
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An unprecedented 1,4-diphospha-2,3-disila butadiene (–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) Si–SiP–) derivative and a 1,3-diphospha-2-silaallyl anion, each stabilized by the amidinate ligand†
Si–SiP–) derivative and a 1,3-diphospha-2-silaallyl anion, each stabilized by the amidinate ligand†
Subrata
Kundu
a,
Chandrajeet
Mohapatra
a,
Prinson P.
Samuel
a,
Johannes
Kretsch
a,
M. G.
Walawalkar
a,
Regine
Herbst-Irmer
a,
Dietmar
Stalke
*a,
Sriman
De
b,
Debasis
Koley
*b and
Herbert W.
Roesky
*a
aInstitut für Anorganische Chemie, Georg-August-Universität, Tammannstraße 4D-37077, Göttingen, Germany. E-mail: hroesky@gwdg.de; dstalke@chemie.uni-goettingen.de
bDepartment of Chemical Sciences, IISER-Kolkata, Mohanpur Campus, Mohanpur 741252, India. E-mail: koley@iiserkol.ac.in
First published on 24th November 2016
Abstract
The first acyclic 4π-electron –P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si–SiP– motif with two four coordinate silicon substituents supported by the amidinate ligand and two coordinate phosphorus has been synthesized from the reaction of heteroleptic chlorosilylene LSiCl (1), TripPCl2 (Trip = 2,4,6-iPr3C6H2) and KC8 in a 1
Si–SiP– motif with two four coordinate silicon substituents supported by the amidinate ligand and two coordinate phosphorus has been synthesized from the reaction of heteroleptic chlorosilylene LSiCl (1), TripPCl2 (Trip = 2,4,6-iPr3C6H2) and KC8 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 ratio. The same reaction in a 1:2:6 ratio in the presence of one equivalent of 18-crown-6 ether affords the 1,3-diphospha-2-silaallyl anion.
:1:3 ratio. The same reaction in a 1:2:6 ratio in the presence of one equivalent of 18-crown-6 ether affords the 1,3-diphospha-2-silaallyl anion.
In 1998, Dillon et al. published a book with the title, phosphorus: the carbon copy.1 For decades this leitmotiv strongly influenced the research in phosphorus chemistry. In the meantime silylenes have attracted much attention due to their unique structures and bonding properties. A number of silylenes have been synthesized and structurally characterized.2 Recently scientists have been fascinated with synthesizing more complex molecules using stable silylenes to unearth their interesting properties, including catalysis.2b In recent years the results in silicon chemistry justify a new leitmotiv, silicon: the phosphorus copy. Compounds containing silicon and phosphorus are attracting attention due to their surprising bonding properties and their potential applications as semiconducting materials.3 After the first successful isolation of a compound with a Si
P double bond by Smit, Lock and Bickelhaupt in 1984, several derivatives having a double bond between silicon and phosphorus have been reported.4 In most of the cases, it has been observed that either the compounds possess a monomeric SiP unit or a cyclic dimeric (Si2P2) or trimeric (Si3P3) arrangement.5 So far, only the reaction of P4 with PhC(N(tBu))2SiN(TMS)2 resulted in the formation of an acyclic SiP–PP–PSi chain.4d Recently we reported an acyclic 4π electron delocalized butadiene analogue containing a Si–Si bonding motif (1,4-diamino-2,3-disila-1,3-butadiene).6 The successful isolation of a CSi–SiC chain inspired us to investigate whether it is possible to isolate a RPSi–SiPR chain, as R–P is isolobal to C.7 In this communication, we report the successful synthesis of an acyclic PSi–SiP chain and a PSi–P anion supported by an amidinate ligand [L = PhC(NtBu)2]. It is known that the SiP bond might form when three coordinate silicon and two coordinate phosphorus are involved, although this bond is highly unstable.4a So we chose amidinate silylene (1) because it will form a four coordinate silicon after the reaction with TripPCl2. Moreover to stabilize the highly reactive SiP bond, a bulky Trip group has been selected at the phosphorus atom. The reduction of LSiCl and TripPCl2 with KC8 in a molar ratio of 1:1:3 in THF resulted in compound 2 [(TripPSi–L)2] (Scheme 1), whereas the same reaction in a 1:2:6 ratio in the presence of one equivalent of 18-crown-6 affords compound 3 [(THF)K(18-crown-6)]+ [(TripP)2SiL]− (Scheme 1). To the best of our knowledge, a mixed phosphorus and silicon-centered chain of a stable and isolable 1,4-bisphosphino-2,3-disila butadiene has not been reported so far. In the past a 1,3-diphospha-2-silaallylic anion [{Li(15-crown-5)+tBuSi(2,4,6-tBu3C6H2)2}−]8 has been reported by Niecke et al., where a tertiary butyl group is attached to the silicon atom. In the present case we have applied a different synthetic strategy to isolate the 1,3-diphospha-2-silaallylic anion. In comparison to the literature reported 1,3-diphospha-2-silaallylic anion where the silicon center is three coordinate with two phosphorus and one carbon, in compound 3, the silicon center is four coordinate with two nitrogen and two phosphorus atoms. The composition is therefore quite different.
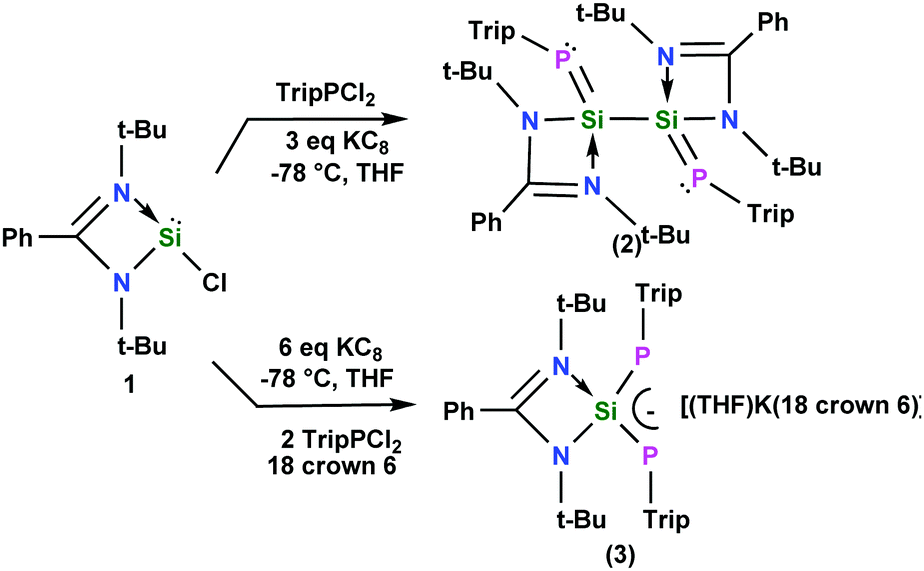 | ||
| Scheme 1 Syntheses of compounds 2 and 3. | ||
Compounds 2 and 3 were stable in an inert atmosphere for three months at room temperature both in the solid state and in solution. However, in the open atmosphere both compounds immediately decomposed with the formation of a mixture of products. In the 29Si NMR spectrum, compound 2 displays a doublet of doublets at δ = 23.3 ppm (1JSiP = 231 Hz, 2JSiP = 44 Hz) thus indicating the presence of two phosphorus atoms in the molecule and the kinetic stability in the solution. The 1JSiP (231 Hz) coupling constant is comparable with the highest coupling constant (234.8 Hz)9 reported for any multiple bonded phosphorus–silicon compound in the literature supporting a more stronger and less polarized SiP bond. As expected, the 29Si NMR spectrum of 3 displays a triplet at δ = 43.4 ppm (1JSiP = 181 Hz). The 31P NMR spectra of 2 and 3 each exhibit a singlet at δ = −133.9 and −161.5 ppm, respectively. These data are in good agreement with a symmetrical environment of the molecules.
The molecular structures of 2 and 3 were determined by single-crystal X-ray diffraction studies (Fig. 1, Fig. S3, ESI† and Fig. 2, Fig. S4, ESI†).‡ The P1–Si1–Si2–P2 skeleton of 2 has a trans-bent geometry (torsion angle 2.67°). The average Si–P bond length in 2 is 2.123(1) Å, hence it is shorter than a Si–P single bond distance previously reported for a e.g. LSiP(i-Pr)2 (2.307(8) Å)10 derivative but only slightly longer than the SiP double bond lengths found for phosphasilenes, e.g. as in LSi(SiMe3)PSiMe3 (2.095(3) Å).4c The average Si–P bond length of 2 and quantum chemical calculations (vide infra) indicate that the Si–P double bonds are significantly polarized. The Si–Si bond distance in 2 is 2.383 Å, which is slightly shorter than the parent bis-silylene L–Si–Si–L (2.413(2) Å).11
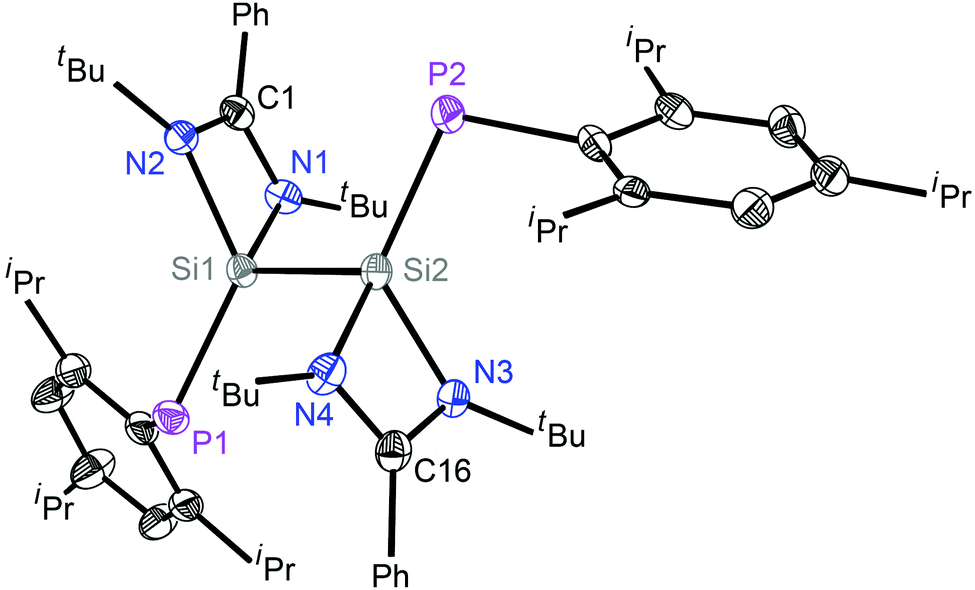 | ||
| Fig. 1 Molecular structure of 2; hydrogen atoms are omitted for clarity. Anisotropic displacement parameters are depicted at the 50% probability level. Selected experimental [calculated at R-M06-2X/def2-SVP for the singlet state] bond lengths (Å) and angles (deg): Si1–P1 2.1201(9) [2.133]; Si1–Si2 2.3825(11) [2.375]; Si2–P2 2.1256(9) [2.125]; Si1–N1 1.8331(16) [1.861]; Si1–N2 1.8763(16) [1.890]; Si2–N3 1.8378(16); Si2–N4 1.8787(16); P1–Si1–Si2 110.87(4) [114.4]; P2–Si2–Si1 111.29(4) [114.4]. | ||
The structure of 3 (Fig. 2) consists of a 1,3-diphospha-2-silaallyl anion and a potassium cation, which is bonded to an 18-crown-6 ether and a THF molecule in one of the axial positions. The Si(1)–P(1) and Si(1)–P(2) bond lengths are 2.166(7) and 2.168(6) Å, respectively, indicating the extensive delocalization of the anion. Thus, the Si–P bond distances in 3 are significantly shorter than an average Si–P single bond distance (2.307(8) Å)10 but longer than the SiP double bond lengths found in the literature for phosphasilenes, (2.053–2.095 Å).4c,12 The P1–Si1–P2 angle in the free anion in 3 is 103.12(2)°, which is more acute than the P–Si–P angle of 125.7(1) found in [{Li(15-crown-5)+tBuSi(2,4,6-tBu3C6H2)2}−].8
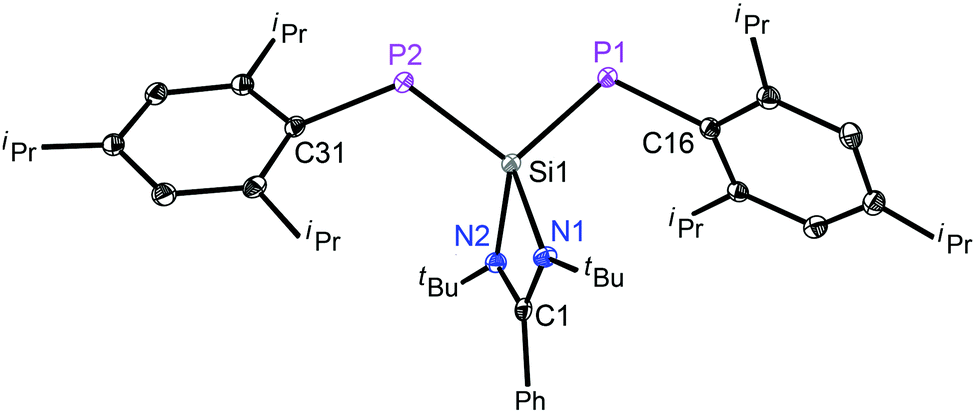 | ||
| Fig. 2 Molecular structure of the anion in 3; the t-butyl groups, the cationic part, the isopropyl groups and hydrogen atoms are omitted for clarity. Anisotropic displacement parameters are depicted at the 50% probability level. Selected experimental [calculated at R-M06-2X/def2-SVP for the singlet state] bond lengths (Å) and angles (deg): Si1–P1 2.1662(7) [2.146]; Si1–P2 2.1677(6) [2.166]; Si1–N1 1.8870(12) [1.906]; Si1–N2 1.8829(12) [1.890]; P1–Si1–P2 103.12(2) [108.5]. | ||
In order to explain the electronic structure and bonding scenario of 2 and 3, DFT calculations were performed at the M06-2X/def2-SVP level of theory (see Computational details, ESI†). Computed singlet and triplet states of 2 and 3 showed that the singlet is the ground electronic state with energy differences of (ΔES→T) 42.3 and 33.7 kcal mol−1, respectively. The geometrical parameters are in good agreement with the X-ray crystal structures as seen from the alignment and superposition of the conformers (Fig. S1 and Table S1, ESI†).
The formation of both complexes 2 and 3 from precursor 1 is highly exergonic with energy values (ΔGSL) −363.0 and −390.5 kcal mol−1, respectively, suggesting their favorable formation. To gain insight into the bonding nature of the Si–Si, Si–P and Si–N bonds in 2, we carried out natural bond orbital analysis at the BP86/TZ2P//M06-2X/def2-SVP level of theory implemented in the ADF2013.01 program suite.13 The Si–Si bond exhibits a σ-occupancy of 1.878e with equal contributions from the bonding partners (Si ∼ 49%). The Si–P covalent bond shows a double bond character with σ and π occupancies of 1.940 and 1.847e, respectively. Both the bonded electron densities (σ and π) of Si–P are polarized towards the P atom [P(σ) ∼ 57%, P(π) ∼ 79%], as pictorially represented by natural bond orbitals (Fig. S2 and Table S2, ESI†). The σ-bond is formed mainly from the sp hybridized orbital of Si and the almost pure p-orbital of the P atom. The NBO (NBO = Natural Bond Orbital) also locates a lone pair with an occupancy of 1.886e at the P atom. The Si atom is connected to one N atom via a single bond with an electron occupancy of 1.862e, where the electron density of this bond is mostly localized on the N center (∼87%), which indicates that it is a very polar electron sharing bond (Table S7, ESI†). In contrast, the other N atom contains a lone pair which suggests a closed shell interaction between the Si and N atoms. The lone pair of electrons on the N atom donates an electron to the Si center as is evident from NRT calculations.13 An accumulation of positive and negative charges on the LSi and TripP fragments (qLSi = 0.76e; qTripP = −0.76e) (Table S3, ESI†) indicates a significant Si → P σ donation; a similar observation was reported for carbene–dichlorosilylene stabilized phosphinidenes.14 The NBO proposed electronic scenario is further studied by QTAIM15 calculations. The important topological parameters at the (3,−1) bond critical points (BCP) are given in Table S4 (ESI†). The electron density at the BCP of the Si–P bond [ρ(r) = 0.111] along with the respective Laplacian [∇2ρ(r) = −0.059] indicates a covalent interaction. The calculated ellipticity of the Si–P bond [εBCP = 0.335] is much higher than that of the Si–P covalent single bond [εBCP = 0.14] previously reported,16 indicating a significant double bond character in this case.
The Laplacian value, ∇2ρ(r), of −0.155 for the Si–Si bond clearly suggests its covalent nature. The Wiberg bond indices (WBI) of the Si–Si and Si–P bonds are calculated to be 0.85 and 1.40, suggesting a single bond and a partial double bond, respectively. Similar to 2, the phosphorus atom in 3 provides a major contribution towards the formation of both Si–P σ- and π-bonds [P(σ) ∼ 59%, P(π) ∼ 81%]. These bonds show occupancies of 1.891 and 1.828e, respectively (Table S2, ESI†). Moreover, similar to 2, the AIM calculations of 3 show slight covalency of the Si–P bond with respect to the Laplacian value [∇2ρ(r) = −0.044]. Here also the ellipticity value (εBCP = 0.334) and Wiberg bond indices (WBI = 1.30) correspond fairly to a double bond character. The KS-HOMOs of compounds 2 and 3 show the π-bonding in the Si–P bonds (Fig. 3). Thus overall the observations have confirmed that both of the compounds have significantly polarized Si–P bonds with a notable double bond character.
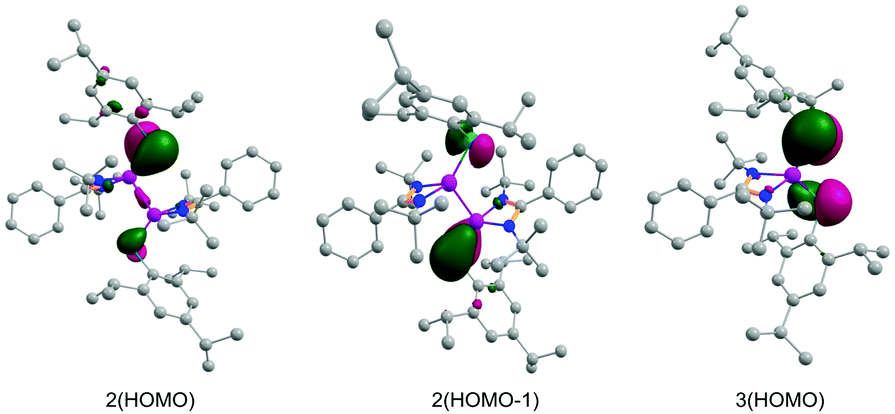 | ||
| Fig. 3 Selected KS-MOs of 2 and 3 (isosurface = 0.05 a.u.). H atoms are omitted for clarity. | ||
NMR calculations at the PBE0/TZ2P17 level reveal the 29Si chemical shift of 2 at 30.0 ppm, which is more up-field shifted when compared with that of 3 (42.4 ppm). The 31P NMR spectra of 2 and 3 also show chemical shifts at −141.5 and −177.8 ppm, respectively (Table S5, ESI†). These values demonstrate similar trends with the experimental findings.
In conclusion an unprecedented acyclic 4π-electron –PSi–SiP– motif and a 1,3-diphospha-2-silaallyl anion have been isolated and structurally characterized. The theoretical investigations of compounds 2 and 3 indicate that both of the products have polarized Si–P bonds with a significant double bond character.
H. W. R. is thankful to the DFG for financial support (RO 224/64-1). D. S. is grateful to the DNRF funded Center of Materials Crystallography (DNRF93) and we appreciate the chemical donations from Rockwood Lithium Albemarle. S. D. thanks UGC for a JRF fellowship, and D. K. acknowledges IISER-Kolkata and the CSIR project fund (01(2770)/13/EMR-II) for financial support. Dedicated to Professor Ionel Haiduc on the occasion of his 80th birthday.
Notes and references
- K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: the carbon copy, Wiley-VCH, 1998 Search PubMed.
- (a) H. W. Roesky, Efficient Methods for Preparing Silicon Compounds, Elsevier, 1st edn, 2016 Search PubMed; (b) B. Blom, D. Gallego and M. Driess, Inorg. Chem. Front., 2014, 1, 134 RSC and references therein.
- (a) C. Perrier, H. Vincent, P. Chaudouët, B. Chenevier and R. Madar, Mater. Res. Bull., 1995, 30, 357 CrossRef CAS; (b) A. Correia, B. Pichaud, A. Lhorte and J. B. Quoirin, J. Appl. Phys., 1996, 79, 2145 CrossRef CAS.
- (a) C. N. Smit, F. M. Lock and F. Bickelhaupt, Tetrahedron Lett., 1984, 25, 3011 CrossRef CAS; (b) N. C. Breit, T. Szilvási and S. Inoue, Chem. Commun., 2015, 51, 11272 RSC; (c) S. Inoue, W. Wang, C. Präsang, M. Asay, E. Irran and M. Driess, J. Am. Chem. Soc., 2011, 133, 2868 CrossRef CAS PubMed; (d) S. Khan, R. Michel, S. S. Sen, H. W. Roesky and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 11786 CrossRef CAS PubMed; (e) P. Willmes, M. J. Cowley, M. Hartmann, M. Zimmer, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2014, 53, 2216 CrossRef CAS PubMed; (f) B. Li, T. Matsuo, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, J. Am. Chem. Soc., 2009, 131, 13222 CrossRef CAS PubMed; (g) N. C. Breit, T. Szilvási, T. Suzuki, D. Gallego and S. Inoue, J. Am. Chem. Soc., 2013, 135, 17958 CrossRef CAS PubMed; (h) K. Hansen, T. Szilvási, B. Blom and M. Driess, Angew. Chem., Int. Ed., 2015, 54, 15060 CrossRef CAS PubMed; (i) S. Yao, S. Block, M. Brym and M. Driess, Chem. Commun., 2007, 3844 RSC; (j) K. Hansen, T. Szilvási, B. Blom, S. Inoue, J. Epping and M. Driess, J. Am. Chem. Soc., 2013, 135, 11795 CrossRef CAS PubMed; (k) M. Driess, Coord. Chem. Rev., 1995, 145, 1 CrossRef CAS.
- (a) S. S. Sen, S. Khan, H. W. Roesky, D. Kratzert, K. Meindl, J. Henn, D. Stalke, J.-P. Demers and A. Lange, Angew. Chem., Int. Ed., 2011, 50, 2322 CrossRef CAS PubMed; (b) Y.-F. Yang, G.-J. Cheng, J. Zhu, X. Zhang, S. Inoue and Y.-D. Wu, Chem. – Eur. J., 2012, 18, 7516 CrossRef CAS PubMed; (c) A. E. Seitz, M. Eckhardt, A. Erlebach, E. V. Peresypkina, M. Sierka and M. Scheer, J. Am. Chem. Soc., 2016, 138, 10433 CrossRef CAS PubMed.
- K. C. Mondal, H. W. Roesky, B. Dittrich, N. Holzmann, M. Hermann, G. Frenking and A. Meents, J. Am. Chem. Soc., 2013, 135, 15990 CrossRef CAS PubMed.
- R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 711 CrossRef.
- D. Lange, E. Klein, H. Bender, E. Niecke, M. Nieger and R. Pietschnig, Organometallics, 1998, 17, 2425 CrossRef CAS.
- S. Cradock, E. A. V. Ebsworth, D. W. H. Rankin and W. J. Savage, J. Chem. Soc., Dalton Trans., 1976, 1661 RSC.
- C.-W. So, H. W. Roesky, P. M. Gurubasavaraj, R. B. Oswald, M. T. Gamer, P. G. Jones and S. Blaurock, J. Am. Chem. Soc., 2007, 129, 12049 CrossRef CAS PubMed.
- S. S. Sen, A. Jana, H. W. Roesky and C. Schulzke, Angew. Chem., Int. Ed., 2009, 48, 8536 CrossRef CAS PubMed.
- Multiple Bonds and Low Coordination in Phosphorus Chemistry, ed. M. Regitz and O. J. Scherer, Thieme, Stuttgart, 1990 Search PubMed.
- (a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS; (b) C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Towards an order-N DFT method, Theor. Chem. Acc., 1998, 99, 391 Search PubMed.
- S. Roy, P. Stollberg, R. Herbst-Irmer, D. Stalke, D. M. Andrada, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2015, 137, 150 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules. A Quantum Theory, Clarendon Press, Oxford, 1990 Search PubMed.
- S. Roy, B. Dittrich, T. Mondal, D. Koley, A. C. Stückl, B. Schwederski, W. Kaim, M. John, S. K. Vasa, R. Linser and H. W. Roesky, J. Am. Chem. Soc., 2015, 137, 6180 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- (a) D. Stalke, Chem. Soc. Rev., 1998, 27, 171 RSC; (b) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615 CrossRef.
- Bruker AXS Inc., Bruker Apex CCD, SAINT v8.30C, Bruker AXS Inst. Inc., WI, USA, Madison, 2013 Search PubMed.
- L. Krause, R. Herbst-Irmer and D. Stalke, J. Appl. Crystallogr., 2015, 48, 1907 CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed information about the NMR measurements, preparation, computational calculations and X-ray structure determination. CCDC 1507708 and 1507709. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc09171g |
‡ Crystal data for 2 at 100(2) K: C63.50H96N4P2Si2, Mr = 1033.56 g mol−1, 0.490 × 0.200 × 0.150 mm, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 12.044(5) Å, b = 14.466(6) Å, c = 19.396(8) Å, α = 81.95(2)°, β = 77.12(2)°, γ = 69.98(2)°, V = 3088(2) Å3, Z = 2, μ(MoKα) = 0.150 mm−1, θmax = 25.4°, 130813 reflections measured, 11414 independent (Rint = 0.0565), R1 = 0.0382 [I > 2σ(I)], wR2 = 0.1043 (all data), res. density peaks: 0.299 to −0.244 e Å−3, CCDC 1507708. Crystal data for 3 at 100(2) K: C61H101KN2O7P2Si, Mr = 1103.56 g mol−1, 0.424 × 0.279 × 0.152 mm, triclinic, P, a = 10.771(3) Å, b = 13.848(3) Å, c = 22.433(5) Å, α = 101.050(10)°, β = 103.860(10)°, γ = 100.660(10)°, V = 3093.4(13) Å3, Z = 2, μ(AgKα) = 0.114 mm−1, θmax = 20.3°, 127873 reflections measured, 12250 independent (Rint = 0. 0419), R1 = 0. 0331 [I > 2σ(I)], wR2 = 0.0813 (all data), res. density peaks: 0.471 to −0.247 e Å−3, CCDC 1507709. All crystals were selected under cold protective conditions using the X-Temp2 device.18 The data were integrated with SAINT.19 A multi-scan absorption correction and a 3λ correction20 were applied using SADABS.21 The structures were solved by SHELXT22 and refined on F2 using SHELXL23 in the graphical user interface SHELXLE.24 , a = 12.044(5) Å, b = 14.466(6) Å, c = 19.396(8) Å, α = 81.95(2)°, β = 77.12(2)°, γ = 69.98(2)°, V = 3088(2) Å3, Z = 2, μ(MoKα) = 0.150 mm−1, θmax = 25.4°, 130813 reflections measured, 11414 independent (Rint = 0.0565), R1 = 0.0382 [I > 2σ(I)], wR2 = 0.1043 (all data), res. density peaks: 0.299 to −0.244 e Å−3, CCDC 1507708. Crystal data for 3 at 100(2) K: C61H101KN2O7P2Si, Mr = 1103.56 g mol−1, 0.424 × 0.279 × 0.152 mm, triclinic, P, a = 10.771(3) Å, b = 13.848(3) Å, c = 22.433(5) Å, α = 101.050(10)°, β = 103.860(10)°, γ = 100.660(10)°, V = 3093.4(13) Å3, Z = 2, μ(AgKα) = 0.114 mm−1, θmax = 20.3°, 127873 reflections measured, 12250 independent (Rint = 0. 0419), R1 = 0. 0331 [I > 2σ(I)], wR2 = 0.0813 (all data), res. density peaks: 0.471 to −0.247 e Å−3, CCDC 1507709. All crystals were selected under cold protective conditions using the X-Temp2 device.18 The data were integrated with SAINT.19 A multi-scan absorption correction and a 3λ correction20 were applied using SADABS.21 The structures were solved by SHELXT22 and refined on F2 using SHELXL23 in the graphical user interface SHELXLE.24 |
| This journal is © The Royal Society of Chemistry 2017 |