
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the activity of a polyazine bridged Ru(II)–Pt(II) supramolecule in F98 rat malignant glioma cells†
Jie
Zhu
a,
José Á.
Rodríguez-Corrales
a,
Reece
Prussin
b,
Zongmin
Zhao
c,
Anthony
Dominijanni
b,
Samantha L.
Hopkins
a,
Brenda S. J.
Winkel
 *d,
John L.
Robertson
b and
Karen J.
Brewer‡
*d,
John L.
Robertson
b and
Karen J.
Brewer‡
aDepartment of Chemistry, Virginia Tech., Blacksburg, Virginia 24061-0212, USA
bSchool of Biomedical Engineering and Sciences, Virginia Tech., Blacksburg, Virginia 24061-0212, USA
cDepartment of Biological Systems Engineering, Virginia Tech., Blacksburg, Virginia 24061-0212, USA
dDepartment of Biological Sciences, Virginia Tech., Blacksburg, Virginia 24061-0406, USA. E-mail: winkel@vt.edu
First published on 17th November 2016
Abstract
The mixed-metal supramolecular complex, [(Ph2phen)2Ru(dpp)PtCl2]2+, displays significant DNA modification, cell growth inhibition, and toxicity towards F98 malignant glioma cells following visible light irradiation. The design of this complex affords superior cellular uptake and antiproliferative activity compared to the classic chemotherapeutic agent, cisplatin.
Cancer is a worldwide public health problem, a leading cause of death across all age groups.1 Aggressive infiltrative malignant glioma (MG), which is estimated to make up approximately 1.41% of the total cancer cases in 2016,1 is among the most devastating. The median survival rate for MG is approximately one year and less than 5% of patients diagnosed with MG live longer than five years, regardless of the type of therapy used.2,3 New treatments for high-grade MG are urgently needed to address and alleviate the limitations of current therapies and improve patient survival rates.
Photodynamic therapy (PDT) is a combination of chemo- and radiation therapy that uses a photosensitizer (PS), low energy light, and in some cases molecular oxygen (3O2) to produce cytotoxic reactive oxygen species (ROS) to damage neoplastic cells.4,5 This minimally-invasive, light-activated therapeutic treatment allows accurate targeting of tumor sites and reduces off-site effects associated with systemic chemotherapies.6 It is particularly promising for cancers such as MG that occur well within the 1 cm tissue penetration limit of light.
Conventional PDT agents, e.g. porphyrin-based PSs, suffer from dark toxicity, prolonged skin sensitivity, and hepatotoxicity, which limit their application as therapeutic agents.7 Transition metal complexes have emerged as promising candidates for the next generation of PDT agents due to their tunable coordination environments and varied spectral and redox properties.8–14 Recently, we reported two Ru/Os polyazine monometallic complexes as photoactivated-oxygen-dependent DNA and protein cleaving reagents.15 The complexes show enhanced cytotoxicity towards F98 rat MG cells upon irradiation with visible light. However, the oxygen dependence of these monometallic complexes limits their potential use as PDT agents due to the hypoxic environments that commonly characterize MGs and many other tumors.16
Extensive research has focused on efforts to conjugate PSs to known chemotherapeutic agents, such as cisplatin analogues, to develop a new class of potential PDT agents.17–20 Among these, mixed-metal supramolecules stand out because of their ability to interact with biomolecules (e.g. DNA and protein) simultaneously via multiple means.21,22 A promising yet challenging design is the incorporation of photochemically activated bioactive sites (PAB) with thermally activated bioactive sites (TAB) through bridging ligands (BL). These novel supramolecular architectures could be triggered by multiple pathways to react with biological targets and lead to cancer cell necrosis and/or apoptosis.
Bimetallic Ru–Pt complexes are a class of PAB-BL-TAB molecules in which the ruthenium center plays a dual role as light absorber and PAB, while the cisplatin-like moiety acts as a TAB.23–25 It has been shown that Ru–Pt heteronuclear complexes are potential PDT agents where Ru and Pt work synergistically and in a light-dependent manner, thus leading to covalent binding to DNA (platination) through the TAB, and DNA photocleavage through the PAB.26,27 Furthermore, the TAB-directed binding of DNA would localize the generation of singlet oxygen (1O2) in proximity to the target biomolecule, which could circumvent the short lifetime and root mean square (RMS) diffusion distance, 4 μs and 125 nm in water.28,29 Recent DFT/TDDFT studies have provided further evidence that the synergistic effect between the metals makes the designed Ru–Pt complexes promising multi-target anticancer drugs.30 Supramolecular Ru(II)–Pt(II) complexes thus appear to be promising new multifunctional PDT agents for the treatment of MGs and other intractable cancers.
We previously reported the synthesis and preliminary characterization of [(Ph2phen)2Ru(dpp)PtCl2]2+ (Ph2phen = 4,7-diphenyl-1,10-phenanthroline; dpp = 2,3-bis(2-pyridyl) pyrazine), herein referred to as Ru(II)–Pt(II) (Fig. 1).26,27 This complex tethers a Ru(II) PS to a DNA targeting cis-Pt(II)Cl2 motif, making it an unprecedented 3MLCT-activated DNA photobinding agent that also produces ROS. By harnessing the properties of the individual components in a supramolecular architecture, this compound opens interesting new prospects for a combined anticancer approach. In fact, we showed that excitation with visible light leads to population of a Ru(dπ) → dpp(π*) MLCT state, which affords enhanced electron density on the dpp ligand, while reducing the Lewis acidity of the platinum center with consequent photolabilization of the Pt–Cl bond.27,30 The photorelease of such ligands is expected to facilitate its reaction with biological targets and thus represents a new pathway of activation of Pt(II) compounds to the classical hydrolysis mechanism. Furthermore, preliminary experiments using blue light activation provided evidence for DNA cleavage even after oxygen removal through six freeze–pump–thaw cycles.26
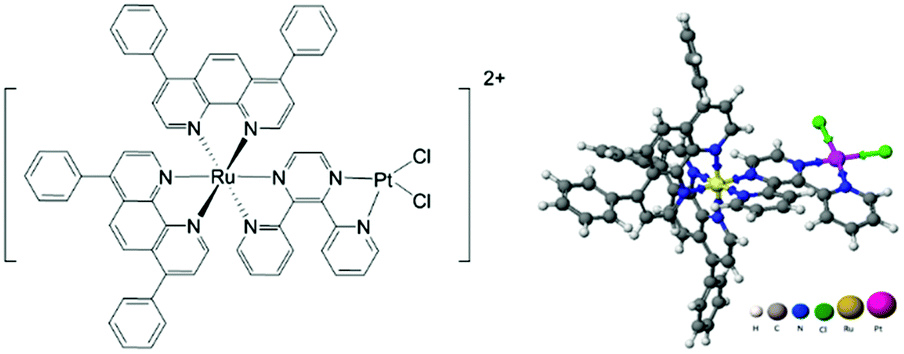 | ||
| Fig. 1 Structure of [(Ph2phen)2Ru(dpp)PtCl2]2+. | ||
In this study we demonstrate the photodynamic activity of Ru(II)–Pt(II) both in vitro with purified plasmid DNA and for the first time with rat MG F98 cells, a model for human glioblastoma. The complex exhibited low toxicity in the dark, but substantially higher cytotoxicity than the classic chemotherapeutic drug cisplatin following exposure to blue light. We also uncovered evidence that the effective cytotoxicity of Ru(II)–Pt(II) at least in part reflects the higher cell membrane permeability and cellular uptake of the complex relative to cisplatin and two other classical chemotherapeutic drugs.
The Ru(II)–Pt(II) title complex was prepared using the previously described building block method and validated by electrochemical analyses (Fig. S1 and S2, ESI†).27 To examine the bioactivity of this molecule in further detail, Ru(II)–Pt(II) was reacted with plasmid DNA in vitro under dark and irradiated conditions (455 and 625 nm). The samples were then examined using agarose gel electrophoresis, which provides a quick and facile evaluation of the complex for both binding and cleavage of DNA.31 As presented in Fig. 2, plasmid DNA incubated with the complex in the dark (lane D) showed a slight decrease in electrophoretic mobility of the supercoiled form (SC) compared to the control containing plasmid DNA alone (lane C), likely due to covalent binding of the complex to the biomolecule. This change is enhanced upon incubation at 37 °C (lane T) and is therefore attributed to thermal aquation of the cis-PtCl2 moiety, a well-known behavior of cisplatin, followed by substitution of an aquo ligand for a purine base in DNA.32 Both blue (lane B) and red (lane R) light activation enhanced the interaction of the complex with DNA compared to dark conditions. The increase in the relative amount of open circular (OC) DNA in these samples is a clear indication of enhanced cleavage, which could arise from DNA oxidation by singlet oxygen or other ROS originating from dissolved O2 or H2O, respectively.27 Since both cleavage and binding were promoted upon photolysis, the two metals play a synergistic role in the multifunctional modification of the biomolecule: the Ru-based PS generates oxidative stress while the Pt-based moiety promotes DNA platination. Importantly, the ability of Ru(II)–Pt(II) to bind and cleave DNA was preserved in deoxygenated solutions (lanes B-Ar and R-Ar) that were purged with argon for 20 min prior to photolysis, which would deplete the majority of the dissolved oxygen in these samples. This remarkable behavior reveals that the ability of Ru(II)–Pt(II) to cleave DNA is not limited by its low singlet oxygen quantum yield, determined to be 0.06(7) in methanol solution.26 Given the short lifetime and diffusion limited activity of 1O2,28,29 we propose that the formation of covalent bonds between Ru(II)–Pt(II) and DNA may serve as a crucial localization step that leads to generation of singlet oxygen or other ROS in the immediate proximity of the biomolecule, facilitating DNA cleavage even under oxygen-depleted conditions. This postulate would explain why DNA cleavage was abolished when photolysis solutions were spiked with NaN3,26 since it is likely that those conditions would result in substitution of the labile ligands and inhibit covalent binding to DNA.
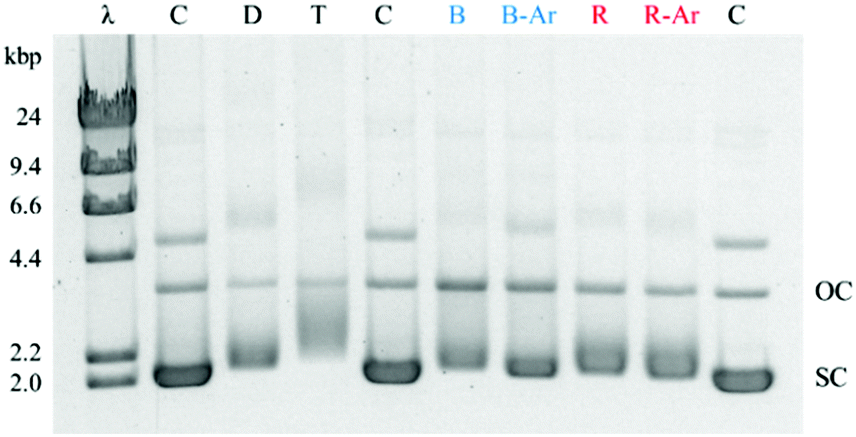 | ||
| Fig. 2 DNA gel shift assay for Ru(II)–Pt(II). λ: DNA weight marker, C: pUC19 DNA (2686 bp), D = DNA/complex incubated at room temperature in the dark, T = DNA/complex incubated at 37 °C, B = DNA/complex exposed to a blue (455 nm) LED with O2, B-Ar = DNA/complex exposed to a blue (455 nm) LED after Ar purging, R = DNA/complex exposed to a red (625 nm) LED with O2, R-Ar = DNA/complex exposed to a red (625 nm) LED after Ar purging, OC = open circular DNA, SC = supercoiled DNA. All incubation steps were 1 h. | ||
We also investigated the reactivity of Ru(II)–Pt(II) with bovine serum albumin (BSA), a prototypical protein. Similar to the DNA experiment, samples containing the complex and BSA (in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 10:1 molar ratios) were incubated in the dark or photolyzed under blue or red light for up to 3 h and analysed by electrophoresis.
:1 and 10:1 molar ratios) were incubated in the dark or photolyzed under blue or red light for up to 3 h and analysed by electrophoresis.
Interestingly, there was no detectable difference in the electrophoretic mobility of the treated and untreated samples under any of the conditions tested, including up to 3 h of photolysis (see Fig. S4, ESI†). This is in striking contrast to the parent compound, [(Ph2phen)2Ru(dpp)]2+, which cleaves BSA even with 1 h of photolysis through an oxygen-dependent mechanism.15 The monometallic complex shows a 10-fold longer lived excited state than Ru(II)–Pt(II), which results in a higher 1O2 quantum yield and significant BSA cleavage.26
The difference in activity between BSA and DNA likely reflects the number of covalent binding sites in these molecules. Covalent binding of cisplatin to DNA occurs primarily at purine bases,32 which account for 47% of the nitrogen bases in pUC19 DNA. In contrast, covalent binding to proteins occurs predominantly at exposed methionine, cysteine, and histidine residues, which make up less than 4% of the amino acids in BSA.33 We hypothesize that without efficient covalent binding with BSA, cleavage of BSA by Ru(II)–Pt(II) is limited by its short-lived excited state and low 1O2 quantum yield upon irradiation, while the cleavage of DNA is promoted by the highly efficient and covalent binding of Ru(II)–Pt(II) to this biomolecule. Although further experiments are needed to determine the affinity of Ru(II)–Pt(II) for protein in vivo, the apparent selectively for DNA over BSA suggests that this complex could exhibit decreased drug loss and off-target toxicity relative to other drugs.
The octanol/water partition coefficient of Ru(II)–Pt(II) was determined to evaluate the hydrophobicity and lipophilicity of the complex, which are correlated with the ability to traverse and distribute throughout the body, the blood brain barrier, and cell membranes (the detailed experimental procedure presented in ESI†). The complex exhibited a logPo/w of 0.55, which falls in the range for optimal oral absorption and/or central nervous system penetration (+1.5 ± 1.0).34 Based on logPo/w values (Table 1), the title complex demonstrates increased potential for cell membrane permeability compared to cisplatin, carboplatin, and TMZ, which are FDA approved drugs for MG treatment. This is hypothesized to arise from the lipophilicity of the aromatic ligands within the Ru(II) PS subunit.35
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Po/w values for FDA approved anticancer agents and [(Ph2phen)2Ru(dpp)PtCl2]Cl2
Po/w values for FDA approved anticancer agents and [(Ph2phen)2Ru(dpp)PtCl2]Cl2
The potential for enhanced permeability was further corroborated through cellular uptake studies in which ICP-MS analysis was used to quantify intracellular platinum content. After 2 h incubation of F98 MG cells with Ru(II)–Pt(II) or cisplatin (75 μM) in the dark, the internalization of Ru(II)–Pt(II) was 18.75 ± 1.25 pmol per cell, which is 10 times higher than the corresponding value for cisplatin (see ESI†). Hence, coupling of the ruthenium PS dramatically increased the cellular uptake of the cisplatin moiety by F98 MG cells within a short period of exposure.
The cytotoxicity of Ru(II)–Pt(II) was evaluated relative to cisplatin in F98 rat MG cells by the trypan blue dye exclusion assay using an automated Vi-CELL® Cell Viability Analyzer. As shown in Fig. 3, in the absence of light Ru(II)–Pt(II) displayed no detectable cytotoxicity after 48 h of exposure at up to 50 μM concentration, with approximately 20% reduction in viability remaining at 75 μM. This was in contrast to cisplatin, which exhibited detectable effects on viability even at 25 μM. The observed difference could be due to the increased steric hindrance around the Pt moiety induced by the covalently-conjugated PS [(Ph2phen)2Ru(dpp)]2+. Additionally, the PS unit decreases electronic density at the Pt subunit, thus slowing down its aquation and allowing controlled photoactivation of the supramolecule. The low cytotoxicity of Ru(II)–Pt(II) in the dark is a critical feature of this molecule with regard to its potential as a PDT agent.
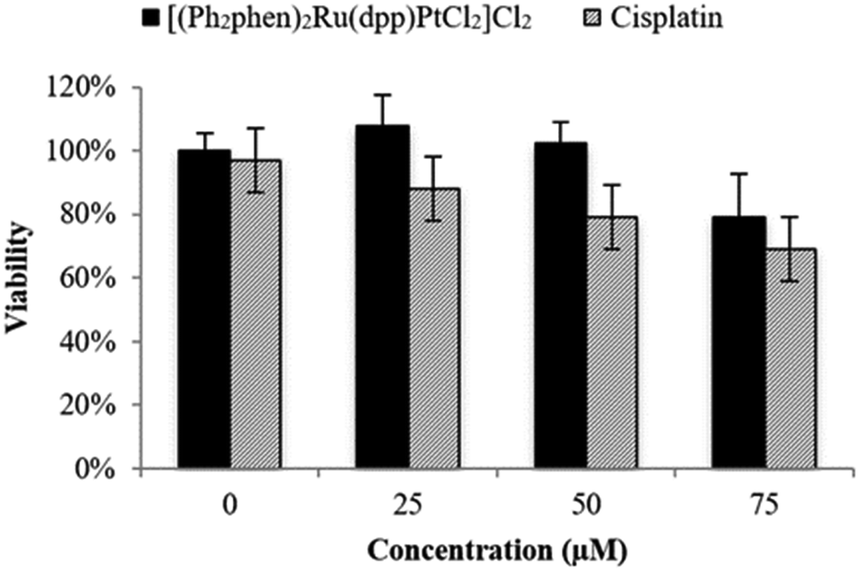 | ||
| Fig. 3 Cytotoxicity in the dark graphed as percent viability (treated/untreated control F98 MG cells) vs. concentration (μM) of Ru(II)–Pt(II) and cisplatin following 48 h incubation in the dark. | ||
The cytotoxic activity of Ru(II)–Pt(II) increased dramatically after blue light irradiation (Fig. 4). Two identical sets of samples were prepared and either incubated in the dark or irradiated in a lab-built LED array that emits blue light (470 nm, 4.35 J cm−2). For the photolysis treatment, cells were incubated for 15 min with 0 to 75 μM Ru(II)–Pt(II), photolyzed for 30 min, and left in the incubator for a final 15 min before the complex solution was removed and replaced with fresh medium. Photolysis had no effect on cell growth in the absence of the complex. However, in the presence of even the lowest concentration of Ru(II)–Pt(II) a strong antiproliferative activity was observed starting immediately after photolysis was completed (0 h time point). Further cell growth inhibition was observed after 48 h. Cells that were treated with Ru(II)–Pt(II) showed 60% lower viability following photolysis than those incubated in the dark, a dramatically enhanced light-induced toxicity. The effective concentration at which 50% of the cells showed a response (EC50) was approximately 20 μM under photolysis conditions, which is lower than for either [(Ph2phen)2Ru(dpp)]2+ or cisplatin alone.15 Revisiting the results from the DNA gel shift assay, we propose that photoactivation of Ru(II)–Pt(II) leads to population of a Ru(dπ) → dpp(π*) 3MLCT state that produces oxidative stress and DNA platination, finally resulting in cell death. The synergistic effect between the two metals appeared to enhance the antiproliferative activities of each individual component towards aggressive MG cells. This promising result encourages further investigation on bimetallic supramolecular complexes as potential new PDT agents for cancer treatment.
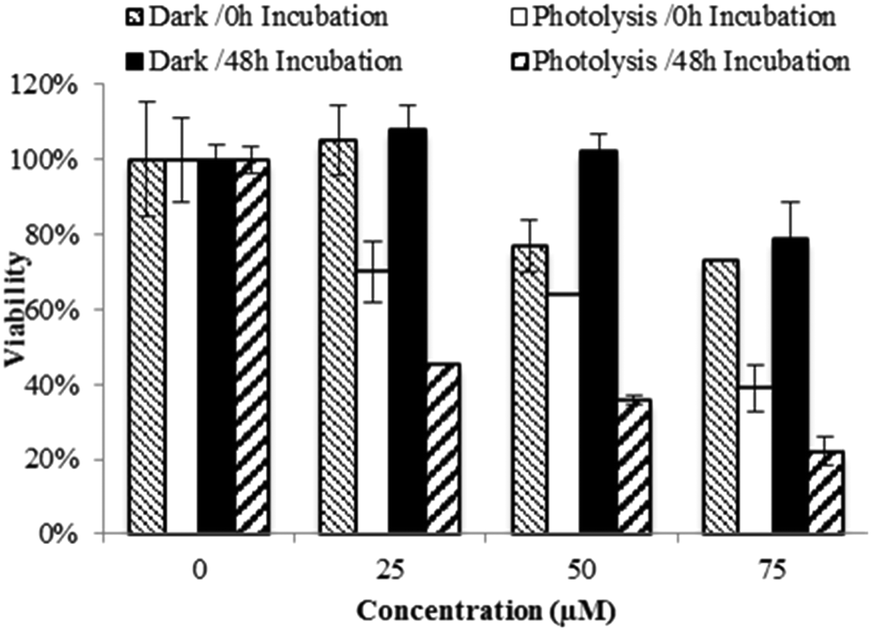 | ||
| Fig. 4 Cytotoxicity graphed as percent viability (treated/untreated control F98 MG cells) vs. concentration (μM) of [(Ph2phen)2Ru(dpp)PtCl2]Cl2 at 0 and 48 h of incubation in darkness (dark) or after photolysis with blue light (photolysis). | ||
In summary, [(Ph2phen)2Ru(dpp)PtCl2]Cl2 appears to exhibit synergistic effects between the metal centers, enabling multiple toxicity pathways (i.e. platination and oxidative stress) that lead to the enhanced cytotoxicity observed herein. In vitro assays showed preferential photoactivated modification of DNA, with little effect on protein under the conditions used here. This potential selective reactivity could minimize off-target binding and cytotoxic side effects in vivo. The present study provides the first example of a blue light photoactivated anticancer platinum(II) complex with activity against F98 rat MG cells following short-term drug exposure. These results suggest that [(Ph2phen)2Ru(dpp)PtCl2]Cl2 could provide the basis for development of an effective multifunctional PDT agent for treatment of human glioblastoma multiforme and other severe cancers.
Acknowledgement is made to VT ICTAS 119552 and the National Science Foundation (grant CHE-1301131) for funding of this work. The authors are grateful to Drs Amanda J. Morris, Brian E. Hanson, and Roberto Padilla for helpful discussions. We thank Dr Jeffrey L. Parks for providing the ICP-MS data. We particularly acknowledge the late Dr Karen J. Brewer, a great mentor and generous friend.
Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- H. R. Kumar, X. Zhong, J. A. Sandoval, R. J. Hickey and L. H. Malkas, Expert Rev. Neurother., 2008, 8, 1497–1506 CrossRef PubMed.
- D. A. Reardon, J. N. Rich, H. S. Friedman and D. D. Bigner, J. Clin. Oncol., 2006, 24, 1253–1265 CrossRef CAS PubMed.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed.
- G. Stochel, A. Wanat, E. Kulis and Z. Stasicka, Coord. Chem. Rev., 1998, 171, 203–220 CrossRef CAS.
- M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897–3915 CrossRef CAS PubMed.
- R. Bonnett, Chemical aspects of photodynamic therapy, CRC Press, 2000 Search PubMed.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS PubMed.
- V. H. S. van Rixel, B. Siewert, S. L. Hopkins, S. H. C. Askes, A. Busemann, M. A. Siegler and S. Bonnet, Chem. Sci., 2016, 7, 4922–4929 RSC.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649–9651 RSC.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089–1097 CrossRef CAS PubMed.
- S. A. Poteet, M. B. Majewski, Z. S. Breitbach, C. A. Griffith, S. Singh, D. W. Armstrong, M. O. Wolf and F. M. MacDonnell, J. Am. Chem. Soc., 2013, 135, 2419–2422 CrossRef CAS PubMed.
- C. Mari, H. Huang, R. Rubbiani, M. Schulze, F. Würthner, H. Chao and G. Gasser, Eur. J. Inorg. Chem., 2016 DOI:10.1002/ejic.201600516.
- U. Basu, I. Khan, A. Hussain, P. Kondaiah and A. R. Chakravarty, Angew. Chem., Int. Ed., 2012, 51, 2658–2661 CrossRef CAS PubMed.
- J. Zhu, A. Dominijanni, J. Á. Rodríguez-Corrales, R. Prussin, Z. Zhao, T. Li, J. L. Robertson and K. J. Brewer, Inorg. Chim. Acta, 2017, 454, 155–161 CrossRef CAS.
- D. A. Reardon, J. N. Rich, H. S. Friedman and D. D. Bigner, J. Clin. Oncol., 2006, 24, 1253–1265 CrossRef CAS PubMed.
- C. Lottner, K.-C. Bart, G. Bernhardt and H. Brunner, J. Med. Chem., 2002, 45, 2064–2078 CrossRef CAS PubMed.
- J. Mao, Y. Zhang, J. Zhu, C. Zhang and Z. Guo, Chem. Commun., 2009, 908–910 RSC.
- I. Manet, F. Manoli, M. P. Donzello, C. Ercolani, D. Vittori, L. Cellai, A. Masi and S. Monti, Inorg. Chem., 2011, 50, 7403–7411 CrossRef CAS PubMed.
- J. T. F. Lau, P.-C. Lo, W.-P. Fong and D. K. P. Ng, J. Med. Chem., 2012, 55, 5446–5454 CrossRef CAS PubMed.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282–283, 110–126 CrossRef CAS PubMed.
- A. G. Weidmann, A. C. Komor and J. K. Barton, Comments Inorg. Chem., 2014, 34, 114–123 CrossRef CAS PubMed.
- R. L. Williams, H. N. Toft, B. Winkel and K. J. Brewer, Inorg. Chem., 2003, 42, 4394–4400 CrossRef CAS PubMed.
- S. L. Hopkins, L. Stepanyan, N. Vahidi, A. Jain, B. S. J. Winkel and K. J. Brewer, Inorg. Chim. Acta, 2017, 454, 229–233 CrossRef CAS.
- T. Yano, S. Hishida, M. Nakai and Y. Nakabayashi, Inorg. Chim. Acta, 2017, 454, 162–170 CrossRef CAS.
- S. L. H. Higgins, T. A. White, B. S. J. Winkel and K. J. Brewer, Inorg. Chem., 2011, 50, 463–470 CrossRef CAS PubMed.
- S. L. H. Higgins, A. J. Tucker, B. S. J. Winkel and K. J. Brewer, Chem. Commun., 2012, 48, 67–69 RSC.
- M. A. J. Rodgers and P. T. Snowden, J. Am. Chem. Soc., 1982, 104, 5541–5543 CrossRef CAS.
- R. W. Redmond and I. E. Kochevar, J. Photochem. Photobiol., 2006, 82, 1178–1186 CrossRef CAS PubMed.
- M. E. Alberto, N. Russo and C. Adamo, Chem. – Eur. J., 2016, 22, 9162–9168 CrossRef CAS PubMed.
- A. J. Prussin, Ii, D. F. Zigler, A. Jain, J. R. Brown, B. S. J. Winkel and K. J. Brewer, J. Inorg. Biochem., 2008, 102, 731–739 CrossRef CAS PubMed.
- S. E. Sherman and S. J. Lippard, Chem. Rev., 1987, 87, 1153–1181 CrossRef CAS.
- G. Ferraro, L. Massai, L. Messori and A. Merlino, Chem. Commun., 2015, 51, 9436–9439 RSC.
- H. Pajouhesh and G. R. Lenz, NeuroRx, 2005, 2, 541–553 CrossRef PubMed.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2006, 129, 46–47 CrossRef PubMed.
- D. Screnci, M. J. McKeage, P. Galettis, T. W. Hambley, B. D. Palmer and B. C. Baguley, Br. J. Cancer, 2000, 82, 966–972 CrossRef CAS PubMed.
- Q. Zhou and J. M. Gallo, Neuro-Oncology, 2009, 11, 301–310 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc07978d |
| ‡ Deceased, October 24, 2014. |
| This journal is © The Royal Society of Chemistry 2017 |