
[3+2]-Annulation of platinum-bound azomethine ylides with distal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C bonds of N-allenamides†
C bonds of N-allenamides†
Indradweep
Chakrabarty
ab,
Suleman M.
Inamdar
a,
Manjur O.
Akram
ab,
Amol B.
Gade
ab,
Subhrashis
Banerjee
c,
Saibal
Bera
c and
Nitin T.
Patil
 *ab
*ab
aDivision of Organic Chemistry, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune – 411008, India. E-mail: n.patil@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), New Delhi – 110025, India
cPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune – 411008, India
First published on 25th November 2016
Abstract
A Pt-catalyzed, highly regioselective reaction between N-allenamides and imino-alkynes leading to pyrrolo[1,2-a]indoles is described. This represents the first example of [3+2]-annulation of Pt-bound azomethine ylides with the distal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of N-allenamides. The mechanism of the reaction was established by computational studies.
C bond of N-allenamides. The mechanism of the reaction was established by computational studies.
Pyrrolo[1,2-a]indoles and their structural analogues are important structural motifs commonly found in numerous bioactive natural products.1 The most promising natural products belonging to this catagory are yuremamine,2 mitomycins1b–d and flinderoles3 (Fig. 1). As a consequence, these scaffolds have received considerable attention over the years and many methods for their preparation have been developed in the literature.4 Accordingly, a general catalytic method5 to access functionalized pyrrolo[1,2-a]indoles is in high demand and would serve to provide a great opportunity to accelerate biological studies of these valuable compounds.6
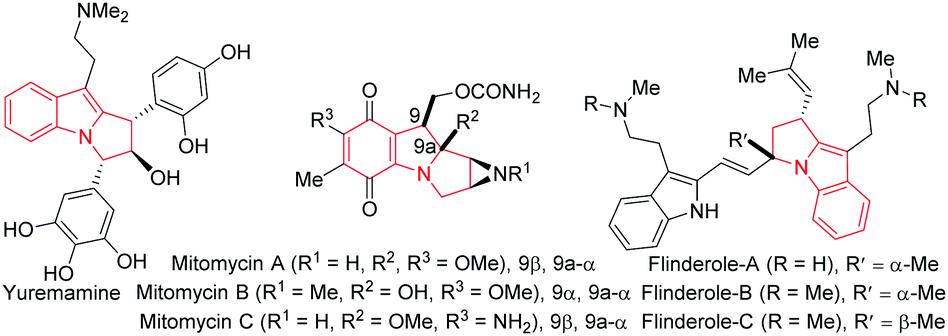 | ||
| Fig. 1 Some natural products containing the pyrrolo[1,2-a]indole motif. | ||
Over the past decade, gold/platinum-catalyzed annulation reactions have emerged as a powerful technique for rapid access to various ring systems in an extremely efficient and stereoselective manner.7 In this context, due to their unique reactivity and ease of accessibility, N-allenamides8 have gained much interest in gold catalyzed cascade annulation, leading to synthetically important carbocyclic or heterocyclic scaffolds (Scheme 1). The research groups of González,9 Chen,10 Mascareñas/López11 and Bandini12 reported gold-catalyzed intermolecular [2+2]-annulation with N-allenamides to obtain cyclobutanes (Scheme 1a, path a). Mascareñas/López,13 Vicente14 and Zhang15 successfully established gold-catalyzed intermolecular [4+2]-annulation of various diene systems (path b) and alkene-tethered ketones16 (path c) with N-allenamides. Similarly, Chen and co-workers reported [3+2]-annulation of N-allenamides with nitrones and azomethine imines (path d).17 Very recently, gold catalyzed regioselective annulation reaction of 2-(1-alkynyl)-2-alken-1-ones with proximal CC bonds of N-allenamides was developed by Zhang and co-workers (Scheme 1b).18 Note that Iwasawa and co-workers, in their pioneering work, reported a series of [3+2] cycloaddition reactions of metal bound azomethine ylides with alkenes.19 Inspired by the above reports and our own interest in the field of π-acid catalysis, we envisioned a scenario wherein annulation of metal-bound azomethine ylides with N-allenamides would lead to pyrrolo[1,2-a]indoles A or Bvia a series of cascade events (Scheme 2). In particular, we became interested in knowing whether the distal bond (path a) or proximal bond (path b) of N-allenamide would participate in the annulation reactions. Herein, we report, for the first time, catalytic [3+2]-annulation of metal-bound azomethine ylides with distal CC bonds of N-allenamides.
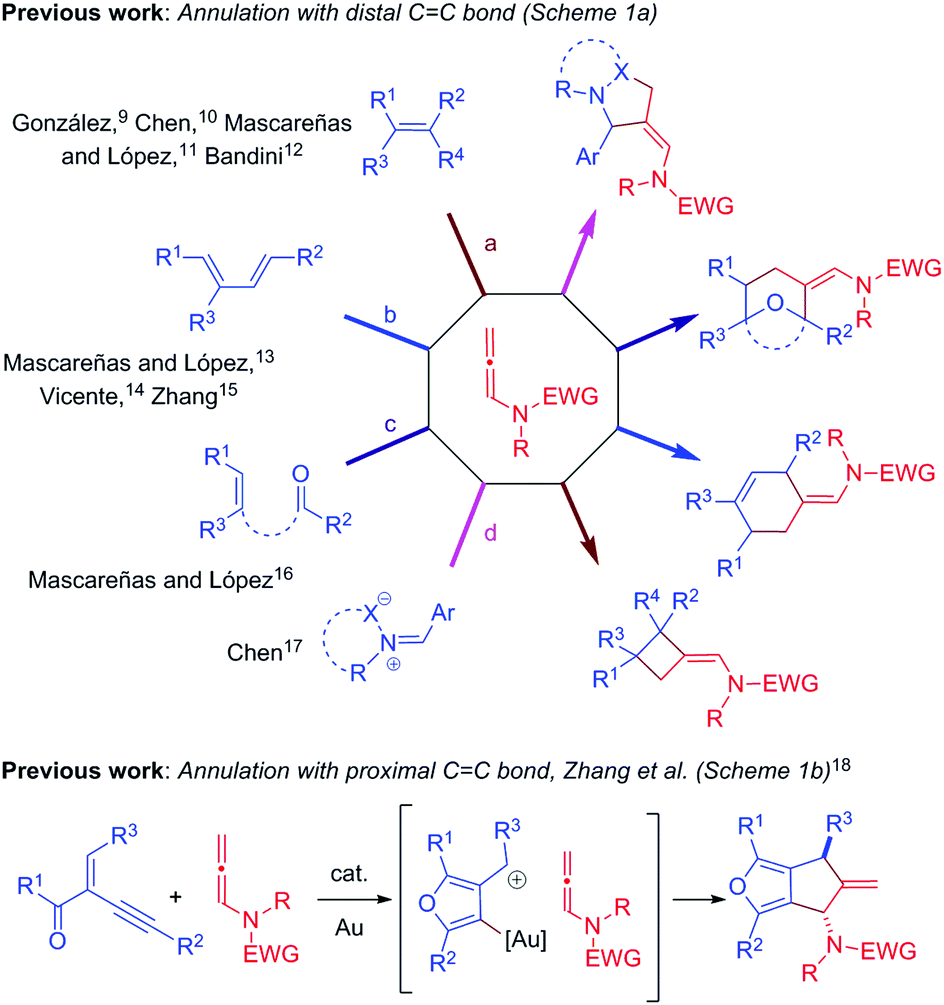 | ||
| Scheme 1 Annulation reactions of N-allenamides with various partners. | ||
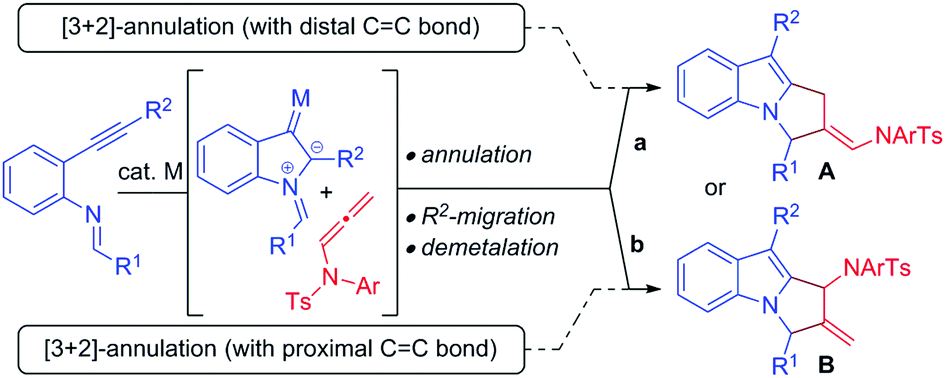 | ||
| Scheme 2 [3+2]-Annulation of N-allenamides with metal-bound azomethine ylides. | ||
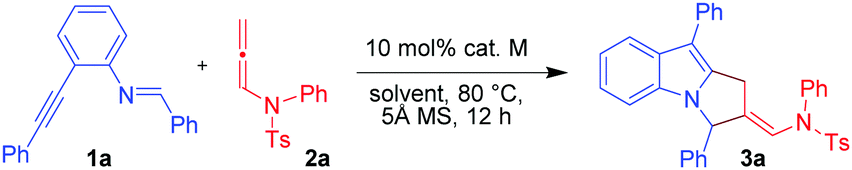
To explore our hypothesis, initial efforts were directed towards finding an appropriate metal catalyst for the proposed reaction using (E)-N-benzylidene-2-(phenylethynyl)aniline (1a) and N-allenamide (2a) as model substrates (Table 1). Unfortunately, the reaction did not occur when AuCl, AuCl3, Ph3PAuOTf and Ph3PAuNTf2 were used as catalysts (entries 1–4) despite their proven abilities to activate alkynes. Next, the reaction was conducted using PtI2 as a catalyst (10 mol%) in toluene at 80 °C. Gratifyingly, product 3a was obtained in 51% yield (entry 5). Product 3a was found to be the result of [3+2]-annulation reaction between Pt-bound azomethine ylides and the distal CC bonds of N-allenamides. The use of the PtBr2 catalyst gave a slightly better result affording 3a in 54% yield (entry 6). The yield was further improved to 66% with the use of PtCl2 (entry 7). A Pt(IV) catalyst (e.g. PtCl4) proved to be inferior and 3a was isolated in 40% yield (entry 8). The reaction was found to be dependent on the solvent used (Table 1, entries 9–13). For instance, in the case of non-polar, non-coordinating solvents such as benzene and m-xylene (entries 9 and 10), the yields of 3a were not much affected; while, in the case of chlorobenzene and fluorobenzene, 3a was obtained in low yields (entries 11 and 12). A drastic reduction in yield was observed when nitrobenzene was used as a solvent (entry 13). Lowering the catalyst loading to 5 mol% resulted in incomplete conversion leading to the isolation of 3a in 38% yield (entry 14). When the temperature of the reaction was decreased to 50 °C, the reaction was not completed and 3a was isolated in 36% yield (entry 15). The use of 5 Å MS was found to be necessary; in its absence, 3a was obtained in only 10% yield (entry 16). The overall optimization studies revealed that the best condition to obtain 3a in acceptable yield is the use of 10 mol% PtCl2 in the presence of 5 Å MS (50 mg) (entry 7).
|
|
|||
|---|---|---|---|
| Entry | Cat. M | Solvent | Yieldb (%) |
| a Reaction conditions: 0.2 mmol of 1a, 0.4 mmol of 2a, 10 mol% metal catalyst, 5 Å MS (50 mg), toluene (2 mL), 80 °C, 12 h. b Isolated yields. c 5 mol% catalyst loading. d Reaction performed at 50 °C for 24 h. e Reaction was carried out without 5 Å MS. | |||
| 1 | AuCl | Toluene | — |
| 2 | AuCl3 | Toluene | — |
| 3 | PPh3AuOTf | Toluene | — |
| 4 | PPh3AuNTf2 | Toluene | — |
| 5 | PtI2 | Toluene | 51 |
| 6 | PtBr2 | Toluene | 54 |
| 7 | PtCl2 | Toluene | 66 |
| 8 | PtCl4 | Toluene | 40 |
| 9 | PtCl2 | Benzene | 52 |
| 10 | PtCl2 | m-Xylene | 55 |
| 11 | PtCl2 | Chlorobenzene | 43 |
| 12 | PtCl2 | Fluorobenzene | 47 |
| 13 | PtCl2 | Nitrobenzene | 16 |
| 14 | PtCl2 | Toluene | 38c |
| 15 | PtCl2 | Toluene | 36d |
| 16 | PtCl2 | Toluene | 10e |
With the optimized conditions in hand (Table 1, entry 7), we explored the scope of the reaction using imino-alkyne 1a with various N-allenamides 2 bearing a variety of electron-deficient as well as electron-rich substituents at the aryl rings (Table 2). The reactions proceeded well to give annulated products 3 in good to excellent yields (61–73%). The presence of electron-rich substituents such as 4-Et, 4-OMe, 2-OPh, 3,4-methylene-di-oxy, and 2,4,6-trimethyl in the phenyl ring of N-allenamide gave the corresponding annulated products 3b, 3c, 3g, 3j and 3l in good yields (65–73%). Even the substrates bearing electron-deficient substituents (e.g. 4-Cl, 4-Br, 4-NO2, 3-CF3 and 3-Br-4-Me) reacted smoothly to give the respective products (3d–f, 3h and 3i) in 61–71% yields. The reaction also proceeded well when the phenyl ring of N-allenamide was replaced by the –Bn group to afford the corresponding product (3k) in 70% yield. Interestingly, 2-oxazolidinone allenamides, which are frequently used in related cycloaddition reactions (Scheme 1), are found to be inert under the present reaction conditions.
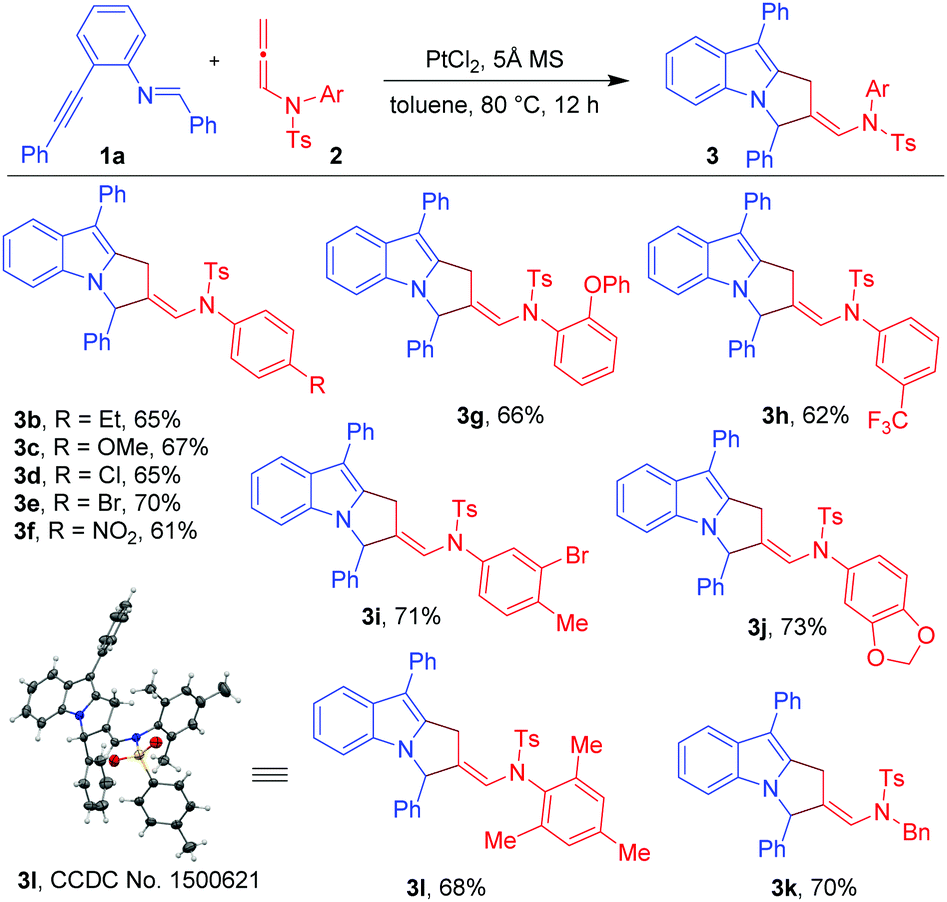
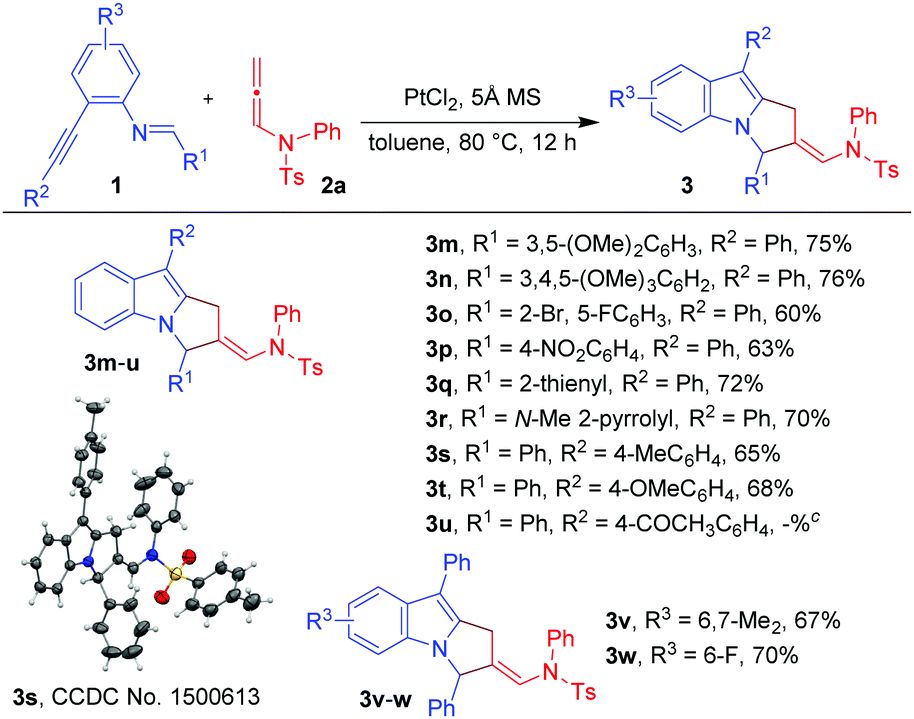
Next, we turned our attention to explore the generality of the [3+2]-annulation reaction of various imino-alkynes 1 with N-allenamide 2a (Table 3). A variety of substrates with electron-donating, electron-withdrawing as well as heterocyclic substituents at R1, R2 and R3 positions were examined to understand the scope and limitations. These results showed that the iminoalkyne possessing electron donating aryl groups as R1 afforded products 3m and 3n in 75 and 76% yields, whereas slightly lower yields were observed in the case of electron withdrawing aryl groups giving 3o and 3p in 60 and 63% yields. The heteroaromatic scaffolds were also well tolerated to give 3q and 3r in 72 and 70% yields, respectively. Next, we were curious to know whether the present strategy is applicable for variation in R2. To this end, appropriate precursors consisting of both electron-donating and electron-withdrawing substituents were subjected to the standard reaction conditions. It was observed that the substituents bearing electron-donating groups (4-Me-Ph and 4-OMe-Ph) as R2 performed well without hampering the yields of products 3s and 3t (65 and 68%), while in the presence of the electron-withdrawing acetyl group the reaction shuts down without formation of 3u. Furthermore, variation of substituents at R3 of iminoalkynes did not affect the efficiency of the reaction affording the desired products 3v and 3w in 67 and 70% yields, respectively. The X-ray crystallography data for 3a, 3l and 3s have also been obtained which unambiguously confirmed the structure.20 Note that the substrates bearing –H or –alkyl as R2 are not viable substrates for the present reactions.
Finally, to probe the synthetic utility of the reaction, transformations of 3a were performed (Scheme 3). Initially, the catalytic hydrogenation of 3a with H2 in the presence of 10% Pd/C led to the formation of aminomethyl-substituted pyrrolo[1,2-a]indole derivative 4 in 83% yield (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). Next, the treatment of 3a with 12 M HCl in EtOAc as a solvent was performed, and the corresponding aldehyde 5 was obtained in 41% yield (9:1 dr). The diastereomer ratio was determined by 1H NMR analysis.20 Finally, compound 3a was subjected to m-CPBA oxidation to furnish dicarbonyl compound 6 in 68% yield.
:1 dr). Next, the treatment of 3a with 12 M HCl in EtOAc as a solvent was performed, and the corresponding aldehyde 5 was obtained in 41% yield (9:1 dr). The diastereomer ratio was determined by 1H NMR analysis.20 Finally, compound 3a was subjected to m-CPBA oxidation to furnish dicarbonyl compound 6 in 68% yield.
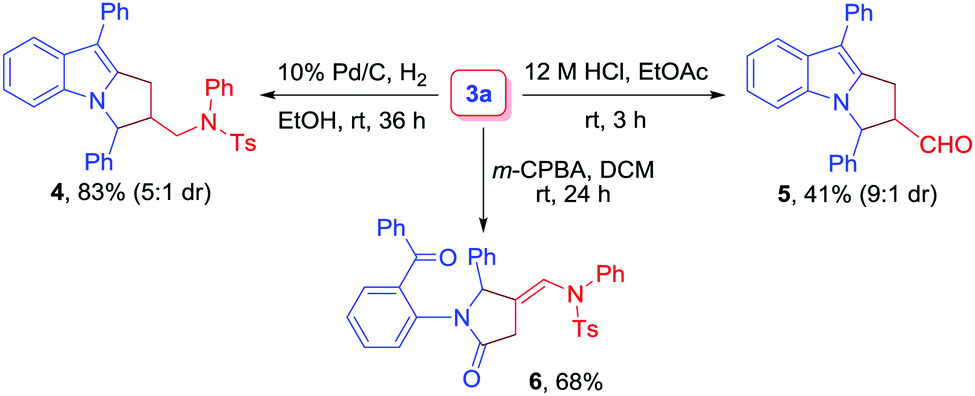 | ||
| Scheme 3 Transformation of 3a. | ||
A plausible mechanism is depicted in Scheme 4. At first, the Pt-catalyst would activate the alkyne moiety of 1a, thereby triggering the attack of imine nitrogen to generate azomethine ylide I. A direct attack of 2a to azomethine intermediate I19c would then occur to form intermediate II. Next, Pt(II) serves to donate electron density into an electron-deficient iminium ion in a 1,4-fashion (cf.II) to generate III. In short, the Pt(II)-catalyst serves both the roles i.e. activating the alkyne moiety in 1a to form I and donating electron density back to an electron-deficient iminium ion (cf.II). Once intermediate III has been generated, 1,2-aryl migration21 would occur to form intermediate IV which subsequently would produce 3a with the regeneration of the Pt-catalyst. Another possibility is that the alkenyl-Pt intermediate V,22 generated in situ from 2a and PtCl2, would combine with I to form intermediate II (shown as dotted lines) which would follow a similar sequence of events to form 3a. Note that such a kind of dual catalysis employing gold as a catalyst is known.23 However, this possibility was ruled out based on computational studies.20
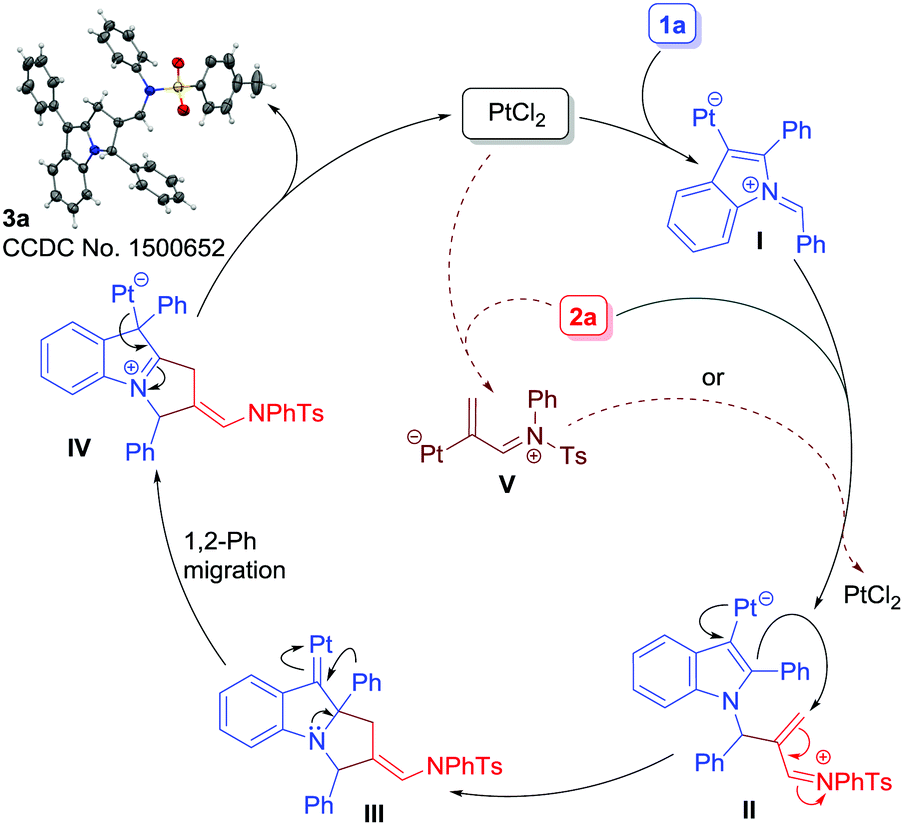 | ||
| Scheme 4 A plausible mechanism (Cl atoms on the Pt-center are omitted for clarity). | ||
In summary, we have demonstrated the first example of intermolecular [3+2]-annulation of Pt-bound azomethine ylides with distal CC bonds of N-allenamides. The ready availability of the starting materials and the great importance of the pyrrolo[1,2-a]indoles make the current methodology particularly interesting. Mechanistic insights gained through density functional theory suggest that the reaction follows a single substrate activation pathway rather than dual activation.20
Generous financial support from the Department of Science and Technology (DST), New Delhi (Grant No. SB/S1/OC-17/2013), and the Council of Scientific and Industrial Research (CSIR), New Delhi (Grant No. CSC0108 and CSC0130), is gratefully acknowledged. We thank Dr Kumar Vanka and Dr Rahul Banerjee for theoretical studies and X-ray crystallographic structure determination, respectively. I. C. thanks UGC for the award of a Junior Research Fellowship (JRF).
Notes and references
- (a) N. Monakhova, S. Ryabova and V. Makarov, J. Heterocycl. Chem., 2016, 53, 685 CrossRef CAS; (b) P. D. Bass, D. A. Gubler, T. C. Judd and R. M. Williams, Chem. Rev., 2013, 113, 6816 CrossRef CAS PubMed; (c) J.-C. Andrez, Beilstein J. Org. Chem., 2009, 5, 1 Search PubMed; (d) U. Galm, M. H. Hager, S. G. V. Lanen, J. Ju, J. S. Thorson and B. Shen, Chem. Rev., 2005, 105, 739 CrossRef CAS PubMed; (e) W. A. Remers and R. T. Dorr, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, John Wiley & Sons, New York, 1988, vol. 6, p. 1 Search PubMed.
- J. J. Vepsäläinen, S. Auriola, M. Tukiainen, N. Ropponen and J. C. Callaway, Planta Med., 2005, 71, 1053 Search PubMed.
- L. S. Fernandez, M. S. Buchanan, A. R. Carroll, Y. J. Feng, R. J. Quinn and V. M. Avery, Org. Lett., 2009, 11, 329 CrossRef CAS PubMed.
- Yuremamine: (a) M. B. Calvert and J. Sperry, Chem. Commun., 2015, 51, 6202 RSC; (b) T. Ohyama, M. Uchida, H. Kusama and N. Iwasawa, Chem. – Asian J., 2015, 10, 1850 CrossRef CAS PubMed ; Mitomycins: ; (c) Z.-Y. Yan, Y. Xiao and L. Zhang, Angew. Chem., Int. Ed., 2012, 51, 8624 CrossRef CAS PubMed; (d) D. A. Gubler and R. M. Williams, Tetrahedron Lett., 2009, 50, 4265 CrossRef CAS PubMed; (e) H. Namiki, S. Chamberland, D. A. Gubler and R. M. Williams, Org. Lett., 2007, 9, 5341 CrossRef CAS PubMed; (f) R. S. Coleman, F.-X. Felpin and W. Chen, J. Org. Chem., 2004, 69, 7309 CrossRef CAS PubMed; (g) T. Fukuyama and L. Yang, J. Am. Chem. Soc., 1989, 111, 8303 CrossRef CAS; (h) T. Fukuyama, F. Nakatsubo, A. J. Cocuzza and Y. Kishi, Tetrahedron Lett., 1977, 18, 4295 CrossRef ; Flinderoles: ; (i) R. Vallakati and J. A. May, J. Am. Chem. Soc., 2012, 134, 6936 CrossRef CAS PubMed; (j) R. M. Zeldin and F. D. Toste, Chem. Sci., 2011, 2, 1706 RSC; (k) D. H. Dethe, R. D. Erande and A. Ranjan, J. Am. Chem. Soc., 2011, 133, 2864 CrossRef CAS PubMed.
- Catalytic approaches to pyrrolo[1,2-a]indoles: (a) N. Saleh and A. Voituriez, J. Org. Chem., 2016, 81, 4371 CrossRef CAS PubMed; (b) K. Bera and C. Schneider, Chem. – Eur. J., 2016, 22, 7074 CrossRef CAS PubMed; (c) D. H. Dethe and R. Boda, Org. Biomol. Chem., 2016, 14, 5843 RSC; (d) D. H. Dethe and R. Boda, Chem. – Eur. J., 2016, 22, 106 CrossRef CAS PubMed; (e) D. H. Dethe, R. Boda and S. Das, Chem. Commun., 2013, 49, 3260 RSC; (f) M. B. Johansen and M. A. Kerr, Org. Lett., 2008, 10, 3497 CrossRef CAS PubMed; (g) H. Ren, Z. Li and P. Knochel, Chem. – Asian J., 2007, 2, 416 CrossRef CAS PubMed.
- Syntheses and biological activities of pyrrolo[1,2-a]indoles and their structural analogues: (a) D. J. Buzard, L. Lopez, J. Moody, A. Kawasaki, T. O. Schrader, M. Kasem, B. Johnson, X. Zhu, L. Thoresen, S. H. Kim, T. Gharbaoui, D. Sengupta, L. Calvano, A. Krishnan, Y. Gao, G. Semple, J. Edwards, J. Barden, M. Morgan, K. Usmani, C. Chen, A. Sadeque, W. Chen, R. J. Christopher, J. Thatte, L. Fu, M. Solomon, K. Whelan, H. Al-Shamma, J. Gatlin, I. Gaidarov, T. Anthony, M. Le, D. J. Unett, S. Stirn, A. Blackburn, D. P. Behan and R. M. Jones, ACS Med. Chem. Lett., 2014, 5, 1334 CrossRef CAS PubMed; (b) Z. Zheng, M. Touve, J. Barnes, N. Reich and L. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9302 CrossRef CAS PubMed; (c) T. O. Schrader, B. R. Johnson, L. Lopez, M. Kasem, T. Gharbaoui, D. Sengupta, D. Buzard, C. Basmadjian and R. M. Jones, Org. Lett., 2012, 14, 6306 CrossRef CAS PubMed; (d) I. Ngantchou, B. Nyasse, C. Denier, C. Blonski, V. Hannaert and B. Schneider, Bioorg. Med. Chem. Lett., 2010, 20, 3495 CrossRef CAS PubMed; (e) M. Tanaka, M. Ubukata, T. Matsuo, K. Yasue, K. Matsumoto, Y. Kajimoto, T. Ogo and T. Inaba, Org. Lett., 2007, 9, 3331 CrossRef CAS PubMed; (f) R. M. Wilson, R. K. Thalji, R. G. Bergman and J. A. Ellman, Org. Lett., 2006, 8, 1745 CrossRef CAS PubMed; (g) M. Tanaka, S. Sagawa, J.-I. Hoshi, F. Shimoma, K. Yasue, M. Ubukata, T. Ikemoto, Y. Hase, M. Takahashi, T. Sasase, N. Ueda, M. Matsushita and T. Inaba, Bioorg. Med. Chem., 2006, 14, 5781 CrossRef CAS PubMed.
- Reviews: (a) L. Liu and J. Zhang, Chem. Soc. Rev., 2016, 45, 506 RSC; (b) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed; (c) T. Cañeque, F. M. Truscott, R. Rodriguez, G. Maestri and M. Malacria, Chem. Soc. Rev., 2014, 43, 2916 RSC; (d) D. Garayalde and C. Nevado, ACS Catal., 2012, 2, 1462 CrossRef CAS; (e) B.-L. Lu, L. Dai and M. Shi, Chem. Soc. Rev., 2012, 41, 3318 RSC; (f) F. López and J. L. Mascareñas, Beilstein J. Org. Chem., 2011, 7, 1075 CrossRef PubMed; (g) M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2011, 47, 6536 RSC; (h) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236 CrossRef CAS PubMed; (i) M. Bandini, Chem. Soc. Rev., 2011, 40, 1358 RSC; (j) S. M. A. Sohel and R.-S. Liu, Chem. Soc. Rev., 2009, 38, 2269 RSC; (k) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed.
- For general reviews on N-allenamides, see: (a) E. Manoni and M. Bandini, Eur. J. Org. Chem., 2016, 3135 CrossRef CAS; (b) T. Lu, Z. Lu, Z.-X. Ma, Y. Zhang and R. P. Hsung, Chem. Rev., 2013, 113, 4862 CrossRef CAS PubMed; (c) L.-L. Wei, H. Xiong and R. P. Hsung, Acc. Chem. Res., 2003, 36, 773 CAS.
- (a) S. Suárez-Pantiga, C. Hernández-Díaz, M. Piedrafita, E. Rubio and J. M. González, Adv. Synth. Catal., 2012, 354, 1651 CrossRef; (b) S. Suárez-Pantiga, C. Hernández-Díaz, E. Rubio and J. M. González, Angew. Chem., Int. Ed., 2012, 51, 11552 CrossRef PubMed.
- X.-X. Li, L.-L. Zhu, W. Zhou and Z. Chen, Org. Lett., 2012, 14, 436 CrossRef CAS PubMed.
- (a) P. Bernal-Albert, H. Faustino, A. Gimeno, G. Asensio, J. L. Mascareñas and F. López, Org. Lett., 2014, 16, 6196 CrossRef CAS PubMed; (b) H. Faustino, P. Bernal, L. Castedo, F. López and J. L. Mascareñas, Adv. Synth. Catal., 2012, 354, 1658 CrossRef CAS.
- M. Jia, M. Monari, Q.-Q. Yang and M. Bandini, Chem. Commun., 2015, 51, 2320 RSC.
- (a) J. Francos, F. Grande-Carmona, H. Faustino, J. Iglesias-Sigüenza, E. Díez, I. Alonso, R. Fernández, J. M. Lassaletta, F. López and J. L. Mascareñas, J. Am. Chem. Soc., 2012, 134, 14322 CrossRef CAS PubMed; (b) H. Faustino, F. López, L. Castedo and J. L. Mascareñas, Chem. Sci., 2011, 2, 633 RSC.
- V. Pirovano, L. Decataldo, E. Rossi and R. Vicente, Chem. Commun., 2013, 49, 3594 RSC.
- Y. Wang, P. Zhang, Y. Liu, F. Xia and J. Zhang, Chem. Sci., 2015, 6, 5564 RSC.
- (a) H. Faustino, I. Varela, J. L. Mascareñas and F. López, Chem. Sci., 2015, 6, 2903 RSC; (b) H. Faustino, I. Alonso, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2013, 52, 6526 CrossRef CAS PubMed.
- (a) W. Zhou, X.-X. Li, G.-H. Li, Y. Wu and Z. Chen, Chem. Commun., 2013, 49, 3552 RSC; (b) G.-H. Li, W. Zhou, X.-X. Li, Q.-W. Bi, Z. Wang, Z.-G. Zhao, W.-X. Hu and Z. Chen, Chem. Commun., 2013, 49, 4770 RSC.
- Y. Wang, P. Zhang, D. Qian and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14849 CrossRef CAS PubMed.
- (a) J. Takaya, Y. Miyashita, H. Kusama and N. Iwasawa, Tetrahedron, 2011, 67, 4455 CrossRef CAS; (b) H. Kusama, Y. Suzuki, J. Takaya and N. Iwasawa, Org. Lett., 2006, 8, 895 CrossRef CAS PubMed; (c) H. Kusama, Y. Miyashita, J. Takaya and N. Iwasawa, Org. Lett., 2006, 8, 289 CrossRef CAS PubMed; (d) J. Takaya, H. Kusama and N. Iwasawa, Chem. Lett., 2004, 33, 16 CrossRef CAS; (e) H. Kusama, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2002, 124, 11592 CrossRef CAS PubMed.
- See the ESI† for details.
- (a) A. Baralle, H. Yorimitsu and A. Osuka, Chem. – Eur. J., 2016, 22, 10768 CrossRef CAS PubMed; (b) W. Kong, Y. Qiu, X. Zhang, C. Fu and S. Ma, Adv. Synth. Catal., 2012, 354, 2339 CrossRef CAS; (c) G. Li, X. Huang and L. Zhang, Angew. Chem., Int. Ed., 2008, 47, 346 CrossRef CAS PubMed; (d) J. Takaya, S. Udagawa, H. Kusama and N. Iwasawa, Angew. Chem., Int. Ed., 2008, 47, 4906 CrossRef CAS PubMed.
- Formation of such alkenyl–metal intermediates from N-allenamides is known, see: ref. 9b, 10–12, 13a and 14–18.
- Review: A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1500613, 1500621, 1500652. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc07874e |
| This journal is © The Royal Society of Chemistry 2017 |