cannot answered using peptide depletion assays alone.
NMR spectroscopy is often perceived as an insensitive analytical technique; however, the concentrations of reactants in the NMR experiments were similar to those used in peptide reactivity assays30,34–37 and spectrophotometric assays.32,33 The concentrations of reactants in MIEs in vivo remain unknown; indeed it is possible that very localized high concentrations of electrophile are required at the site of reaction to initiate a MIE. We have shown that NMR spectroscopy can be used to readily obtain reaction rate constants which, as they contain a concentration term, provide a means of comparing relative reactivity of compounds that is applicable to any absolute concentrations. Therefore, although NMR data may have been obtained at reactant concentrations greater than may occur at the MIE level in vivo, the rate constant data derived from the NMR data are applicable at the MIE level.
Uniquely, NMR data can also provide, as evidenced in the example of 2,4-dimethyl-3-cyclohexenecarboxaldehyde, a direct measure of material purity and solubility immediately prior to reaction; such information cannot be directly obtained by other techniques. NMR data also report directly on whether any side reactions, such as reactions with buffer components, have occurred. Furthermore, NMR measurements are direct and non-destructive; they can be made in ‘real-time’, they do not affect reaction outcomes and concentrations of chemicals at similar levels to those used in peptide depletion assays can be detected. The property of NMR data that is of greatest importance for the generation of reaction rates, however, is the intrinsically quantitative nature of NMR spectroscopy data; allowing both reactant and product concentrations to be measured directly and concomitantly, without need to determine response factors.
Using chemical reactivity data obtained by NMR spectroscopy, differentiation between adduct formation through the reaction of a test compound with a nucleophile and, for example, the oxidation or hydrolysis of the test compound is possible; such distinctions are not always possible with other assays which could, therefore, provide misleading data. The potential for the detection of decomposition of unstable test materials and/or reaction products, as well as the detection and identification of adducts, which may be “missed” by other techniques, is also enhanced by using NMR spectroscopy, as this technique inherently detects and measures soluble organic components of a reaction mixture directly, simultaneously and non-selectively.
Conclusions
We have demonstrated, through the straightforward measurement of reaction rates and the simultaneous characterisation of reaction mechanisms, the potential of NMR spectroscopy for providing direct mechanistic insights into MIEs that are driven by chemical reactions. Such NMR spectroscopy analyses can be of particular benefit in early stage screening of compounds for reactivity, prior to subsequent in-depth investigations in line with TT21C approaches. The mechanistic insights that can be obtained from NMR spectroscopy are complementary to, and can provide additional information above that available from existing techniques for providing chemical reactivity information used to inform toxicological risk assessments.Acknowledgements
The authors would like to acknowledge helpful discussions with Nora Aptula and Beate Nicol.References
- National Research Council, Toxicity Testing in the 21st Century: a Vision and a Strategy, The National Academic Press, Washington, DC, 2007 Search PubMed.
- F. Reynolds, C. Westmoreland and J. Fentem, Biochemist, 2014, 36, 19–25 CAS.
- G. T. Ankley, R. S. Bennett, R. J. Erickson, D. J. Hoff, M. W. Hornung, R. D. Johnson, D. R. Mount, J. W. Nichols, C. L. Russom, P. K. Schmieder, J. A. Serrrano, J. E. Tietge and D. L. Villeneuve, Environ. Toxicol. Chem., 2010, 29, 730–741 CrossRef CAS PubMed.
- V. J. Kramer, M. A. Etterson, M. Hecker, C. A. Murphy, G. Roesijadi, D. J. Spade, J. A. Spromberg, M. Wang and G. T. Ankley, Environ. Toxicol. Chem., 2011, 30, 64–76 CrossRef CAS PubMed.
- M. Vinken, Toxicology, 2013, 312, 158–165 CrossRef CAS PubMed.
- K. E. Tollefsen, S. Scholz, M. T. Cronin, S. W. Edwards, J. de Knecht, K. Crofton, N. Garcia-Reyero, T. Hartung, A. Worth and G. Patlewicz, Reg. Toxicol. Pharmacol., 2014, 70, 629–640 CrossRef PubMed.
- M. P. Dent, P. L. Carmichael, K. C. Jones and F. L. Martin, Environ. Int., 2015, 83, 94–106 CrossRef CAS PubMed.
- C. Westmoreland, P. Carmichael, I. Malcomber, G. Maxwell, O. Price and J. Fentem, 21st Century Safety Science and Non-Animal Approaches at Unilever, AltTox.org, 2013 Search PubMed.
- T. Allen, J. Goodman, S. Gutsell and P. Russell, Chem. Res. Toxicol., 2014, 27, 2100–2112 CrossRef CAS PubMed.
- OECD, Report of the Workshop on Using Mechanistic Information in Forming Chemical Categories. 138. ENV/JM/MONO(2011)8, OECD Environment, Health and Safety Publications Series on Testing and Assessment, 2011.
- J. G. Bessems and N. P. Vermeulen, Crit. Rev. Toxicol., 2001, 31, 55–138 CrossRef CAS PubMed.
- V. R. Thompson and A. P. DeCaprio, Chem. Res. Toxicol., 2013, 26, 1263–1271 CrossRef CAS PubMed.
- A. V. Wisnewski, M. Mhike, J. M. Hettick, J. Liu and P. D. Siegel, Toxicol. In Vitro., 2013, 27, 662–671 CrossRef CAS PubMed.
- F. Gerberick, M. Aleksic, D. Basketter, S. Casati, A. T. Karlberg, P. Kern, I. Kimber, J. P. Lepoittevin, A. Natsch, J. M. Ovigne, C. Rovida, H. Sakaguchi and T. Schultz, ATLA, Altern. Lab. Anim., 2008, 36, 215–242 CAS.
- S. D. Cohen, N. R. Pumford, E. A. Khairallah, K. Boekelheide, L. R. Pohl, H. R. Amouzadeh and J. A. Hinson, Toxicol. Appl. Pharmacol., 1997, 143, 1–12 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- R. Mah, J. R. Thomas and C. M. Shafer, Bioorg. Med. Chem. Lett., 2014, 24, 33–39 CrossRef CAS PubMed.
- D. Asturiol and A. Worth. The Use of Chemical Reactivity Assays in Toxicity Prediction. EUR24870 EN-2011. 2011. JRC Scientific and Technical Reports.
- K. Chan and P. J. O'Brien, J. Appl. Toxicol., 2008, 28, 1004–1015 CrossRef CAS PubMed.
- D. W. Roberts, B. F. Goodwin, D. L. Williams, K. Jones, A. W. Johnson and J. C. Alderson, Food Chem. Toxicol., 1983, 21, 811–813 CrossRef CAS PubMed.
- E. L. Roggen, Basic Clin. Pharmacol. Toxicol., 2014, 115, 32–40 CrossRef CAS PubMed.
- J. A. Schwobel, Y. K. Koleva, S. J. Enoch, F. Bajot, M. Hewitt, J. C. Madden, D. W. Roberts, T. W. Schultz and M. T. Cronin, Chem. Rev., 2011, 111, 2562–2596 CrossRef CAS PubMed.
- C. MacKay, M. Davies, V. Summerfield and G. Maxwell, ALTEX, 2013, 30, 473–486 CrossRef PubMed.
- D. Krewski, D. Acosta Jr., M. Andersen, H. Anderson, J. C. Bailar III, K. Boekelheide, R. Brent, G. Charnley, V. G. Cheung, S. Green Jr., K. T. Kelsey, N. I. Kerkvliet, A. A. Li, L. McCray, O. Meyer, R. D. Patterson, W. Pennie, R. A. Scala, G. M. Solomon, M. Stephens, J. Yager and L. Zeise, J. Toxicol. Environ. Health, Part B, 2010, 13, 51–138 CAS.
- M. K. Dennehy, K. A. Richards, G. R. Wernke, Y. Shyr and D. C. Liebler, Chem. Res. Toxicol., 2006, 19, 20–29 CrossRef CAS PubMed.
- E. Parkinson, P. Boyd, M. Aleksic, R. Cubberley, D. O'Connor and P. Skipp, Toxicol. Sci., 2014, 42, 239–249 CrossRef PubMed.
- M. Tzouros and A. Pahler, Chem. Res. Toxicol., 2009, 22, 853–862 CrossRef CAS PubMed.
- A. Vila, K. A. Tallman, A. T. Jacobs, D. C. Liebler, N. A. Porter and L. J. Marnett, Chem. Res. Toxicol., 2008, 21, 432–444 CrossRef CAS PubMed.
- D. C. Liebler, Chem. Res. Toxicol., 2008, 21, 117–128 CrossRef CAS PubMed.
- M. Aleksic, E. Thain, D. Roger, O. Saib, M. Davies, J. Li, A. Aptula and R. Zazzeroni, Toxicol. Sci., 2009, 108, 401–411 CrossRef CAS PubMed.
- A. O. Aptula, G. Patlewicz, D. W. Roberts and T. W. Schultz, Toxicol. In Vitro, 2006, 20, 239–247 CrossRef CAS PubMed.
- I. Chipinda, R. O. Ajibola, M. K. Morakinyo, T. B. Ruwona, R. H. Simoyi and P. D. Siegel, Chem. Res. Toxicol., 2010, 23, 918–925 CrossRef CAS PubMed.
- I. Chipinda, W. Mbiya, R. A. Adigun, M. K. Morakinyo, B. F. Law, R. H. Simoyi and P. D. Siegel, Toxicology, 2014, 315, 102–109 CrossRef CAS PubMed.
- G. F. Gerberick, J. D. Vassallo, R. E. Bailey, J. G. Chaney, S. W. Morrall and J. P. Lepoittevin, Toxicol. Sci., 2004, 81, 332–343 CrossRef CAS PubMed.
- G. F. Gerberick, J. D. Vassallo, L. M. Foertsch, B. B. Price, J. G. Chaney and J. P. Lepoittevin, Toxicol. Sci., 2007, 97, 417–427 CrossRef CAS PubMed.
- A. Natsch, H. Gfeller, M. Rothaupt and G. Ellis, Toxicol. In Vitro, 2007, 21, 1220–1226 CrossRef CAS PubMed.
- A. Natsch and H. Gfeller, Toxicol. Sci., 2008, 106, 464–478 CrossRef CAS PubMed.
- A. Natsch, R. Emter and G. Ellis, Toxicol. Sci., 2008, 107, 106–121 CrossRef PubMed.
- D. W. Roberts and A. Natsch, Chem. Res. Toxicol., 2009, 22, 592–603 CrossRef CAS PubMed.
- D. W. Roberts, A. O. Aptula, G. Patlewicz and C. Pease, J. Appl. Toxicol., 2008, 28, 443–454 CrossRef CAS PubMed.
- S. Flavien, D. Smith and A. Codina, Org. Process Res. Dev., 2012, 61–64 Search PubMed.
- A. O. Aptula and D. W. Roberts, Chem. Res. Toxicol., 2006, 19, 1097–1105 CrossRef CAS PubMed.
- M. D. Barratt, D. A. Basketter and D. W. Roberts, Toxicol. In Vitro., 1994, 8, 823–826 CrossRef CAS PubMed.
- E. Eder, D. Henschler and T. Neudecker, Xenobiotica, 1982, 12, 831–848 CrossRef CAS PubMed.
- E. Eder, C. Deininger and D. Muth, Mutagenesis, 1991, 6, 261–269 CrossRef CAS PubMed.
- E. Helaskoski, O. Kuuliala and K. Aalto-Korte, Contact Dermatitis, 2009, 60, 214–221 CrossRef CAS PubMed.
- T. J. McCarthy, E. P. Hayes, C. S. Schwartz and G. Witz, Fundam. Appl. Toxicol., 1994, 22, 543–548 CrossRef CAS PubMed.
- A. Natsch, H. Gfeller, F. Kuhn, T. Granier and D. W. Roberts, Chem. Res. Toxicol., 2010, 23, 1913–1920 CrossRef CAS PubMed.
- G. Y. Patlewicz, D. A. Basketter, C. K. Pease, K. Wilson, Z. M. Wright, D. W. Roberts, G. Bernard, E. G. Arnau and J. P. Lepoittevin, Contact Dermatitis, 2004, 50, 91–97 CrossRef CAS PubMed.
- D. W. Roberts, Contact Dermatitis, 1987, 17, 281–289 CrossRef CAS PubMed.
- D. W. Roberts, A. O. Aptula and G. Patlewicz, Chem. Res. Toxicol., 2007, 20, 44–60 CrossRef CAS PubMed.
- K. M. Venables, Br. J. Ind. Med., 1989, 46, 222–232 CAS.
- M. Vocanson, M. Valeyrie, A. Rozieres, A. Hennino, F. Floc'h, A. Gard and J. F. Nicolas, Contact Dermatitis, 2007, 57, 361–364 CrossRef CAS PubMed.
- H. Wang, Y. Mao, A. Y. Chen, N. Zhou, E. J. LaVoie and L. F. Liu, Biochemistry, 2001, 40, 3316–3323 CrossRef CAS PubMed.
- P. W. Atkins, Physical Chemistry, Oxford University Press, Oxford, 1978 Search PubMed.
- G. O. Andres, A. M. Granados and R. H. de Rossi, J. Org. Chem., 2001, 66, 7653–7657 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |