
Polymer-mediated ternary supramolecular interactions for sensitive detection of peptides†
Mahalia A. C.
Serrano‡
,
Huan
He‡
,
Bo
Zhao
,
Rajasekhar R.
Ramireddy
,
Richard W.
Vachet
* and
S.
Thayumanavan
*
Department of Chemistry, University of Massachusetts, Amherst, MA 01003-9336, USA. E-mail: rwvachet@chem.umass.edu; thai@chem.umass.edu
First published on 2nd November 2016
Abstract
A combination of donor–acceptor and electrostatic interactions in a three-component supramolecular system has been shown to form the basis for selective and sensitive detection of peptides. Different substituents in the polymer and the detection matrix were compared to demonstrate that the favorable donor–acceptor interactions explain the observed signal enhancement. The ternary supramolecular interactions discovered in this work are enabled by the self-packing behavior of amphiphilic homopolymers and their ability to mediate interactions between the detection matrix and peptide that facilitate sensitive detection of peptides.
Executing a specific recognition event through ternary supramolecular interactions is quite common in Nature. Prominent examples involve adaptor proteins and enzymes that carry out coupling reactions precisely by first recognizing and binding to two different biomolecules and bringing them together.1 Artificial molecular assemblies with such capabilities are relatively limited.2,3 In this manuscript, we show that amphiphilic homopolymers can act as a ‘molecular glue’ to effectively and specifically bring together two complementary molecules using disparate non-covalent interactions with each of the molecules. We show that these subtle supramolecular interactions manifest themselves in a macroscopic clustering phenomenon, which in turn has a significant effect on the highly sensitive detection of analyte peptides by matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS) (Fig. 1). Formation of densely packed morphologies in the solid state through donor–acceptor supramolecular interactions has found applications in electronic materials,4 but the work here is the first study, to our knowledge, that tackles its role in mass spectrometric signal enhancement for sensitive molecular detection. Others have shown that matrix additives, analyte labelling or derivatization, and/or surface modification of the detection target can improve MALDI-MS signals.5 Our approach is different in that it relies on the inherent properties of the analyte and requires no such types of derivatizations or target surface modifications. Moreover, the same agent used for extraction enhances analyte detection. We report here on how both selectivity and sensitivity can be achieved through favourable ternary supramolecular interactions that can be tuned by variations in the structure of the interacting components.
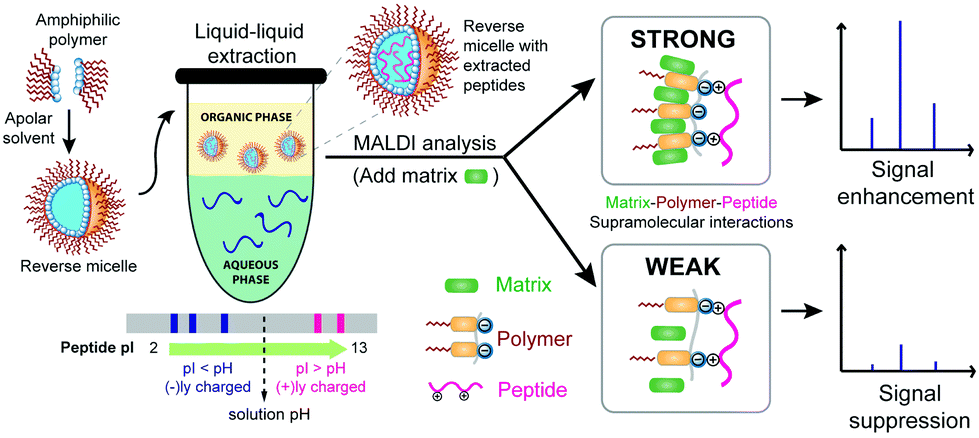 | ||
| Fig. 1 Schematic representation of amphiphilic polymers self-assembling into reverse micelles for the selective extraction of peptides based on pI. The ternary interactions involving the amphiphilic homopolymer assembly with the analyte peptide through ionic complementarity and with the detection matrix through aromatic donor–acceptor interactions are shown. Stronger ternary molecular interactions result in significant enhancement in the peptide detection signal. | ||
Recently, our group has developed amphiphilic homopolymers (Chart 1) that form kinetically-trapped reverse micelle assemblies in apolar solvents.6 We have shown previously that these assemblies are capable of selectively sequestering specific peptides based on their isoelectric point (pI) and solution pH.7,8 This electrostatically driven interaction causes peptides, which are otherwise insoluble in apolar solvents, to be selectively moved to the apolar phase of a biphasic mixture (Fig. S1†). The versatility of this method has been illustrated by the highly sensitive MALDI-MS detection of peptides in complex mixtures such as protein digests.7 Furthermore, the general applicability of this approach in real samples has been demonstrated by the detection of pM concentrations of selected peptides in serum.8 These low detection limits are achieved not only from the selective enrichment that occurs upon extracting the peptides from an aqueous phase to an organic phase, but also from more than an order of magnitude enhancement observed in the MALDI ion signal after extraction. While it is understood that the amphiphilic homopolymer, exemplified by the polydialkoxystyrene (PDAS) as shown in Chart 1, might play some role in this enhancement,7b the underlying mechanism for such an enhancement is not understood, and is thus the subject of this study. Elucidating the molecular interactions responsible for this signal enhancement paves the way for improving detection sensitivities and can have tremendous impact on the analysis of complex peptide mixtures and the detection of low-level peptide and protein biomarkers.
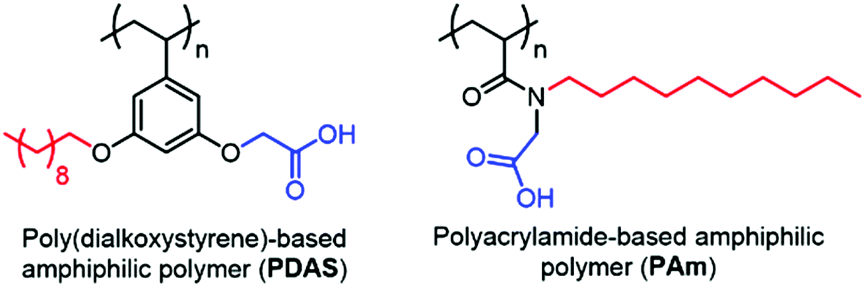 | ||
| Chart 1 Chemical structures of poly(dialkoxystyrene)-based and polyacrylamide-based amphiphilic polymers. | ||
The first clue about the molecular basis for this signal enhancement came from the observation that a polyacrylamide, PAm as shown in Chart 1, does not provide any signal enhancement (Fig. 2). This polymer has been shown to form reverse micelle assemblies,9 similar to those formed by PDAS. We find that this tendency to form an assembly translates to selective sequestration of peptides based on pI in a manner similar to that of PDAS (characterization of the polymers in terms of reverse micelle formation and selectivity of extraction are shown in Fig. S6–S12†). The key difference is that there is MS signal enhancement only in the case of PDAS (Fig. 2). Interestingly, there is an associated macroscopic cluster formation in the ternary mixture formed between PDAS, the analyte peptide, and the MALDI matrix, α-cyano-4-hydroxycinnamic acid (CHCA) that does not occur in the presence of PAm (Fig. 2). This led us to hypothesize that the electron-rich aromatic ring of the PDAS polymer is engaged in a donor–acceptor interaction with the electron-poor aromatic ring of CHCA, while the polymer's carboxylate unit is involved in interaction with the electrostatically complementary peptide. Although these interactions can presumably exist in the presence of just the monomer bearing the aromatic ring and the carboxylate moiety, the results from experiments where the monomer was added to the peptide–matrix mixture in place of its corresponding polymer reveal no significant enhancement in the signal, suggesting that the multivalent structure of the polymer is needed for signal enhancement (Fig. S2†). Likewise, using the polymer as merely a matrix additive without doing any extraction does not lead to signal enhancement, as we demonstrated previously.7a Thus, upon extraction the polymer structure acts as an effective mediator between the matrix molecule and the peptide.
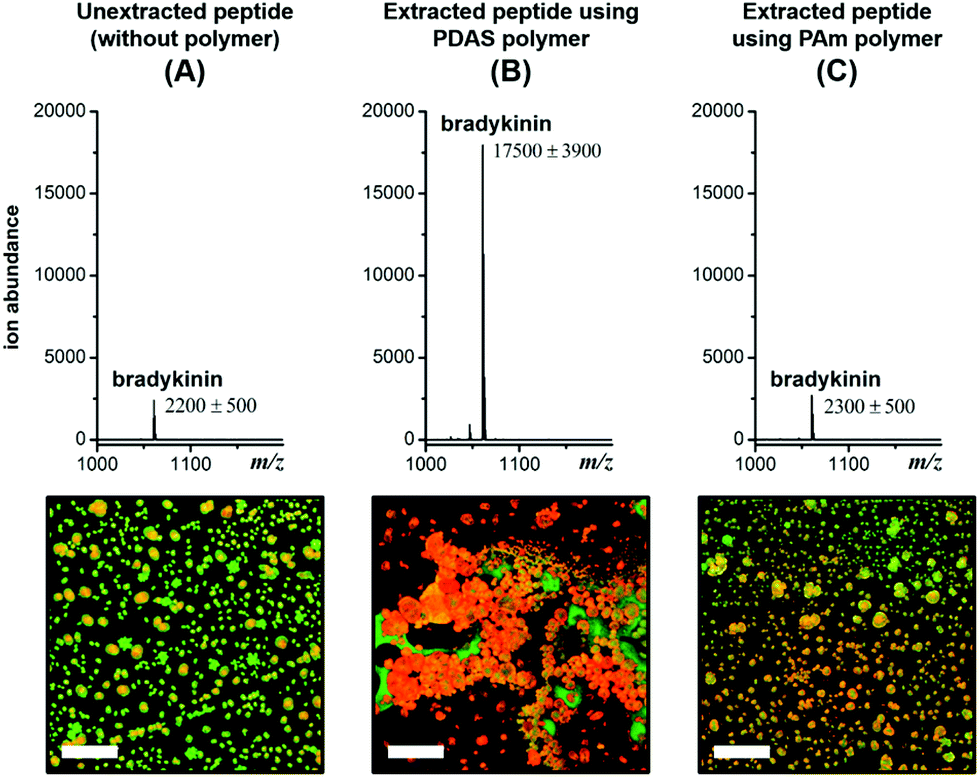 | ||
Fig. 2 MALDI mass spectra for unextracted 1 μM* bradykinin (A), and 10 nM* bradykinin (RPPGFSPFR, m/z 1060.6) extracted using the PDAS polymer (B), and the PAm polymer (C). Corresponding fluorescence images showing the degree of clustering and co-crystallization of the TAMRA-labeled bradykinin peptide (red) and CHCA matrix (green). Scale bar = 100 μm. Fluorescence images of individual and merged channels to illustrate co-localization of the three components are shown in Fig. S3.† *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100-fold higher concentration in the unextracted sample (1 μM) relative to the extracted sample (10 nM) to account for the 100-fold enrichment during extraction. 100-fold higher concentration in the unextracted sample (1 μM) relative to the extracted sample (10 nM) to account for the 100-fold enrichment during extraction. | ||
To test this hypothesis, we first varied the matrix from CHCA to α-cyanocinnamic acid (CCA) and α-cyano-4-chlorocinnamic acid (CClCA). By replacing the –OH group with –H or –Cl, the electron-withdrawing ability of the substituent is varied in the order –OH < –H < –Cl. Upon extraction and detection of the positively charged bradykinin peptide (RPPGFSPFR), we find that the MS signal is enhanced by 20% with CCA as the matrix, and more than 100% with CClCA, compared to that of CHCA as the matrix (Fig. 3). Moreover, corresponding fluorescence images reveal that clustering of the extracted peptide is the densest with the CClCA matrix (Fig. 3). This observation is consistent with the hypothesis that an electron-rich PDAS polymer would interact better with more electron-poor matrices, which in turn causes more clustering and affords greater signal enhancement.
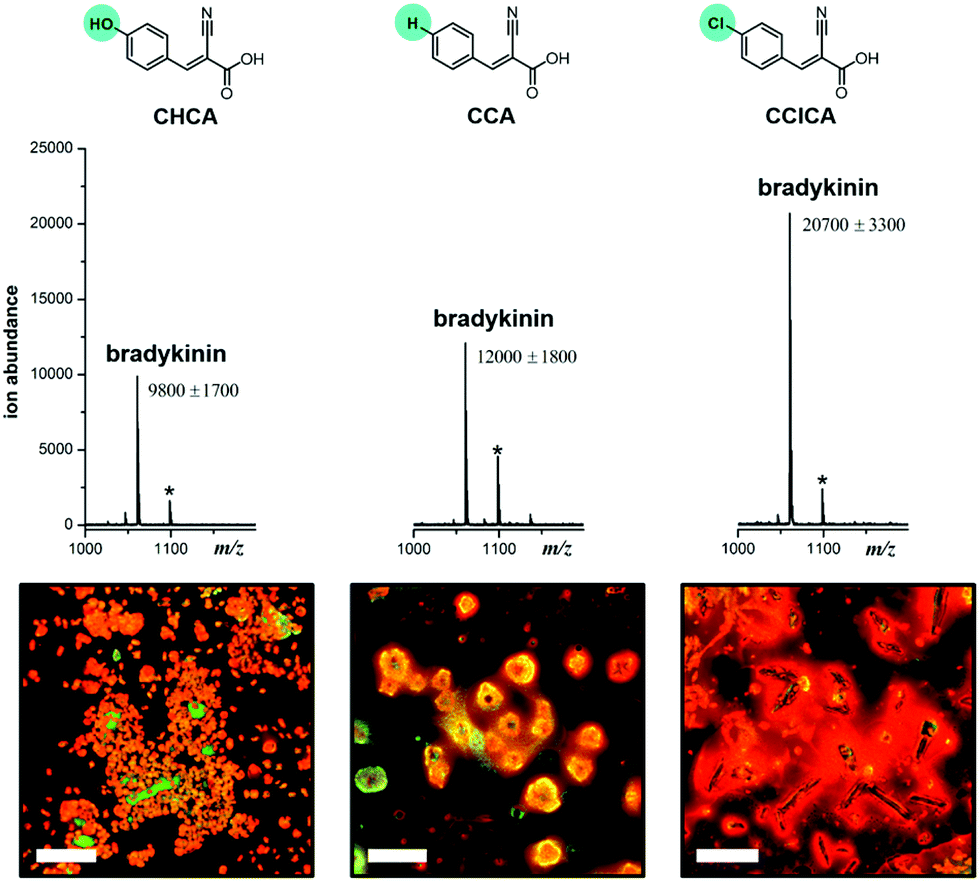 | ||
| Fig. 3 Chemical structures of the different MALDI matrices in the order of increasing electron deficiency on the ring, along with the corresponding MALDI mass spectra and fluorescence images obtained for 100 nM bradykinin extracted using the PDAS polymer (Chart 1) and analysed using these MALDI matrices. Peaks with asterisks are potassium adducts of the peptides, [M + K]+. Fluorescence images of individual and merged channels of the peptide with the matrix to illustrate co-localization are shown in Fig. S4.† | ||
To further test our hypothesis, we investigated how varying the electron density on the aromatic ring of the polymer affects MALDI signal enhancement and hotspot formation, while keeping the MALDI matrix constant. In this case, we introduced aromatic rings with electron-donating or electron-withdrawing substituents in an amphiphilic homopolymer, shown as PTLAm in Fig. 4 (synthetic details in the ESI†). These polymers also form reverse micelles and selectively extract positively-charged peptides (Fig. S6 and S7†). Consistent with our hypothesis, using the PTLAm polymer with the electron-donating methoxy substituent (PTLAm-OMe) resulted in a two-fold enhancement in the MALDI signal relative to the unextracted peptide (Fig. 4). Interestingly, the signal was significantly suppressed, when PTLAM-NO2, with its electron-withdrawing nitro group, was used. The unsubstituted PTLAm polymer (PTLAm-H) did not show a significant difference in the MALDI signal relative to the unextracted one. Furthermore, clustering of the peptide and matrix into hotspots is apparent in the presence of the PTLAm-OMe, but not for either PTLAm-H or PTLAm-NO2, suggesting that a polymer with an electron-rich aromatic ring is needed for hotspot formation and signal enhancement.
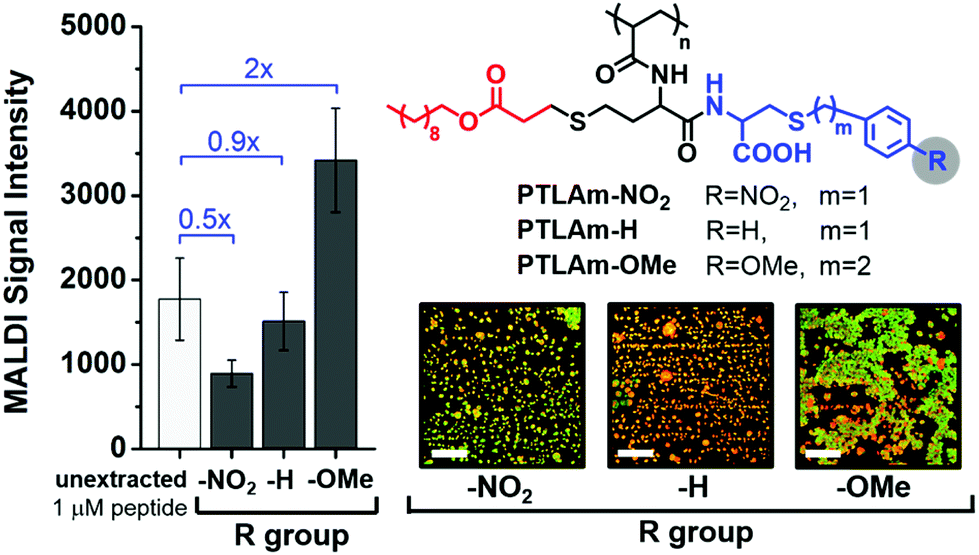 | ||
| Fig. 4 MALDI signal intensity of 10 nM bradykinin extracted by using polythiolactone amide (PTLAm)-based carboxylate polymers with different R groups compared with 1 μM of unextracted bradykinin (100-fold higher concentration to account for 100-fold enrichment during extraction). Fluorescence images show the degree of clustering of the peptide (red) and CHCA matrix (green) after extraction using each PTLAm polymer. Fluorescence images of individual and merged channels to illustrate co-localization of the three components are shown in Fig. S5.† | ||
While the PTLAm polymers provided access to amphiphilic polymers with aromatic units, we were interested in a scaffold that could provide the opportunity to conveniently introduce subtle changes in the aromatic unit, such as a wide range of electron-donating or -withdrawing substituents on the ring, while keeping the overall structure consistent with the PAm structure as shown in Chart 1. Accordingly, we designed and synthesized (see the ESI† for details) a new set of amphiphilic homopolymers with a polyacrylamide (PAm) scaffold (Chart 2). These polymers provide the opportunity to further test our hypothesis that a ternary supramolecular interaction between the polymer, matrix and the peptide is essential for enhanced detection capabilities. Six polymers with different electron-withdrawing and electron-donating substituents were synthesized (Chart 2). All these polymers self-assemble into nanometer-sized reverse micelles (Fig. S8–S10†) and selectively extract oppositely charged peptides (Fig. S11 and S12†), just like the PDAS and PAm polymers.
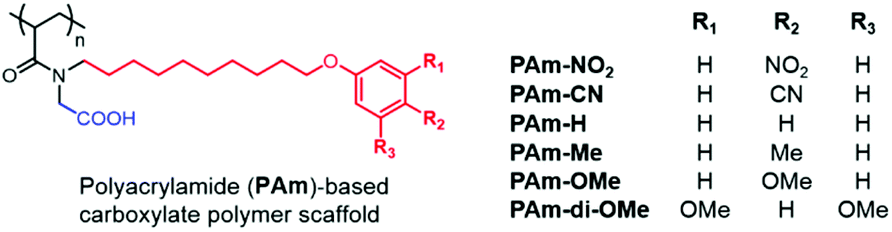 | ||
| Chart 2 Chemical structures and labels of the polyacrylamide-based amphiphilic homopolymers with the carboxylate hydrophilic moiety and variable hydrophobic units containing electron-donating or electron-withdrawing functionalities. | ||
The degree of matrix–peptide clustering and MALDI signal enhancement after extraction was monitored by fluorescence microscopy and MS, respectively. Through mere visual inspection (Fig. 5), it is evident that a higher degree of clustering is present in samples extracted by using polymers with electron-donating groups (PAm-H, PAm-Me, PAm-OMe, PAm-di-OMe) compared with those extracted by using polymers with electron-withdrawing groups (PAm-NO2 and PAm-CN). At first glance, we would expect PAm-H to behave similarly to the control (unextracted) sample, because of the –H substituent. Note, however, that the base aromatic ring is based on an alkoxyarene, making PAm-H a relatively electron-rich aromatic ring. We quantitatively assessed the degree of hotspot formation by measuring the fluorescence intensity per area using the software ImageJ (Fig. S14†) and correlated this with the MALDI-MS signal obtained for samples extracted by using each polymer. Fig. 5 reveals that an approximately 3-fold enhancement in the signal is obtained when polymers with electron-donating substituents are used for extraction, whereas no appreciable enhancement in the signal is observed when samples are extracted by using polymers with electron-withdrawing groups. Correspondingly, higher degrees of hotspot formation were calculated for samples extracted using PAm-H, PAm-Me, PAm-OMe and PAm-di-OMe compared to PAm-NO2 and PAm-CN. This correlation shows that the clustering phenomenon indeed translates to enhanced signals in MALDI-MS. Because good co-crystallization between the peptide and the MALDI matrix is also crucial to obtaining strong MS signals, we measured the degree of co-localization between the TAMRA-labeled peptide and the MALDI matrix for each of the fluorescence micrographs (Fig. S15†) and found an index of correlation of >0.800 for all images. This high correlation suggests very good co-localization between the peptide and the matrix regardless of the polymer used. Thus, the signal enhancement is not simply due to better analyte/matrix co-crystallization. Instead, the formation of “hotspots” through the use of polymers with favorable donor–acceptor interaction with the MALDI matrix is key to obtaining signal enhancement.
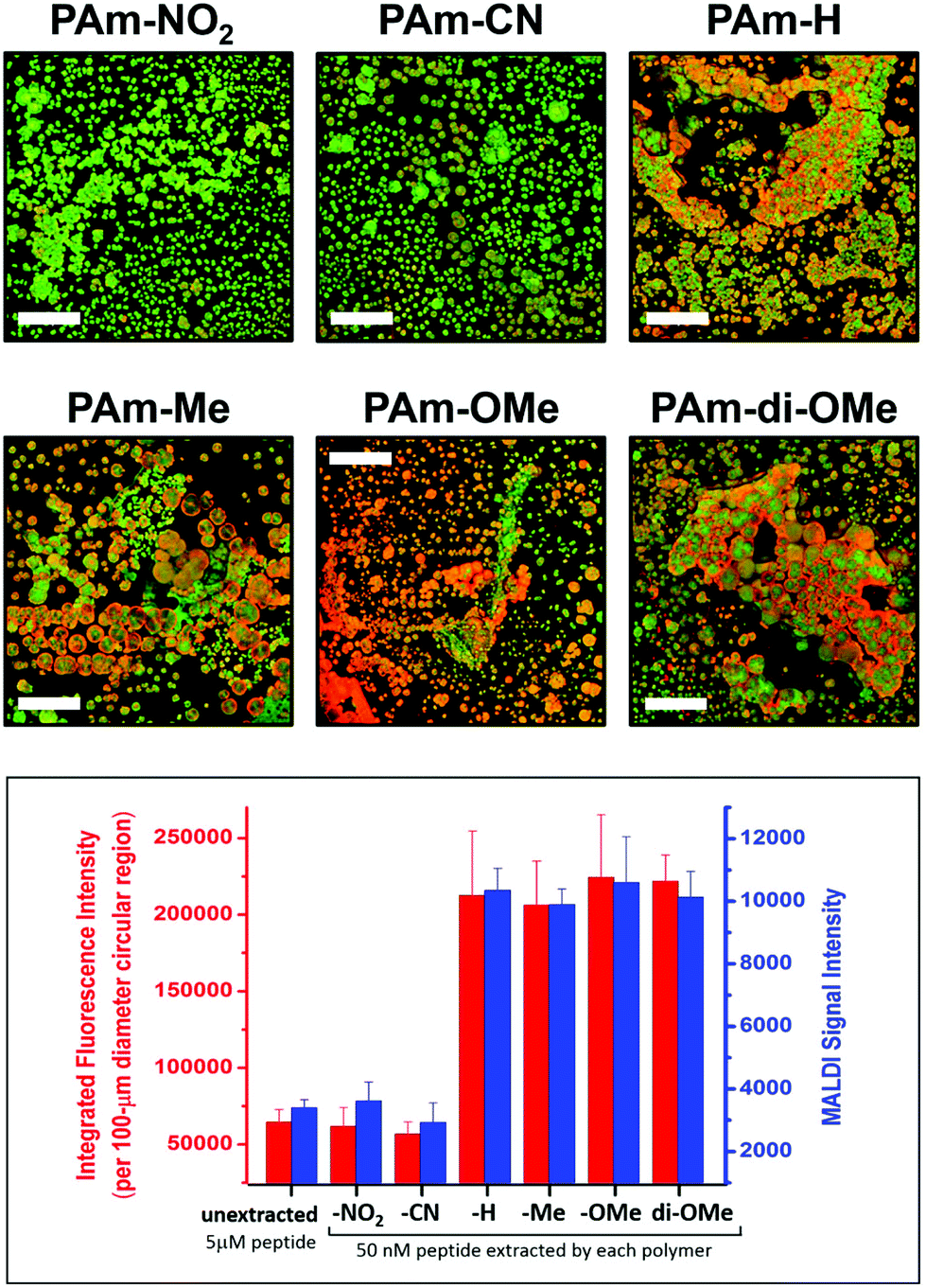 | ||
| Fig. 5 Fluorescence images (top panels; scale bar = 100 μm) showing the degree of clustering of the peptide (red) and CHCA matrix (green) upon extraction using the PAm-based polymer indicated, and correlation of the degree of clustering with the MALDI-MS signal (bottom graph). Each blue bar represents the average ± SEM of 90 spectra obtained from 3 replicate extractions with 3 spots each. Fluorescence images of individual and merged channels to illustrate co-localization of the three components are shown in Fig. S13.† | ||
Conclusions
In summary, we have investigated the molecular basis for the observed signal enhancement in peptide detection by MALDI-MS, after extraction using reverse micelles of amphiphilic polymers. Through systematic variations in the electronic character of the aromatic ring in the amphiphilic polymer and matrix, we find that favourable donor–acceptor interactions are necessary to form a ternary assembly of the polymer, peptide, and matrix, which is essential for producing peptide-rich zones that maximize the ion signal in the mass spectrum. An important point to recognize from all the data presented here is that although the electrostatic interaction between the polymer and the peptide is required to selectively extract peptides from the aqueous phase into the organic phase, these interactions alone are not sufficient to cause hotspot formation and signal enhancement. All these polymers bear the carboxylate moiety that can conceivably cause the peptides to coalesce into hotspots, but evidently, a favourable aromatic donor–acceptor interaction between the polymer and the matrix is necessary for hotspots to form and signal enhancement to manifest. Thus, the ternary supramolecular interaction formed between the polymer and the peptide through electrostatics, and between the polymer and the matrix through aromatic donor–acceptor interactions, forms the basis for the selective enrichment and sensitive detection of peptides. Overall, these results reveal that amphiphilic polymers can self-pack in such a way that they mediate favourable interactions between peptides and the matrix. Such interactions are reminiscent of proteins that upon folding, position other biomolecules in just the right way to perform chemistry that is impossible without the ternary interaction.Acknowledgements
This work was supported by the National Institutes of Health (R01 CA169140). M. A. C. S. was partially supported through the UMass Chemistry–Biology Interface (CBI) Fellowship Program (NIH NRSA T32 GM008515).Notes and references
- (a) L. N. Johnson, M. E. M. Noble and D. J. Owen, Cell, 1996, 85, 149 CrossRef CAS PubMed; (b) S. Cusack, Curr. Opin. Struct. Biol., 1997, 7, 881 CrossRef CAS PubMed; (c) D. C. Flynn, Oncogene, 2001, 20, 6270 CrossRef CAS PubMed; (d) A. J. Pratt and I. J. MacRae, J. Biol. Chem., 2009, 284, 17897 CrossRef CAS PubMed; (e) G. Meister, Nat. Rev. Genet., 2013, 14, 447 CrossRef CAS PubMed.
- For examples with heteroternary complexation in cucurbiturils, see: (a) S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320 CrossRef CAS PubMed; (b) H. J. Kim, J. Heo, W. S. Jeon, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 1526 CrossRef CAS; (c) E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, RSC Adv., 2012, 2, 1213 RSC.
- For specific examples of the self-assembly of dendrimers and polymers, see: (a) K. Moon, J. Grindstaff, D. Sobransingh and A. E. Kaifer, Angew. Chem., Int. Ed., 2004, 43, 5496 CrossRef CAS PubMed; (b) J. Zhang, R. J. Coulston, S. T. Jones, J. Geng, O. A. Scherman and C. Abell, Science, 2012, 335, 690 CrossRef CAS PubMed; (c) E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251 CrossRef CAS PubMed.
- (a) P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009 CrossRef CAS PubMed; (b) H. Li, S. Sun, S. Mhaisalkar, M. T. Zin, Y. M. Lam and A. C. Grimsdale, J. Mater. Chem. A, 2014, 2, 17925 RSC; (c) S. R. Peurifoy, C. X. Guzman and A. B. Braunschweig, Polym. Chem., 2015, 6, 5529 RSC.
- (a) Y. Fukuyama, R. Tanimura, K. Maeda, M. Watanabe, S. Kawabata, S. Iwamoto, S. Izumi and K. Tanaka, Anal. Chem., 2012, 84, 4237 CrossRef CAS PubMed; (b) H. Kuyama, K. Sonomura and O. Nishimura, Rapid Commun. Mass Spectrom., 2008, 22, 1109 CrossRef CAS PubMed; (c) A. Pashkova, E. Moskovets and B. L. Karger, Anal. Chem., 2004, 76, 4550 CrossRef CAS PubMed; (d) J. Li, H. Ma, X. Wang, S. Xiong, S. Dong and S. Wang, Rapid Commun. Mass Spectrom., 2007, 21, 2608 CrossRef CAS PubMed; (e) J.-S. Kim, J.-H. Kim and H.-J. Kim, Rapid Commun. Mass Spectrom., 2008, 22, 495 CrossRef CAS PubMed; (f) X. Qiao, L. Sun, L. Chen, Y. Zhou, K. Yang, Z. Liang, L. Zhang and Y. Zhang, Rapid Commun. Mass Spectrom., 2011, 25, 639 CrossRef CAS PubMed; (g) M. Schuerenberg, C. Luebbert, H. Eickhoff, M. Kalkum, H. Lehrach and E. Nordhoff, Anal. Chem., 2000, 72, 3436 CrossRef CAS PubMed; (h) Y. Xu, M. L. Bruening and J. T. Watson, Anal. Chem., 2004, 76, 3106 CrossRef CAS PubMed; (i) J. D. Dunn, E. A. Igrisan, A. M. Palumbo, G. E. Reid and M. L. Bruening, Anal. Chem., 2008, 80, 5727 CrossRef CAS PubMed.
- S. Basu, D. R. Vutukuri and S. Thayumanavan, J. Am. Chem. Soc., 2005, 127, 16794 CrossRef CAS PubMed.
- (a) M. Y. Combariza, E. N. Savariar, D. R. Vutukuri, S. Thayumanavan and R. W. Vachet, Anal. Chem., 2007, 79, 7124 CrossRef CAS PubMed; (b) N. Rodthongkum, Y. Chen, S. Thayumanavan and R. W. Vachet, Anal. Chem., 2010, 82, 3686 CrossRef CAS PubMed; (c) N. Rodthongkum, Y. Chen, S. Thayumanavan and R. W. Vachet, Anal. Chem., 2010, 82, 8686 CrossRef CAS PubMed.
- N. Rodthongkum, R. Ramireddy, S. Thayumanavan and R. W. Vachet, Analyst, 2012, 137, 1024 RSC.
- E. N. Savariar, S. V. Aathimanikandan and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 16224 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an01591c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |