
Assay for Listeria monocytogenes cells in whole blood using isotachophoresis and recombinase polymerase amplification†
Charbel
Eid
and
Juan G.
Santiago
*
Department of Mechanical Engineering, Stanford University, 440 Escondido Mall, Bldg 530, room 225, Stanford, CA 94305, USA. E-mail: juan.santiago@stanford.edu; Fax: +1 650-723-5689; Tel: +1 650-723-7657
First published on 1st December 2016
Abstract
We present a new approach which enables lysis, extraction, and detection of inactivated Listeria monocytogenes cells from blood using isotachophoresis (ITP) and recombinase polymerase amplification (RPA). We use an ITP-compatible alkaline and proteinase K approach for rapid and effective lysis. We then perform ITP purification to separate bacterial DNA from whole blood contaminants using a microfluidic device that processes 25 μL sample volume. Lysis, mixing, dispensing, and on-chip ITP purification are completed in a total of less than 50 min. We transfer extracted DNA directly into RPA master mix for isothermal incubation and detection, an additional 25 min. We first validate our assay in the detection of purified genomic DNA spiked into whole blood, and demonstrate a limit of detection of 16.7 fg μL−1 genomic DNA, the equivalent of 5 × 103 cells per mL. We then show detection of chemically-inactivated L. monocytogenes cells spiked into whole blood, and demonstrate a limit of detection of 2 × 104 cells per mL. Lastly, we show preliminary experimental data demonstrating the feasibility of the integration of ITP purification with RPA detection on a microfluidic chip. Our results suggest that ITP purification is compatible with RPA detection, and has potential to extend the applicability of RPA to whole blood.
Bacteremia is the presence of viable bacteria in the bloodstream, and this condition can be life-threatening.1,2 The gold standard for bacteremia diagnosis is blood culture. Though capable of resolving very low bacterial counts, this method requires trained personnel and specialized microbiology infrastructure, and typically takes several days to produce a result. The time-critical nature of certain bacterial infections makes bacterial cultures a suboptimal diagnostic approach.3 Nucleic acid amplification has gained traction in bacterial infection detection. The most widespread and adopted technique is polymerase chain reaction (PCR). PCR is highly sensitive, capable of detecting as little as a single copy of a bacterial genome. However, PCR requires extensive sample preparation, and is highly vulnerable to inhibitors.4,5
Recombinase Polymerase Amplification (RPA) is an isothermal amplification technique that is a promising alternative to PCR.6 RPA requires no thermal cycling and can be completed in less than 30 min. RPA is also likely more robust to inhibitors compared to PCR.7 For example, Kersting et al.8 demonstrated successful RPA in the presence of several known PCR inhibitors. Furthermore, RPA has been successfully demonstrated with minimal sample preparation in serum,8 saliva,9 and urine.10 Though RPA shares several similarities with other isothermal amplification methods, such as loop-mediated isothermal amplification (LAMP), recent studies indicate that RPA might be better suited for integration with complex samples.11
Despite these advantages, RPA is incompatible with whole blood, an important matrix in infectious disease diagnostics. Kersting et al.8 tested RPA in the presence of blood components like serum and hemoglobin, and showed successful amplification. However, experiments with whole blood were unsuccessful. They hypothesized that RPA inhibition was due to other blood components or the use of sodium fluoride (NaF) as anticoagulant. Rohrman et al.12 discussed the inhibitory effect of large quantities of background DNA present in 50–100 μL of white blood cells, and suggested selective capture of target DNA would improve assay sensitivity. Though the inhibitory effect of blood on RPA is consistently observed, the full set of causes is not yet established.
In this work, we leverage isotachophoresis (ITP) purification to enhance RPA compatibility with blood. ITP is an electrophoretic technique that preconcentrates and separates ions based on their electrophoretic mobilities.13,14 ITP uses a heterogeneous buffer system consisting of a high-mobility leading electrolyte (LE) and a low-mobility trailing electrolyte (TE) to create an interface with a sharp electric field gradient. Sample ions with effective mobilities greater than the TE (when in the TE buffer) and less than the LE (when in the LE buffer) focus at the TE–LE interface. ITP has been applied to the rapid extraction of nucleic acids from a variety of complex samples including blood,15 serum,16 urine,17 and cell culture.18 Further, ITP sample preparation of nucleic acids has been shown to be compatible with downstream detection.15,19–21 Until somewhat recently, nucleic acid purification assays using ITP were limited to small volumes on the order of 100 nL, due to geometrical constraints of (convenient) commercially available chips. Marshall et al.22 developed an injection-molded chip that processes 25 μL sample, greatly increasing processed volume and yield. We use the latter device in the current assay to increase sensitivity.
ITP-based sample preparation is a promising alternative to conventional solid-phase or liquid-phase extractions of nucleic acids. ITP can extract nucleic acids from complex sample without centrifugation, filtering, or other time-consuming steps. ITP provides an automated buffer exchange and preconcentration and elution of purified nucleic acid. Further, ITP has significant recovery efficiency when working with small amounts of DNA (sub-nanogram) compared with traditional SPE methods.15,19 These features make ITP well compatible to miniaturization and automation. Disadvantages of ITP-based purification include Joule heating limitations on sample volume23 and restrictions on lysing and elution chemistry imposed by the requirements of ITP (see Rogacs et al.24 for further discussion).
Borysiak et al.25 recently developed an assay combining ITP and LAMP for the detection of E. coli bacteria in milk. They used electromigration and heat-induced pressure driven flow to direct purified DNA into an amplification reservoir. Their assay demonstrated two orders of magnitude improvement over tube-based LAMP assays in milk. We know of no other work that combines ITP with isothermal amplification. We know of no other microfluidic assay (e.g., ITP) for RPA for blood.
We here combine rapid cell lysis, ITP purification, and RPA for the detection of inactivated L. monocytogenes cells in whole blood. Presence of L. monocytogenes cells in whole blood, known as Listeriosis, is a condition particularly hazardous for pregnant women and immunocompromised patients.26L. monocytogenes is a Gram-positive bacteria, and is difficult to lyse due to its thick peptidoglycan cell wall. This work differs from Borysiak et al., as we here address a more difficult to lyse Gram-positive bacteria from a significantly more complex sample, blood. We also employ a different isothermal amplification technique, and for simplicity and speed use ITP for all transport (e.g., no valving or pressure driven flows). Our results suggest that ITP purification can be integrated with RPA detection even for difficult-to-lyse species in whole blood.
Materials and methods
A schematic of our protocol is shown in Fig. 1. We performed two versions of the assay. First, we performed a controlled experiment where we spiked L. monocytogenes genomic DNA into whole blood. In the second, we spiked L. monocytogenes inactivated cells into blood. In both cases, we chemically lysed the blood using the HotSHOT lysis method, which relies on high pH (12.5–13) for rapid and effective lysis.27 We quenched and reduced this pH down to 8.1 with LE buffer, and loaded the 25 μL mixture into the microfluidic chip. We then applied electric field to initiate ITP. Following ITP purification, we pipetted the purified DNA from the extraction reservoir, and transferred directly to the RPA master mix for 25 min off-chip amplification with RPA.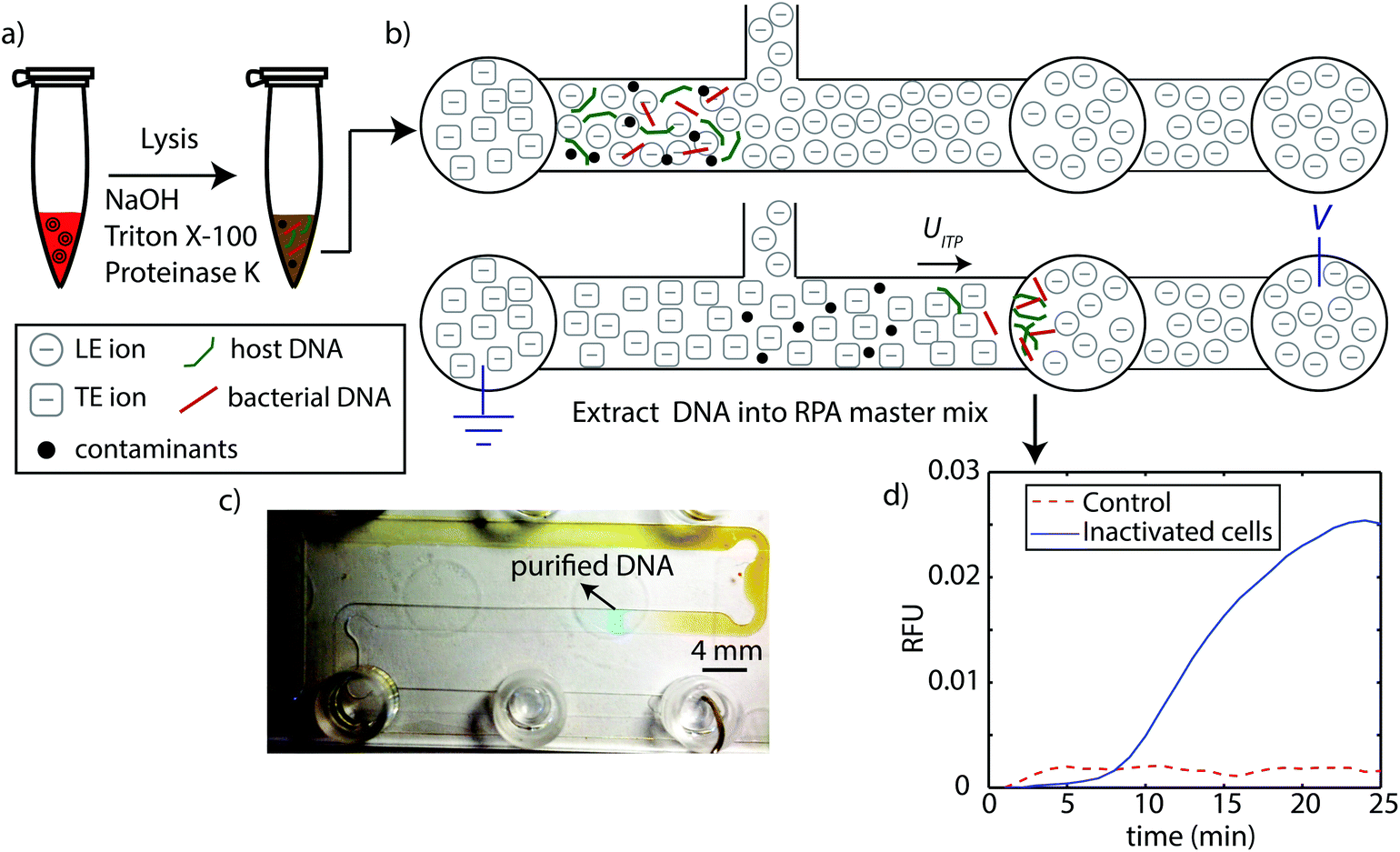 | ||
| Fig. 1 Schematic of ITP-RPA assay protocol summarizing lysis, extraction, and detection steps. (a) We lyse whole blood spiked with L. monocytogenes using NaOH, Triton X-100, and proteinase K. This method is rapid, extremely effective, and ITP-compatible. We quench the high pH (12.5–13) with LE buffer, then transfer and load the 25 μL mixture into the high-throughput microfluidic chip. (b) We apply electric field and initiate constant-current ITP purification of bacterial DNA and host DNA from whole blood. The current ITP extraction requires about 40 min to complete. (c) Image of ITP process in chip. The ITP zone (containing purified total nucleic acids) and its separation from contaminants in whole blood is clearly visible by eye. The ITP zone is tracked/visualized using AlexaFluor 647 as an ITP peak tracking dye. The chip contains channels with nominal widths of 2 mm, 0.15 mm depth, and a total channel length of 200 mm (see Marshall et al.22). (d) After purified DNA reaches the extraction reservoir, we pipette and transfer this fraction directly into standard RPA master mix. RPA is performed at 40 °C for 25 min in a thermal cycler. | ||
Whole blood and L. monocytogenes samples
Human blood samples from a healthy donor were collected in heparin tubes at the Stanford Blood Center. We prepared aliquots of 1 mL and stored them at −80 °C. Purified L. monocytogenes genomic DNA was purchased from ATCC (no. 19115D-5, ATCC, VA) and suspended in 1× Tris-EDTA buffer (Sigma-Aldrich, MO). Chemically-inactivated L. monocytogenes cells were purchased from ZeptoMetrix (NY) and suspended in their proprietary purified protein matrix.Lysis protocol
We lysed 2.5 μL of whole blood with suspended genomic DNA or L. monocytogenes cells with 2.5 μL lysis buffer consisting of 125 mM NaOH (Sigma-Aldrich, MO), 1% Triton X-100 (Sigma-Aldrich, MO), and 1 mg mL−1 proteinase K (Invitrogen, CA). We incubated the mixture for 2 min at room temperature when using genomic DNA, and for 10 min at 65 °C when using inactivated cells, as shown in Brewster and Paoli.28 We then added 20 μL of 40 mM HCl, 50 mM Tris (Sigma-Aldrich, MO), 0.1% PVP (Sigma-Aldrich, MO), and 1% Triton X-100 to reduce pH to ∼8 and dilute the cell lysate mixture 10-fold.Channel preparation
Prior to first use, we rinsed the channel with methanol, deionized (DI) water, NaOH, and HCl for 2 min each. Following each experiment, we rinsed the channels with 10% bleach for 10 min to minimize cross-contamination between experiments. This was followed by rinses with DI water, NaOH, HCl, and then DI water again, each for 2 min. We then dried the channel under vacuum for 10 min. We note that these inter-experiment washes would not be required for implementation of the injection molded chip as a disposable device.ITP extraction
We used a buffering LE (loaded into the LE reservoir) consisting of 200 mM HCl, 400 mM Tris, and 0.1% PVP (Sigma-Aldrich, MO). Our channel LE (loaded into the channel) consisted of 35 mM HCl, 70 mM Tris, and 0.1% PVP (Sigma-Aldrich, MO) (predicted pH = 8.1). Buffering TE consisted of 100 mM HEPES, 230 mM Tris, and 0.1% PVP (predicted pH = 8.4). At the start of each experiment, we loaded LE buffer into the extraction reservoir and allowed it to passively fill the separation channel. We then loaded the quenched lysate mixture into the TE reservoir and allowed it to fill the sample channel. Finally, we added 20 μL of buffering LE, channel LE, and buffering TE to each of the LE, extraction, and TE reservoirs, respectively. A more detailed description of the loading protocol is included in the ESI.†Our microfluidic device is an injection molded (cyclic olefin copolymer (COC) material) chip, and is described in detail by Marshall et al.22 Briefly, the chip footprint is 25 by 76 mm and contains channels with nominal widths of 2 mm, 0.15 mm depth, and a total channel length of 200 mm. The sample channel segment can has a loading volume of 25 μL and the separation channel a volume of 30 μL. We applied 105 μA of current using a Keithley 2410 current source (Keithley, OH) between the LE and TE reservoirs. We monitored ITP progress by using AlexaFluor 647 (Life Technologies, CA) simply as an ITP tracking dye. We performed experiments (not shown) to confirm the dye did not interfere with downstream RPA detection.
RPA
We used off-chip RPA to demonstrate the compatibility of ITP purification with RPA detection. We added 5 μL of extracted DNA from ITP to RPA mastermix (TwistDx, Cambridge, UK). Mastermix includes a pellet containing enzymes, a rehydration buffer, and magnesium acetate to activate the enzymes. We used the TwistAmp exo + ListeriaM kit.29 This kit provides real-time detection of target DNA. Exo probes are single stranded DNA that contain an abasic nucleotide analogue (tetrahydrofuran, or THF), as well as a fluorophore and a quencher in close proximity. Upon binding of an exo probe to a target DNA molecule, exonucleases cleave the probe at the THF position, and separate the fluorophore from quencher, resulting in significantly increased fluorescence. The secondary structure of exo probes grants them significantly increased specificity, thereby reducing formation of primer–dimer pairs and other false positive signals.30 Primers and probe were provided as part of the kit from TwistDx. Sequences were published in Schuler et al.31 Forward primer was TTCAATTTCATCCATGGCAC, reverse primer was CTTTGTAACCTTTTCTTGGC, and the exo probe sequence was [FAM]ACGCCAATCGAAAAGAAACACGC[BHQ-1]. We performed RPA using a real-time PCR thermocycler (MiniOpticon, Bio-Rad, CA), set at 40 °C for 25 min.On-chip RPA experiments
For on-chip RPA experiments, we performed ITP purification and RPA detection on commercially-available glass chips (NS12 Caliper chips, Perkin Elmer, CA). After elution of the ITP-focused DNA into the reservoir, we used a pipette to dispense 2 μL of 280 mM of magnesium acetate into the reservoir and mixed the contents using the same pipette. We achieved the required incubation temperature by placing an indium-tin-oxide (ITO) heater under the microfluidic device. The ITO heater was feedback-controlled using a microcontroller (mTCII, Cell MicroControls, VA) and set to 41 °C. We used an inverted epifluorescent microscope (Eclipse TE200, Nikon, NY) equipped with a fluorescein isothiocyanate (FITC) filter cube (XF100-2, Omega Optical, VT), and connected to a coupled charge device (CCD) camera (Coolsnap, Roper Scientific, Trenton, NJ). Illumination was provided by a 100 W short-arc mercury lamp (102DH, Ushio, Japan). We acquired images of the reservoir at regular intervals of 30 s during 10 min of incubation, and computed the area-integrated fluorescence intensity using custom MATLAB (R2012a, Mathworks, MA) scripts.Results and discussion
Assay operation
The ability to process a relatively large volume of sample is important in infectious disease detection assays, as bacterial or viral species are often present in trace amounts. As mentioned above, we used the chip of Marshall et al.22 which is an injection-molded device that can process 25 μL of sample. We used this device in our experiments to increase our assay's extraction efficiency and sensitivity. Scaling up microfluidic systems implies several challenges, including Joule heating, limited buffering capacity, and increased susceptibility to hydrodynamic pressure effects.23 As shown in Fig. 1, the chip contains features designed to support pH buffering of large volumes.32 A downstream-most reservoir accommodates a high-concentration (200 mM) Tris-HCl LE buffer (predicted pH = 8.1). After loading of sample channel section, the loading reservoir is filled with a high concentration (100 mM) TE buffer (predicted pH = 8.4). The LE loaded into the 30 μl separation channel has a concentration of 35 mM to achieve extraction time of approximately 40 min. In the ESI,† we analytically derive expected assay times. In their work, Marshall et al.22 used Pluronic F-127, a temperature-sensitive gel that is liquid at cold temperatures but solidifies into a gel at room temperature, in their two buffering reservoirs in order to minimize pressure-driven flow. While elegant, this approach requires the presence of nearby refrigerator and swift handling of the gel. We here successfully mitigated pressure-driven effects without Pluronic F-127, through careful loading and balancing volumes dispensed into the reservoirs.Lysis
Gram-positive bacteria are harder to lyse than Gram-negative species due to their thicker peptidoglycan wall. Methods such as gentle thermal lysis, detergent-based lysis, and even lysozyme lysis, are either ineffective or require overnight incubation. We therefore used a more aggressive chemical method to lyse the L. monocytogenes cells. We used an alkaline-based method (HotSHOT), which leverages high pH for rapid and effective lysis. This method was shown by Brewster and Paoli28 to be highly effective in the extraction of genomic DNA for L. monocytogenes, and shown by Rogacs et al.33 to be ITP-compatible. We included proteinase K in our lysis buffer to aid in lysis and degrade DNA-binding proteins. Consistent with the findings of Persat et al.,15 we found that using proteinase K was necessary for high extraction efficiency (we discuss this further in the ESI†). Our lysis buffer avoids chaotropic agents which are used in high ionic strengths (such as guanidine hydrochloride), and require significant dilution to integrate with ITP. We diluted the infected blood in our assay by 10-fold.Detection of purified genomic DNA in whole blood
We first demonstrated this assay using purified L. monocytogenes genomic DNA spiked into whole blood. Fig. 2 presents the results of these experiments. We varied genomic DNA concentration from 16.7 fg μL−1 to 16.7 pg μL−1. Our assay reliably detects as little as 16.7 fg μL−1 of genomic DNA in whole blood. This is a limit of detection (LOD) equivalent to 5 × 103 cells per mL (mL of original undiluted blood), or about 10–15 cells’ worth of genomic DNA loaded into our microfluidic chip. At higher concentrations, the fluorescent signal is relatively constant, as the probe is depleted within the 20 min of incubation. For comparison, we performed control experiments using genomic DNA spiked into whole blood without ITP purification. RPA was severely inhibited by whole blood. This severe inhibition is consistent with previous efforts to apply RPA to blood in the literature.8 Our results suggest that the current LOD is constrained by the dimensions of the current chip (10–15 cells’ worth of DNA) rather than RPA or ITP. At lower concentrations, and accounting for various losses due to adsorption, pipetting, and other handling steps, it is likely that no DNA would be present for detection, which imposes a lower bound on achievable LOD given our current chip and sample handling scheme. We theorize that sensitivity may be further improved by using a device that can process more sample. Rohrman et al.12 found in their experiments that background DNA found in white blood cells had an inhibitory effect on RPA detection. We have no evidence this is the case in our experiments, but hypothesize that sequence-specific target capture34,35 could also improve sensitivity.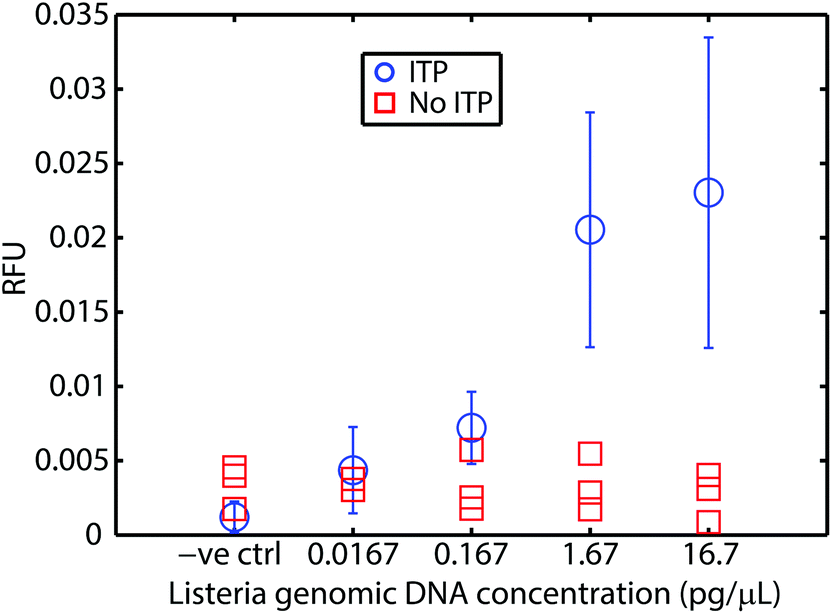 | ||
| Fig. 2 Results of our ITP-RPA assay using purified L. monocytogenes genomic DNA spiked into whole blood. We measured fluorescence following 20 min of incubation. Our assay (circles) has a limit of detection of 16.7 fg μL−1 of spiked genomic DNA in whole blood, which corresponds to approximately 5 × 103 cells per mL (mL of original undiluted blood). This is the equivalent of 10–15 cells’ worth of DNA loaded into the channel. Error bars represent 95% confidence on the mean (from Student's t-distribution). We also plot the results from the corresponding experiments using 10-fold diluted whole blood without ITP purification. As expected, whole blood inhibits RPA (red squares). Results plotted are for N = 6, 5, 10, 7, and 6 repetitions, respectively, in order of increasing concentration. | ||
Detection of L. monocytogenes cells in whole blood
For safety concerns (and limitations of our lab), we tested our approach using chemically-inactivated L. monocytogenes cells spiked into whole blood. These cells are rendered non-infectious by inactivation of proteins at the cell surface but are otherwise reported to be chemically intact.36Fig. 3 shows our assay bacteria in blood had a limit of detection of 2 × 104 cells per mL (mL of original undiluted blood), which corresponds to about 50–60 cells loaded into the microfluidic device. The comparison with parallel experiments wherein RPA was performed without ITP show clearly that RPA is severely inhibited by whole blood. Our assay's LOD for the bacteria lysate DNA in blood is approximately 4-fold worse than the genomic DNA in blood. We hypothesize two reasons for this. First, we suspect our lysis of this Gram-positive bacteria is incomplete. Second, we hypothesize that the cell solution (as received from ZeptoMetrix) may contain contaminants which may lower ITP extraction efficiency (e.g., anions which lower accumulation rate of DNA into ITP zone).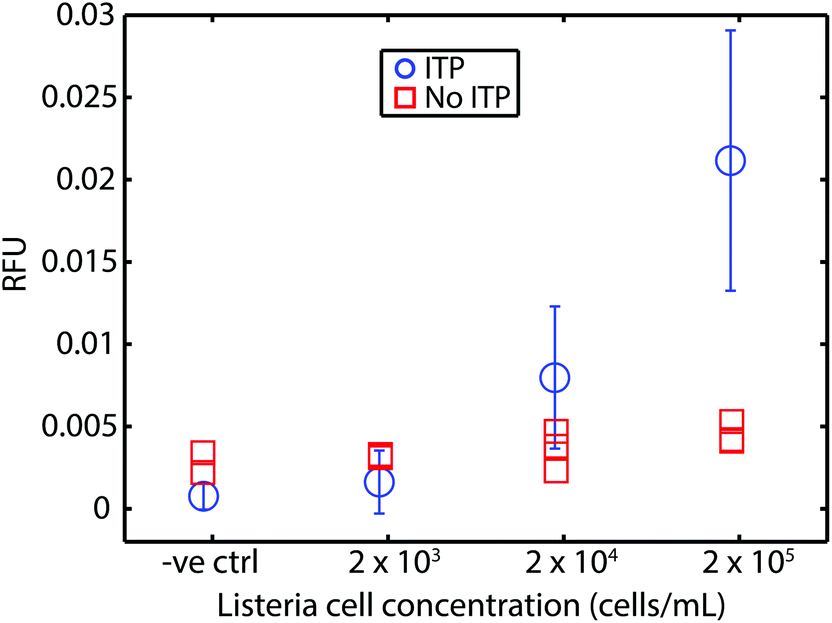 | ||
| Fig. 3 Results of our ITP-RPA assay using chemically inactivated L. monocytogenes cells spiked directly into whole blood. Cells are inactivated at the surface and are thus non-infectious, but otherwise intact. We measured fluorescence (RFU) after 20 min of incubation. We demonstrate the compatibility of our approach with RPA detection from bacterial cells. Our assay (circles) LOD of about 2 × 104 cells per mL (mL of original undiluted blood), which corresponds to about 50–60 cells loaded into our device. Uncertainty bars represent 95% confidence on the mean (Student's t-distribution). We also plot results from experiments without ITP purification. RPA is strongly inhibited by whole blood (red squares). The LOD using cells is approximately 4-fold worse than that of the experiments of Fig. 2. We suspect this is due to incomplete lysis and perhaps potential losses due to contaminants (e.g., in the initial cell solution). Results plotted are from N = 5, 4, 8, and 7 repetitions, respectively, in order of increasing concentration. | ||
The LOD of our assay is comparable to that of other microfluidic assays that used PCR to detect L. monocytogenes in cell culture37 and food and milk samples.38 Cocolin et al. published two studies in 1997 using PCR and agarose gel electrophoresis to detect L. monocytogenes in spiked blood samples.39,40 They achieved an LOD of 10 cells per mL, though these assays were not microfluidic and required up to 8 h of assay time. As noted earlier, the volume of sample processed by the chip used here imposes a lower bound on the achievable LOD in microfluidic assays like ours. We are confident that the LOD can be improved using a higher-throughput device (e.g., processing 200 μL samples versus the current 25 μL). Our results nevertheless suggest that ITP purification is compatible with RPA detection of Gram-positive species from whole blood samples. Lastly, we hypothesize that ITP purification of blood samples and RPA can be combined into a single multi-step process on a single chip.
Toward on-chip integration of ITP and RPA
We here present preliminary experiments toward exploring the feasibility of detection of on-chip RPA. We performed a limited set of experiments wherein we purified DNA on-chip and eluted the ITP peak containing purified DNA into a microfluidic reservoir. In Fig. 4a, we show images of the reservoir prior to and after RPA. We found that fluorescence increased significantly due to RPA, indicating successful amplification. In Fig. 4b, we compare on-chip amplification with data obtained from MiniOpticon thermal cycler. For these experiments, we used 2.5 pg μL−1 purified L. monocytogenes genomic DNA. We normalized each curve by its maximum value, and plot versus amplification time. The two curves have similar shape suggesting that on-chip RPA detection is feasible and can be performed without the use of a dedicated real-time PCR machine. Though these results are promising, there remain several challenges with automating RPA detection on microfluidic devices. Evaporation, though not as significant as in PCR assays, remains an issue. Another challenge is that the current protocol requires a magnesium acetate dispensing and mixing step, which would require further development to be automated.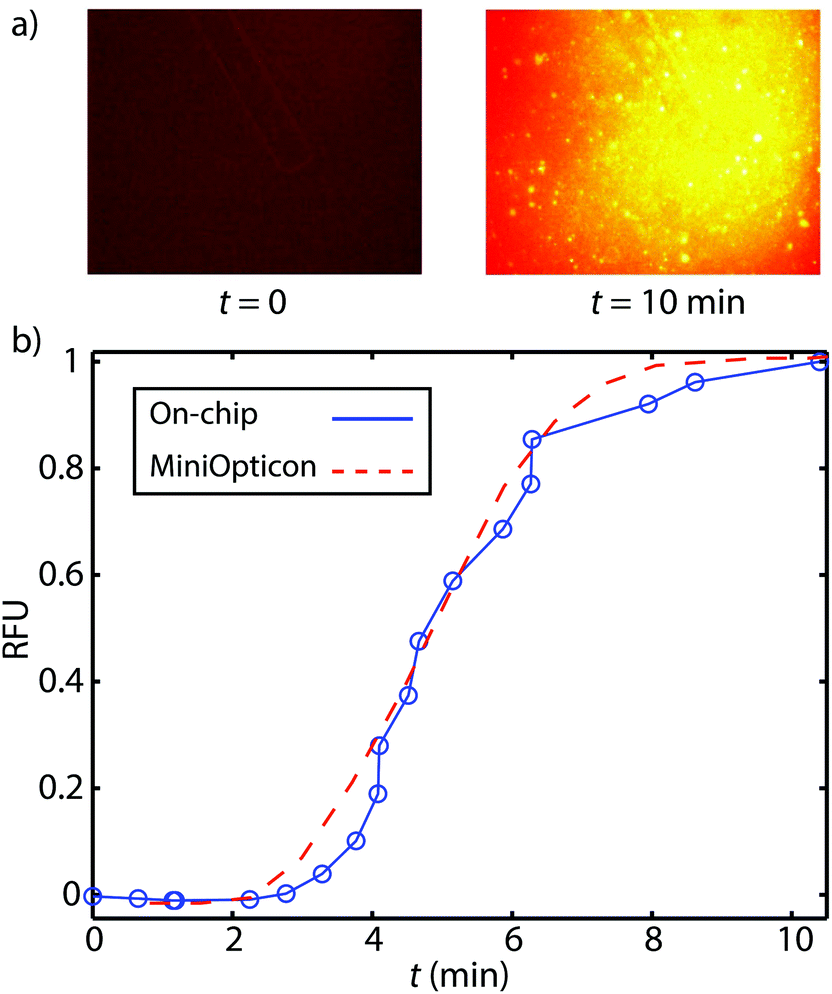 | ||
| Fig. 4 Feasibility demonstration of on-chip amplification using RPA. (a) Images of fluorescence signal within the chip reservoir before and after RPA. Fluorescence increases significantly after amplification. (b) Comparison of on-chip amplification results with those obtained using a commercial PCR thermal cycler, using 2.5 pg μL−1 purified L. monocytogenes genomic DNA. We normalize each curve by its maximum value, and plot versus amplification time. We find good agreement between the shape and time scale of both curves, indicating successful RPA amplification and suggesting feasibility of on-chip RPA detection. | ||
Conclusion
We demonstrated a novel assay for the lysis, extraction, and detection of L. monocytogenes bacteria in whole blood. We used an ITP-compatible lysis method capable of lysing difficult-to-lyse, Gram-positive bacteria. We performed two versions of the assay: first, ITP purification of total DNA from whole blood starting with bacterial DNA spiked into whole blood, and, second, ITP purification starting with L. monocytogenes spiked into whole blood. Our lysis, extraction, and on-chip purification are completed in a total of less than 50 min (about 40 min of that time on chip), and requires minimal user intervention. We then transferred purified DNA to a standard off-chip RPA assay. The LOD for bacterial DNA spiked into blood was 16.7 fg μL−1 (corresponding to about 5 × 103 bacterial cells per ml). The LOD for bacteria spiked into blood was 2 × 104 cells per mL. We attribute the latter higher LOD to imperfect lysing and perhaps lowering of ITP extraction efficiency. Assay sensitivity could be further improved by improving lysis protocol, using a higher-throughput microfluidic device, or perhaps incorporating species-specific target capture/enrichment into the assay. Our assay is amenable to automation and may have potential for point-of-care applications. For example, we hypothesize that both lysis and RPA and detection can be performed within chip reservoirs using an integrated heater.Our assay can be extended for the detection of other species, including Gram-positive and Gram-negative bacteria in whole blood. ITP purification has potential to expand the applicability of RPA to blood samples.
The authors would like to acknowledge support from the National Science Foundation and the industrial members of the Center for Advanced Design and Manufacturing of Integrated Microfluidics (NSF I/UCRC award number IIP-1362165).
Notes and references
- G. M. Bearman and R. P. Wenzel, Arch. Med. Res., 2005, 36, 646–659 CrossRef PubMed.
- H. Seifert, Clin. Infect. Dis, 2009, 48(Suppl 4), S238–S245 CrossRef PubMed.
- L. G. Reimer, M. L. Wilson and M. P. Weinstein, Clin. Microbiol. Rev., 1997, 10, 444–465 CAS.
- C. Schrader, A. Schielke, L. Ellerbroek and R. Johne, J. Appl. Microbiol., 2012, 113, 1014–1026 CrossRef CAS PubMed.
- J. Bessetti, Profiles in DNA, 2007, 9–10 Search PubMed.
- O. Piepenburg, C. H. Williams, D. L. Stemple and N. A. Armes, PLos Biol., 2006, 4, e204 Search PubMed.
- L. Lillis, J. Siverson, A. Lee, J. Cantera, M. Parker, O. Piepenburg, D. A. Lehman and D. S. Boyle, Mol. Cell. Probes, 2016, 30, 74–78 CrossRef CAS PubMed.
- S. Kersting, V. Rausch, F. F. Bier and M. von Nickisch-Rosenegk, Malar. J., 2014, 13, 99 CrossRef PubMed.
- A. Abd El Wahed, A. El-Deeb, M. El-Tholoth, H. Abd El Kader, A. Ahmed, S. Hassan, B. Hoffmann, B. Haas, M. A. Shalaby, F. T. Hufert and M. Weidmann, PLoS One, 2013, 8, e71642 CAS.
- K. Krolov, J. Frolova, O. Tudoran, J. Suhorutsenko, T. Lehto, H. Sibul, I. Mager, M. Laanpere, I. Tulp and U. Langel, J. Mol. Diagn., 2014, 16, 127–135 CrossRef CAS PubMed.
- C. Escadafal, J. T. Paweska, A. Grobbelaar, C. Le Roux, M. Bouloy, P. Patel, A. Teichmann, O. Donoso-Mantke and M. Niedrig, PLoS Neglected Trop. Dis., 2013, 7, e2244 CAS.
- B. Rohrman and R. Richards-Kortum, Anal. Chem., 2015, 87, 1963–1967 CrossRef CAS PubMed.
- G. Garcia-Schwarz, A. Rogacs, S. S. Bahga and J. G. Santiago, J. Visualized Exp., 2012, e3890, DOI:10.3791/3890.
- F. M. Everaerts, J. L. Beckers and T. P. E. M. Verheggen, Isotachophoresis: theory, instrumentation, and applications, Elsevier Scientific Pub. Co., Amsterdam, New York, 1976 Search PubMed.
- A. Persat, L. A. Marshall and J. G. Santiago, Anal. Chem., 2009, 81, 9507–9511 CrossRef CAS PubMed.
- Y. Qu, L. A. Marshall and J. G. Santiago, Anal. Chem., 2014, 86, 7264–7268 CrossRef CAS PubMed.
- M. Bercovici, G. V. Kaigala, K. E. Mach, C. M. Han, J. C. Liao and J. G. Santiago, Anal. Chem., 2011, 83, 4110–4117 CrossRef CAS PubMed.
- R. B. Schoch, M. Ronaghi and J. G. Santiago, Lab Chip, 2009, 9, 2145–2152 RSC.
- L. A. Marshall, C. M. Han and J. G. Santiago, Anal. Chem., 2011, 83, 9715–9718 CrossRef CAS PubMed.
- C. Eid, G. Garcia-Schwarz and J. G. Santiago, Analyst, 2013, 138, 3117–3120 RSC.
- C. Eid, J. W. Palko, E. Katilius and J. G. Santiago, Anal. Chem., 2015, 87, 6736–6743 CrossRef CAS PubMed.
- L. A. Marshall, A. Rogacs, C. D. Meinhart and J. G. Santiago, J. Chromatogr., A, 2014, 1331, 139–142 CrossRef CAS PubMed.
- L. Marshall, PhD, Stanford University, 2013.
- A. Rogacs, L. A. Marshall and J. G. Santiago, J. Chromatogr., A, 2014, 1335, 105–120 CrossRef CAS PubMed.
- M. D. Borysiak, K. W. Kimura and J. D. Posner, Lab Chip, 2015, 15, 1697–1707 RSC.
- B. Swaminathan and P. Gerner-Smidt, Microbes Infect., 2007, 9, 1236–1243 CrossRef PubMed.
- L. Rudbeck and J. Dissing, Biotechniques, 1998, 25, 588–590 CAS.
- J. D. Brewster and G. C. Paoli, Anal. Biochem., 2013, 442, 107–109 CrossRef CAS PubMed.
- T. e. L. Q. Guide, TwistAmp® exo + ListeriaM Quick Guide, (http://www.twistdx.co.uk/images/uploads/docs/QG_EXOLIST_online_RevB.pdf).
- M. S. Forrest and O. Nentwich, International Society for Neglected Tropical Diseases, London, England, 2013, pp. 39–41 Search PubMed.
- F. Schuler, F. Schwemmer, M. Trotter, S. Wadle, R. Zengerle, F. von Stetten and N. Paust, Lab Chip, 2015, 15, 2759–2766 RSC.
- A. Persat, M. E. Suss and J. G. Santiago, Lab Chip, 2009, 9, 2454–2469 RSC.
- A. Rogacs, Y. Qu and J. G. Santiago, Anal. Chem., 2012, 84, 5858–5863 CrossRef CAS PubMed.
- G. Garcia-Schwarz and J. G. Santiago, Anal. Chem., 2012, 84, 6366–6369 CrossRef CAS PubMed.
- V. Shkolnikov and J. G. Santiago, Anal. Chem., 2014, 86, 6229–6236 CrossRef CAS PubMed.
- Z. Corporation, NATtrol – Molecular Controls – ZeptoMetrix Corporation, http://www.zeptometrix.com/store/nattrola-molecular-controls/.
- N. C. Cady, S. Stelick, M. V. Kunnavakkam and C. A. Batt, Sens. Actuators, B, 2005, 107, 332–341 CrossRef CAS.
- D. A. Kanayeva, R. Wang, D. Rhoads, G. F. Erf, M. F. Slavik, S. Tung and Y. Li, J. Food Prot., 2012, 75, 1951–1959 CrossRef CAS PubMed.
- L. Cocolin, M. Manzano, C. Cantoni and G. Comi, Mol. Cell. Probes, 1997, 11, 453–455 CrossRef CAS PubMed.
- L. Cocolin, M. Manzano, C. Cantoni and G. Comi, Res. Microbiol., 1997, 148, 485–490 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an02119k |
| This journal is © The Royal Society of Chemistry 2017 |