
Synthesis and full characterization of an iridium B–H activation intermediate of the monocarba-closo-dodecaborate anion†
Yunjun
Shen
a,
Yani
Pan
a,
Jiyong
Liu
a,
Tosaporn
Sattasathuchana
b,
Kim K.
Baldridge
*c and
Simon
Duttwyler
*a
aDepartment of Chemistry, Zhejiang University, Zheda Road 38, 310027 Hangzhou, P. R. China. E-mail: duttwyler@zju.edu.cn
bDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland
cSchool of Pharmaceutical Science and Technology, Tianjin University, A203/Building 24, 92 Weijin Road, Nankai District, 300072 Tianjin, P. R. China. E-mail: kimb@oci.uzh.ch
First published on 29th November 2016
Abstract
The preparation and full characterization of an iridium complex of the monocarba-closo-dodecaborate anion is reported. It was prepared by B–H bond activation using a tosyl amide directing group. Analysis by spectroscopic methods and X-ray crystallography revealed the presence a direct B–Ir interaction. The carborane acts as a B,N chelating ligand towards the Ir(Cp*)(solvent) fragment, resulting in a monomeric complex that is inert in solution and the solid state. Treatment with N-chlorosuccinimide resulted in selective monochlorination of the B–Ir position. In addition, its structure, spectroscopic features and reactivity were investigated by DFT calculations.
The monocarbaborane anion [CB11H12]− (I) and dicarbaboranes C2B10H12 (II) are icosahedral boron cage compounds in which one or more boron vertices are replaced by a carbon unit (Fig. 1). They feature three-dimensional delocalization of electron density, which leads to exceptional chemical and thermal stability.1,2 Applications of carboranes have been reported in the fields of coordination,3 supramolecular4 and medicinal5 chemistry, as well as fluorescence/phosphorescence6 and materials science.7 The extremely weakly coordinating nature of halogenated derivatives of I has made them the anions of choice as counterions reactive intermediates and of highly active cationic catalysts.8
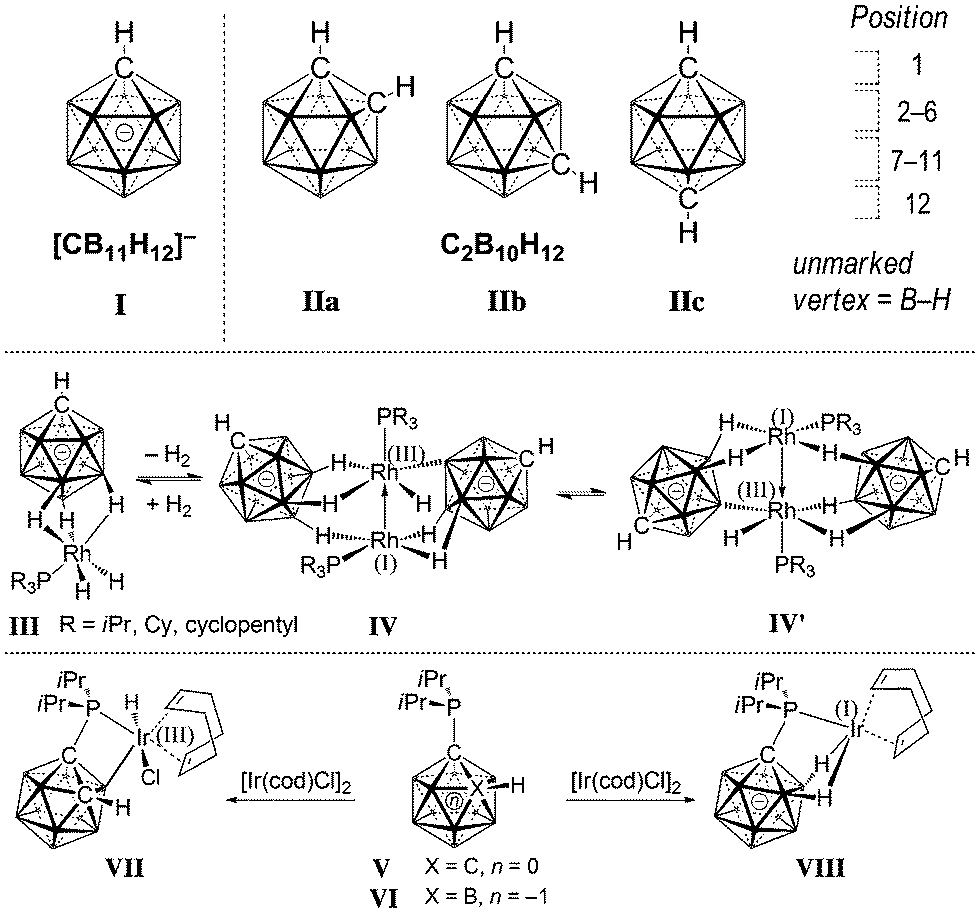 | ||
| Fig. 1 General framework of icosahedral carboranes and selected reported transition metal complexes. | ||
Early studies demonstrated that iridium complexes are capable of activating boron hydride clusters by formation of a B–[TM] intermediate.9 Stoichiometric reactions showed that other metals such as Co, Ru, and Pt can also bring about substitution at boron vertices.10 Recently, catalytic protocols for the functionalization of II have been developed on the basis of Ni, Pd and Ir.11 In contrast to the advances in the chemistry of II, well-controlled B–H activation of I by transition metals has not been reported. Weller and coworkers observed that agostic Rh(III) complexes of the type III can undergo cage ethenylation in the presence of ethene.12 Moreover, placing III in a vacuum resulted in loss of H2 and reversible formation of Rh(I)/Rh(III) species IV, which was in a fast quilibrium with its isomer IV′ on the NMR timescale at room temperature. In a comparative study, Lavallo and coworkers investigated the divergent reactivities of the {CB11} and {C2B10} cages with respect to oxidative addition towards Ir(I).13a,b Phosphine-substituted V readily reacts with [Ir(cod)Cl]2 to afford VII with a B–Ir bond. Conversely, mono-carbaborane VI did not undergo oxidative addition but furnished agostic complex VIII instead. Very recently, Zhang and Ma probed Lavallo's {CB11} and {C2B10} as well as closely related systems by DFT calculations.13c Their results indicated that oxidative addition in the case of VI was not favorable; however, fluorine-containing groups at the phosphorous center or a spacer (CH2, NH or O) between Ccage and P rendered formation of a B–Ir bond energetically feasible.13d Herein, we report on the first preparation and full characterization of an inert {CB11}–iridium intermediate with a direct cage B–Ir interaction, formed by combination of an amide-substituted carborane precursor and an Ir(III) complex.
Cyclometalation is a key step in transition metal-mediated C–H bond functionalization. Diverse directing groups have been used to effect C–H activation followed by coupling to a new group or more complex cascade processes.14 Our goal was to identify a coordinating moiety that would allow for characterization of an intermediate of I with a direct B–metal bond and potential subsequent substitution reactions. For the choice of the directing group, three main points had to be addressed: (a) its efficient introduction to the starting material I, (b) its ability to coordinate to the metal and enable B–H activation beyond agostic interactions, and (c) lack of competitively activated C–H bonds. We found that the sulfonamide functionality (C(O)NHSO2Ar) fulfills these criteria.
Tosyl amide 1 was synthesized from carborane 1-carboxylic acid (Scheme 1). Treatment with oxalyl chloride, followed by reaction with tosyl amine, afforded 1 in 72% yield. This product was fully characterized by spectroscopic methods (see the ESI†). Furthermore, single crystals of 1 enabled the elucidation of its solid-state structure. Observed distances (Å) are C1–C2 1.505(3), C2–O 1.206(2), C2–N 1.378(3) and N–S 1.657(2). The sum of angles around C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is 360.0°, and the torsion angle C1–C2–N–S of 176.6° indicates coplanarity of the entire sulfonamido moiety. All C1–B distances are comparable to those of I, averaging out at 1.714 Å.
O is 360.0°, and the torsion angle C1–C2–N–S of 176.6° indicates coplanarity of the entire sulfonamido moiety. All C1–B distances are comparable to those of I, averaging out at 1.714 Å.
 | ||
| Scheme 1 Synthesis and X-ray crystal structure of carborane amide 1 (C–H hydrogen atoms omitted for clarity; 25% displacement ellipsoids). | ||
Reaction of 1 with a stoichiometric amount of IrCp*(OAc)2 (Cp* = [C5Me5]−) in acetonitrile at 25 °C resulted in the formation of a yellow precipitate, which was collected by filtration (Scheme 2a). Concentration of the filtrate by slow evaporation of ca. 50% of the solvent afforded yellow crystals. Both solids were identified as iridacycle 2, the combined yield being 80%. It is likely that this step proceeds via concerted metalation–deprotonation assisted by acetate as the base. This mechanism has been proposed for C–H activation reactions where Rh(III) or Ir(III) complexes are used to generate the C–[M(III)] intermediate.15 Formation of 2 was also observed in solvents other than acetonitrile, namely, DMSO, dichloromethane and 1,2-dichloroethane, indicating that acetonitrile is not crucial for cyclometalation. Conversely, [Ir(cod)Cl]2 did not cause B–H activation at 25 °C or 60 °C.
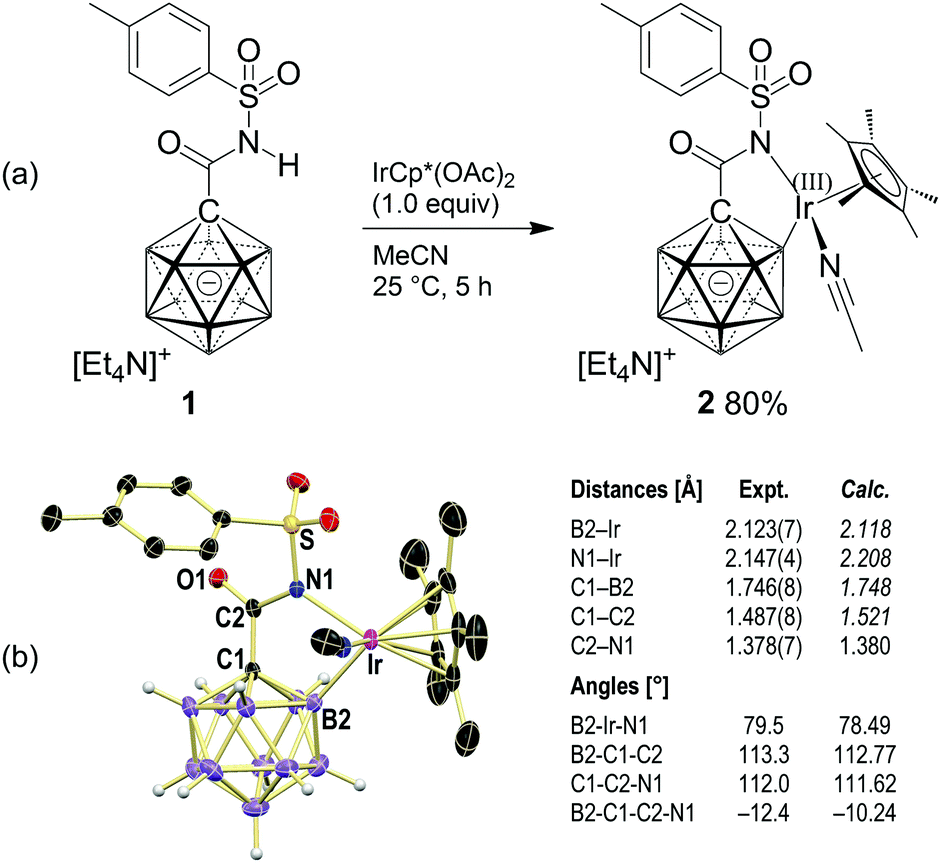 | ||
| Scheme 2 Synthesis and X-ray crystal structure of 2 (cation and C–H hydrogen atoms omitted for clarity; 25% displacement ellipsoids). | ||
X-ray crystallography showed that the anion of 2 consists of the carborane amide coordinated as a bidentate B,N ligand to the IrCp*(MeCN) fragment, with distances of B1–Ir 2.123(7) Å and N1–Ir 2.147(4) Å (Scheme 2b). The former is similar to that found in VII (2.094 Å), and there is no indication of a residual H atom attached to B1 or Ir. The C1–B1 bond (1.746(8) Å) is slightly elongated compared to the average of the other four C1–B distances (1.709 Å) and those of 1, while the C2–N distance (1.378(7) Å) is identical to that observed for 1. The five-membered metallacycle exhibits only slight puckering, with internal angles of B1–Ir–N1 79.5°, B1–C1–C2 113.3°, C1–C2–N1 112.0° and a torsion angle of B1–C1–C2–N1 −12.4°.
Multinuclear NMR analysis of 2 in CD3CN solution at room temperature was fully consistent with the structure found by X-ray crystallography. In the 1H and 1H{11B} NMR spectra, sharp signals were obtained except for the B–H nuclei, which appeared as broad overlapping resonances at 1.9–1.3 ppm even upon decoupling from 11B (Fig. 2 and Fig. S1, ESI†). CH3CN ligand exchange with CD3CN solvent molecules resulted in the liberation of one equivalent of acetonitrile (1.96 ppm, residual CHD2CN referenced to 1.94 ppm). The 11B and 11B{1H} NMR spectra showed resonances around −7.6, −10.2 to −14.7 and −14.7 to −17.2 ppm in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:2, consistent with 11 boron atoms including B–Ir. No other peaks were found in the range of +180 to −80 ppm. A detailed discussion of the 11B NMR data is provided in the ESI.† Based on this analysis, the signal at −7.6 ppm is assigned to B12, while B–Ir and B9 give rise to overlapping signals centered at −16.2 and −15.7 ppm.16
:8:2, consistent with 11 boron atoms including B–Ir. No other peaks were found in the range of +180 to −80 ppm. A detailed discussion of the 11B NMR data is provided in the ESI.† Based on this analysis, the signal at −7.6 ppm is assigned to B12, while B–Ir and B9 give rise to overlapping signals centered at −16.2 and −15.7 ppm.16
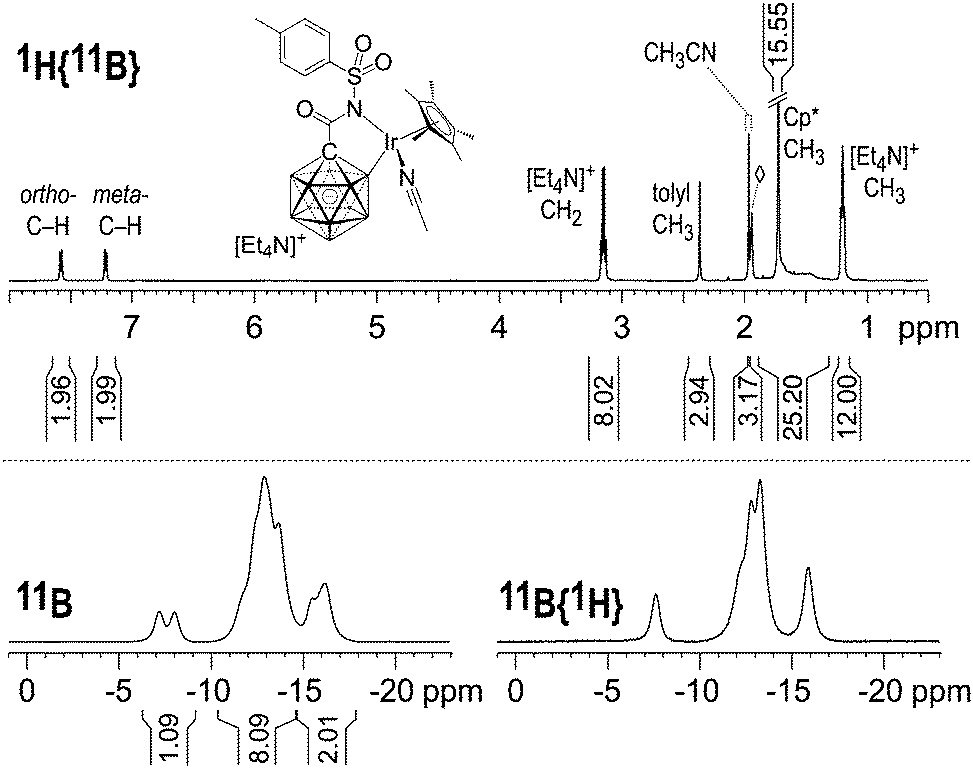 | ||
| Fig. 2 NMR spectra of 2 (500 MHz (1H), 8 mg in 0.6 mL CD3CN, 23 °C). The 1H{11B} integral from 1.9 to 1.3 ppm comprises (15 + 10) protons from Cp* and the carborane cage; ◊ = residual CHD2CN. | ||
The IR spectrum of 2 clearly showed the disappearance of the N–H stretching of 1 at 3291 cm−1. In addition, the intense diagnostic B–H absorptions centered at 2543 cm−1 experienced a shift to 2531 cm−1. In line with this latter observation, signals at 1398 and 1166 cm−1, assigned to SO2 stretchings, moved to 1314 and 1156 cm−1, respectively.
Solutions of 2 in acetonitrile under a nitrogen atmosphere were inert over weeks; heating to 70 °C in a sealed NMR tube for two days did not lead to any decomposition. In the solid state, either as a powder or in crystalline form, 2 remained unchanged in air for two weeks. When acetonitrile solutions were stirred exposed to air, a color change from yellow to brown was observed, along with formation of a brown precipitate. NMR analysis revealed decomposition to a mixture of products, none of which could be identified unambiguously.
DFT calculations performed on 1 and 2 agree well with the observed structural and spectroscopic parameters (for computational details, see the ESI†). Geometry optimization at the B97-D/Def2-TZVPD level of theory gave structures consistent with the results from X-ray crystallography (Scheme 2b and Fig. S10, S12, Tables S2, S4, ESI†). In particular, all key distances and angles of 2 were reproduced with high accuracy. The HOMO of 2 is primarily localized around the iridium fragment as well as N1 and B2; the LUMO is associated with the tolyl–SO2 moiety and the Cp* π system (Fig. 3). Electrophilic and nucleophilic frontier density disclosed the most reactive areas of the molecule (Fig. S14, ESI†). They indicate B2 and Ir as the centers with the highest probability of attack by an electrophile, while attack by a nucleophile is predicted to occur at Cipso of the tolyl ring. 1H, 13C and 11B NMR chemical shifts, calculated at the B97-D/Def2-QZVPPD//B97-D/Def2-TZVPD CSGT level of theory in acetonitrile, are also in agreement with the experimental values for 1 and 2. Specifically, the 11B NMR signals of 2 are predicted to be −6.85 ppm for B12 and −16.99 for B9, while all other B–H vertices fall in the range of −10.52 to −14.41 ppm. For the 11B chemical shift of B2 (+7.26), there is a significant difference from the experimental value by greater than the expected error range. Its origin is unclear given additional investigations into the computational methodology (e.g., DFT-type, basis set extent, NMR method, relativistic corrections, calibration scheme, etc.). However, interestingly, further NPA charge analysis shows that, while the majority of the B atoms in the carborane have partial negative charge, the B attached to Ir carries a partial positive charge, with the B–Ir bond polarized as B(+0.226)–Ir(+0.149). Additionally, NBO bond analysis shows a strong D/A interaction between the B lone pair and the Ir–N1 σ bond. Further considerations into this issue are ongoing.
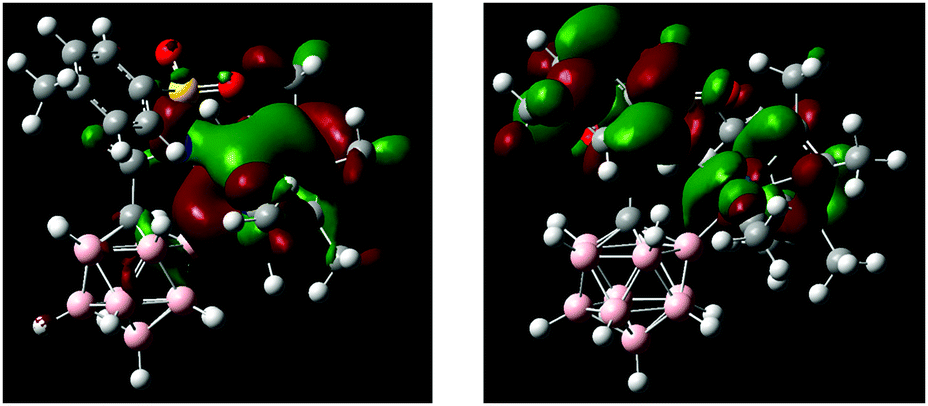 | ||
| Fig. 3 HOMO (left) and LUMO (right) of the anion of 2. | ||
Electrophilic chlorination was used as a model reaction to test the reactivity of 2. Based on the frontier density plots, attack was expected to take place around the B2–Ir moiety. Synthesis of a B2-substituted derivative seemed intruiging provided that selective functionalization at this position is unprecedented. Treatment with N-chlorosuccinimide followed by cation exchangeindeed afforded product 3 in 68% yield (Scheme 3); no other regioisomers were detected in this transformation. In order to probe the reactivity of the iridium center, ligand exchange reactions were performed. Pyridine and dppe (Ph2P–(CH2)2–PPh2) readily and completely replaced MeCN of 2; PPh3 lead to partial exchange under equilibrating conditions.
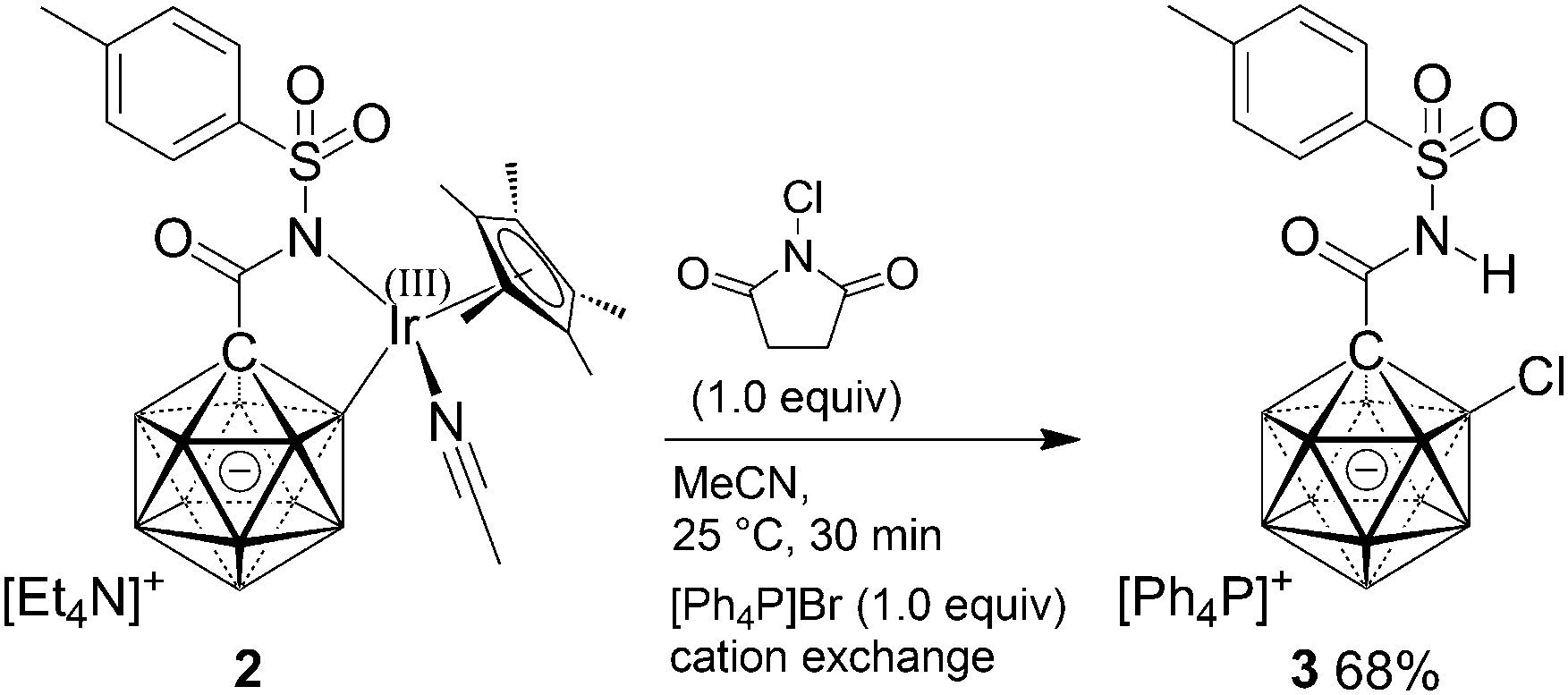 | ||
| Scheme 3 Regioselective monochlorination of 2. | ||
In conclusion, the first iridium-promoted B–H activation of the {CB11} cage is reported, affording a retainable complex with B,N coordination of the cluster. It was characterized by spectroscopic methods and X-ray crystallography and also probed by DFT calculations. Chlorination provided selective functionalization of the B2 position. This study sets the stage for catalytic B–H activation of monocarboranes, and such work is currently pursued in our laboratory.
This work was supported by the Natural Science Foundation of China (grant 107305-N11412), the National Basic Research Program of China (973 project 2015CB856500) and the Chinese “1000 Young Talents Plan”.
Notes and references
- (a) R. N. Grimes, Carboranes, Elsevier, Amsterdam, 2nd edn, 2011 Search PubMed; (b) N. S. Hosmane, Boron Science: New Technologies and Applications, Taylor & Francis Books/CRC, Boca Raton, FL, 2011 Search PubMed.
- (a) C. A. Reed, Acc. Chem. Res., 1998, 31, 133–139 CrossRef CAS; (b) C. Douvris and J. Michl, Chem. Rev., 2013, 113, PR179–PR233 CrossRef PubMed; (c) C. Knapp, Weakly Coordinating Anions: Halogenated Borates and Dodecaborates, Comprehensive Inorganic Chemistry II, Elsevier, Amsterdam, 2013, vol. 1, pp. 651–679 Search PubMed; (d) R. N. Grimes, Dalton Trans., 2015, 44, 5939–5956 RSC; (e) J. Poater, M. Solà, C. Viñas and F. Teixidor, Chem. – Eur. J., 2016, 22, 7437–7443 CrossRef CAS PubMed.
- (a) A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed; (b) M. E. El-Zaria, H. Arii and H. Nakamura, Inorg. Chem., 2011, 50, 4149–4161 CrossRef CAS PubMed; (c) Z.-J. Yao and G.-X. Jin, Coord. Chem. Rev., 2013, 257, 2522–2535 CrossRef CAS; (d) M. J. Asay, S. P. Fisher, S. E. Lee, F. S. Tham, D. Borchardt and V. Lavallo, Chem. Commun., 2015, 51, 5359–5362 RSC; (e) S. P. Fisher, A. El-Hellani, F. S. Tham and V. Lavallo, Dalton Trans., 2016, 45, 9762–9765 RSC; (f) L. E. Riley, A. P. Y. Chan, J. Taylor, W. Y. Man, D. Ellis, G. M. Rosair, A. J. Welch and I. B. Sivaev, Dalton Trans., 2016, 45, 1127–1137 RSC; (g) J. Holmes, C. M. Pask, M. A. Fox and C. E. Willans, Chem. Commun., 2016, 52, 6443–6446 RSC.
- (a) L. Kobr, K. Zhao, Y. Shen, R. K. Shoemaker, C. T. Rogers and J. Michl, Adv. Mater., 2013, 25, 443–448 CrossRef CAS PubMed; (b) R. D. Kennedy, V. Krungleviciute, D. J. Clingerman, J. E. Mondloch, Y. Peng, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, J. T. Hupp, T. Yildirim, O. K. Farha and C. A. Mirkin, Chem. Mater., 2013, 25, 3539–3543 CrossRef CAS; (c) C. E. Housecroft, J. Organomet. Chem., 2015, 798, 218–228 CrossRef CAS.
- (a) A. F. Armstrong and J. F. Valliant, Dalton Trans., 2007, 4240–4251 RSC; (b) Z. J. Lesnikowski, Collect. Czech. Chem. Commun., 2007, 72, 1646–1658 CrossRef CAS; (c) I. B. Sivaev and V. V. Bregadze, Eur. J. Inorg. Chem., 2009, 1433–1450 CrossRef CAS; (d) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed; (e) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (f) B. C. Das, P. Thapa, R. Karki, C. Schinke, S. Das, S. Kambhampati, S. K. Banerjee, P. Van Veldhuizen, A. Verma, L. M. Weiss and T. Evans, Future Med. Chem., 2013, 5, 653–676 CrossRef CAS PubMed; (g) H. S. Ban and H. Nakamura, Chem. Rec., 2015, 15, 616–635 CrossRef CAS PubMed; (h) D. Gabel, Pure Appl. Chem., 2015, 87, 173–179 CrossRef CAS; (i) Z. J. Leśnikowski, J. Med. Chem., 2016, 59, 7738–7758 CrossRef PubMed.
- (a) T. Kim, J. Lee, S. U. Lee and M. H. Lee, Organometallics, 2015, 34, 3455–3458 CrossRef CAS; (b) Y.-J. Cho, S.-Y. Kim, M. Cho, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 18, 9702–9708 RSC; (c) K. O. Kirlikovali, J. C. Axtell, A. Gonzalez, A. C. Phung, S. I. Khan and A. M. Spokoyni, Chem. Sci., 2016, 7, 5132–5138 RSC; (d) Z. Wang, P. Jiang, T. Wang, G. J. Moxey, M. P. Cifuentes, C. Zhang and M. G. Humphrey, Phys. Chem. Chem. Phys., 2016, 18, 15719–15726 RSC; (e) B. H. Choi, J. H. Lee, H. Hwang, K. M. Lee and M. H. Park, Organometallics, 2016, 35, 1771–1777 CrossRef CAS; (f) B. P. Dash, R. Satapathy, E. R. Gaillard, J. A. Maguire and N. S. Hosmane, J. Am. Chem. Soc., 2016, 132, 6578–6587 CrossRef PubMed; (g) X. Li, H. Yan and Q. Zhao, Chem. – Eur. J., 2016, 22, 1888–1898 CrossRef CAS PubMed; (h) S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC.
- (a) K. Tanaka and Y. Chujo, Macromol. Rapid Commun., 2012, 33, 1235–1255 CrossRef CAS PubMed; (b) E. G. Cansu-Ergun and A. Cihaner, Mater. Chem. Phys., 2013, 143, 387–392 CrossRef CAS; (c) J. Pecyna, D. Pociecha and P. Kaszyński, J. Mater. Chem. C, 2014, 2, 1585–1591 RSC; (d) T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizunl, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem., Int. Ed., 2014, 53, 3173–3177 CrossRef CAS PubMed; (e) S. G. McArthur, L. Geng, J. Guo and V. Lavallo, Inorg. Chem. Front., 2015, 2, 1101–1104 RSC; (f) J. Pecyna, P. Kaszyński, B. Ringstrand, D. Pociecha, S. Pakhomov, A. G. Douglass and V. G. Young, Jr., Inorg. Chem., 2016, 55, 4016–4025 CrossRef CAS PubMed; (g) R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC; (h) R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS PubMed.
- (a) C. A. Reed, Acc. Chem. Res., 1997, 31, 325–332 CrossRef; (b) K.-C. Kim, C. A. Reed, D. W. Elliott, L. J. Mueller, F. Tham, L. Lin and J. B. Lambert, Science, 2002, 297, 825–827 CrossRef CAS PubMed; (c) T. Kato and C. A. Reed, Angew. Chem., Int. Ed., 2004, 43, 2908–2911 CrossRef CAS PubMed; (d) C. Douvris and O. V. Ozerov, Science, 2008, 31, 1188–1190 CrossRef PubMed; (e) O. Allemann, S. Duttwyler, P. Romanato, K. K. Baldridge and J. S. Siegel, Science, 2011, 332, 574–577 CrossRef CAS PubMed; (f) C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS PubMed; (g) N. Kordts, C. Borner, R. Panisch, W. Saak and T. Müller, Organometallics, 2014, 33, 1492–1498 CrossRef CAS; (h) Y. Shoji, N. Tanaka, K. Mikami, M. Uchiyama and T. Fukushima, Nat. Chem., 2014, 6, 498–503 CrossRef CAS PubMed.
- (a) E. L. Hoel and M. F. Hawthorne, J. Am. Chem. Soc., 1975, 97, 6388–6395 CrossRef CAS; (b) M. G. L. Mirabelli and L. G. Sneddon, J. Am. Chem. Soc., 1988, 110, 449–453 CrossRef CAS.
- Reviews: (a) Z. Xie, Sci. China: Chem., 2014, 57, 1061–1063 CrossRef CAS; (b) Z. Qiu, Tetrahedron Lett., 2015, 56, 963–971 CrossRef CAS ; specific examples: ; (c) N. Fey, M. F. Haddow, R. Mistry, N. C. Norman, A. G. Orpen, T. J. Reynolds and P. G. Pringle, Organometallics, 2012, 31, 2907–2913 CrossRef CAS; (d) Z. Wang, H. Ye, Y. Li, Y. Li and H. Yan, J. Am. Chem. Soc., 2013, 135, 11289–11298 CrossRef CAS PubMed; (e) Z. J. Yao, W. B. Yu, Y. J. Lin, S. L. Huang, Z. H. Li and G. X. Jin, J. Am. Chem. Soc., 2014, 136, 2825–2832 CrossRef CAS PubMed; (f) H. Dai, G. Liu, X. Zhang, H. Yan and C. Lu, Organometallics, 2016, 35, 1488–1496 CrossRef CAS; (g) B. J. Eleazer, M. D. Smith, A. A. Popov and D. V. Peryshkov, J. Am. Chem. Soc., 2016, 138, 10531–10538 CrossRef CAS PubMed; (h) L. M. A. Saleh, R. M. Dziedzic, S. I. Khan and A. M. Spokoyny, Chem. – Eur. J., 2016, 22, 8466–8470 CrossRef CAS PubMed.
- (a) H. Lyu, Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2015, 54, 10623–10626 CrossRef CAS PubMed; (b) K. Cao, Y. Huang, J. Yang and J. Wu, Chem. Commun., 2015, 51, 7257–7260 RSC; (c) Y. Quan and Z. Xie, J. Am. Chem. Soc., 2015, 137, 3502–3505 CrossRef CAS PubMed; (d) Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 1295–1298 CrossRef CAS PubMed; (e) Y. Quan, C. Tang and Z. Xie, Chem. Sci., 2016, 7, 5838–5845 RSC.
- (a) E. Molinos, S. K. Brayshaw, G. Kociok-Köhn and A. W. Weller, Organometallics, 2007, 26, 2370–2382 CrossRef CAS; (b) E. Molinos, S. K. Brayshaw, G. Kociok-Köhn and A. S. Weller, Dalton Trans., 2007, 4829–4844 RSC.
- (a) A. El-Hellani, C. E. Kefalidis, F. S. Tham, L. Maron and V. Lavallo, Organometallics, 2013, 32, 6887–6890 CrossRef CAS; (b) J. Estrada, S. E. Lee, S. G. McArthur, A. El-Hellani, F. S. Tham and V. Lavallo, J. Organomet. Chem., 2015, 798, 214–217 CrossRef CAS; (c) C. Zhang and D. Ma, J. Organomet. Chem., 2016, 819, 242–247 CrossRef CAS; (d) upon submission of our manuscript, we became aware of a report by Lavallo on the isolation of a bis(carborane) palladium complex with one of the ligands bound by B, P chelation: A. L. Chan, J. Estrada, C. E. Kefalidis and V. Lavallo, Organometallics, 2016, 35, 3257–3260 CrossRef CAS.
- (a) L. Ackermann, Acc. Chem. Res., 2014, 2, 281–295 CrossRef PubMed; (b) F. Zhang and D. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC; (c) C. Zheng and S. You, RSC Adv., 2014, 4, 6173–6214 RSC; (d) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (e) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007–1020 CrossRef CAS PubMed; (f) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- (a) M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS PubMed; (b) Y.-F. Han and G.-X. Jin, Chem. Soc. Rev., 2014, 43, 2799–2823 RSC.
- 11B NMR shifts for B–Ir in {C2B10}–Ir complexes were reported in the range of −19 to −25 ppm: M. Herberhold, H. Yan, W. Milius and B. Wrackmeyer, Chem. – Eur. J., 2002, 8, 388–395 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. CCDC 1508615 and 1508616 for 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08121e |
| This journal is © The Royal Society of Chemistry 2017 |