
Stabilizing the cubic perovskite phase of CsPbI3 nanocrystals by using an alkyl phosphinic acid†
Chujie
Wang
a,
Anthony S. R.
Chesman
b and
Jacek J.
Jasieniak
*a
aDepartment of Materials Science and Engineering, Faculty of Engineering, Monash University, Clayton, VIC 3800, Australia. E-mail: jacek.jasieniak@monash.edu
bCSIRO Manufacturing, Ian Wark Laboratories, Bayview Ave, Clayton, VIC 3168, Australia
First published on 23rd November 2016
Abstract
CsPbI3 nanocrystals suffer from a facile cubic perovskite to orthorhombic phase transformation, which deteriorates their appealing optoelectronic properties. Here, we report a new colloidal synthesis that replaces the conventionally used oleic acid with an alkyl phosphinic acid to grow high-quality, phase-stable cubic perovskite CsPbI3 nanocrystals.
All-inorganic cesium lead halide perovskite (CsPbX3, X = Cl−, Br−, I−) nanocrystals (NCs) exhibit near-unity photoluminescence (PL) quantum yields, narrow emission peak widths,1 and anion-tuneable absorption/emission wavelengths.2 In addition, these materials have been shown to possess negligible electron/hole trapping and undergo ultra-fast charge transfer to suitable acceptor/donor species.3 All these advantages make the CsPbX3 perovskites attractive materials for applications such as light emitting diodes,4 lasers,5 and photodetectors.6
The group of Kovalenko was the first to report the synthesis of nearly monodisperse CsPbX3 NCs using a hot-injection approach,7 with a host of alternative synthetic methods having since been developed.8,9 Despite their promise, a major drawback of CsPbX3 perovskites is their chemical instability when exposed to polar solvents, moisture, or an external halide source.10 The CsPbI3 perovskite, which is a particularly promising candidate for red-emitting LEDs11 and solar cells12 owing to its 1.73 eV optical band-gap,13 also has an inherent phase instability. Unlike CsPbCl3 and CsPbBr3, the functional cubic perovskite (α) phase of CsPbI3 is only stable at temperatures above 315 °C.7 The metastable α-CsPbI3 spontaneously transforms into the non-functional orthorhombic (δ) phase, also known as the “yellow phase” due to its colour, at room temperature (Fig. 1),14 preventing it from being effectively studied and harnessed in practical applications.
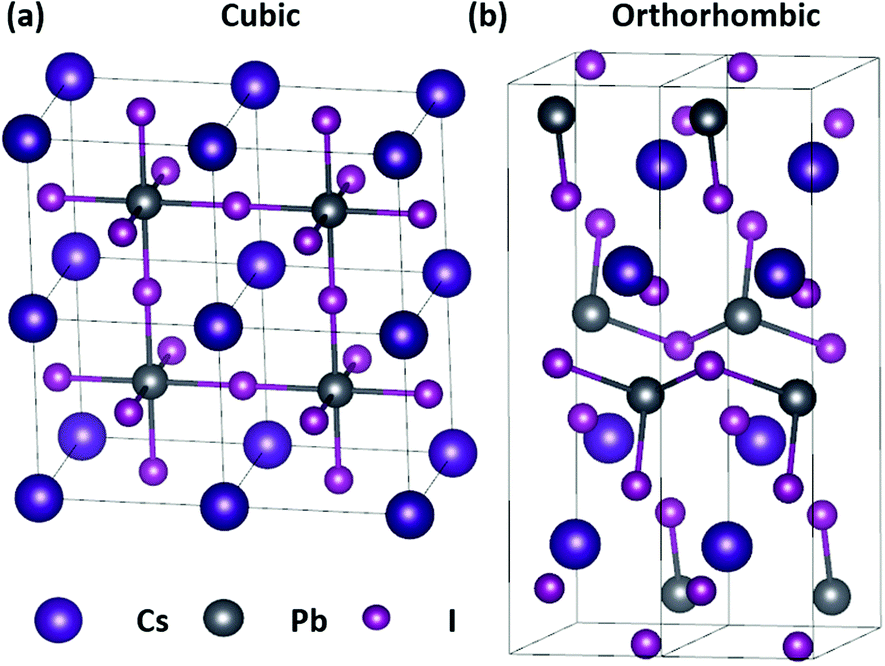 | ||
| Fig. 1 Crystal structures of the CsPbI3 (a) cubic perovskite and (b) orthorhombic phases. | ||
Efforts to stabilize α-CsPbI3 have mainly focussed on thin film coatings. The use of excess hydroiodic acid in its deposition has been shown to yield smaller CsPbI3 grain sizes with an induced lattice strain, which has lowered the α-to-δ phase transformation temperature and, thus, enhanced the α-phase stability at room-temperature.15 Additionally, sintering nanocrystalline mixtures of α-CsPbCl3 with α-CsPbI3 has been shown to create polycrystalline thin films of α-CsPbI3−xClx that are phase-stable at room-temperature.16 Only recently has it been shown that α-CsPbI3 NCs can be stabilized through the purification of the growth solution to remove excess precursors and ligands, which were shown to drive the destabilisation process.17
As a critical component of NC synthesis, the surface capping ligands not only control the nucleation and growth dynamics and prevent NC aggregation, but also determine their physical and chemical behaviour,18 as well as the final NC phase.19,20 Conventionally, oleic acid (OA) and oleylamine (OLA) ligands have been used in the preparation of CsPbX3 NCs. It has been suggested that OLA, oleylammonium halides and oleylammonium oleates are the main surface complexing agents of CsPbX3 NCs, with the level of co-passivation by OA still under debate.21,22 While effective for synthesising high quality NCs, this ligand mixture encourages α-CsPbI3 NCs to undergo a phase transformation when the crude reaction solution is cooled to room temperature. The necessity of removing excess OA and oleate-based precursors from the solution suggests that a synthesis that is free from OA could be conducive towards enhancing phase stability.17
In this communication we report the use of bis-(2,2,4-trimethylpentyl)phosphinic acid (TMPPA) as an OA replacement in the synthesis of CsPbI3 NCs. This novel synthesis is found to yield high quality NC ensembles and, importantly, preserve the phase of α-CsPbI3 to produce stable colloidal dispersions.
Our synthetic protocol for CsPbI3 was adapted from a hot-injection procedure developed by Kovalenko et al.7 PbI2 was dissolved in OLA and OA or TMPPA with 1-octadecene (ODE) as the solvent at 140 °C, which is our hot injection temperature. The Cs-precursor solution was prepared separately by dissolving Cs2CO3 in OA or TMPPA in ODE at 120 °C. This was subsequently injected into the Pb-precursor solution and heated for 5 s, after which it was quenched. Purification of the NCs from the reaction solution was performed via precipitation using tert-butanol as the anti-solvent. The washed NCs were then re-dispersed in toluene. From herein, CsPbI3 synthesised in TMPPA (OA) will be termed CsPbI3–TMPPA (–OA).
The first advantage of using TMPPA is that the respective Cs precursor, Cs–TMPPA, can be readily formed in ODE at room temperature, while the OA-based precursor, Cs–oleate, requires heating up to 100 °C before dissolution occurs (Fig. S1, ESI†). This allows for the convenient storage of Cs–TMPPA. The second major advantage, and the focus of this paper, is that CsPbI3–TMPPA NCs retain the α-phase in both crude and washed solutions (Fig. S2, ESI†).
A comparison of the UV-Vis absorption and PL spectra of CsPbI3–OA and CsPbI3–TMPPA over a period of time are shown in Fig. 2a–d. As-synthesised samples exhibit band-edge absorption in the near-infrared region that approach the α-CsPbI3 bulk optical bandgap of ∼1.73 eV. PL characteristics are also similar, albeit with the CsPbI3–TMPPA samples exhibiting a narrower FWHM of 34 nm versus 50 nm for CsPbI3–OA. The absorption spectra of CsPbI3–OA show a progressively larger scattering contribution upon storage over only 3 days, with yellow precipitates forming in that period, characteristic of δ-CsPbI3. The PL intensity simultaneously declines during this time. In contrast, the absorption and PL properties of CsPbI3–TMPPA are almost perfectly preserved after 20 days of storage under ambient conditions. Similar findings were observed for CsPbI3–TMPPA synthesised at lower reaction temperatures (Fig. S3, ESI†).
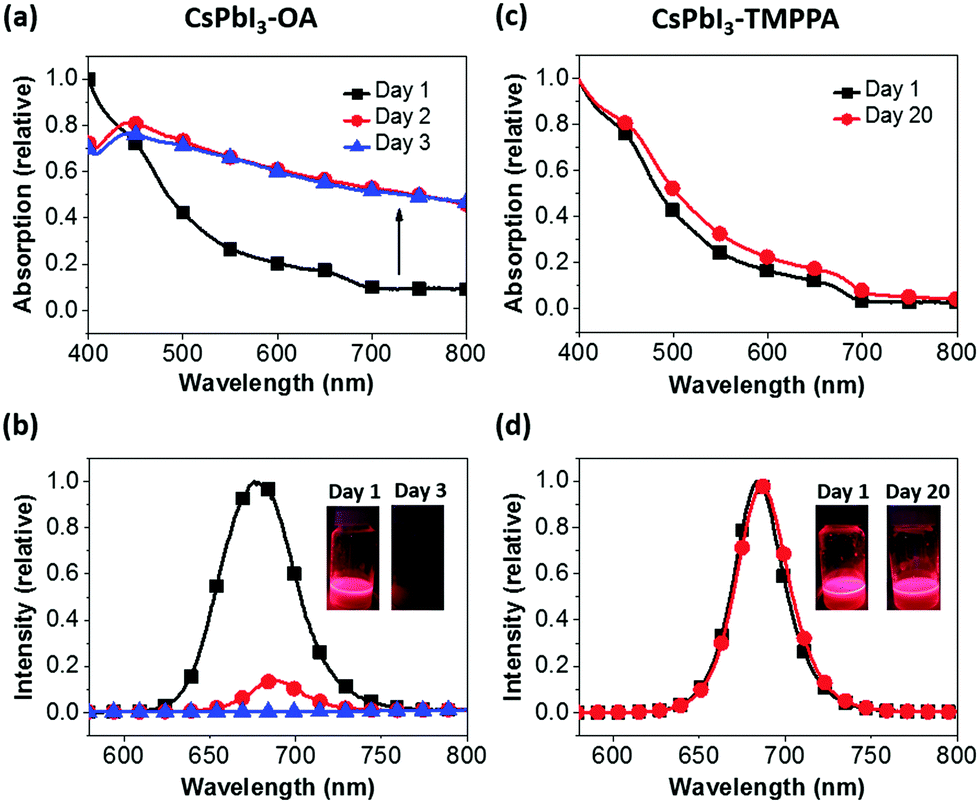 | ||
| Fig. 2 UV-Vis absorption and PL spectra for CsPbI3–OA (a and b) and CsPbI3–TMPPA (c and d). Insets of (b and d): solutions of the respective washed NCs under UV light at different times following synthesis. | ||
To confirm the potential structural stability and changes inferred from optical measurements, powder X-ray diffraction (PXRD) measurements were performed on CsPbI3–TMPPA and CsPbI3–OA NCs (Fig. 3a and b). CsPbI3–TMPPA retained the α phase throughout the seven-day testing period (further measurements show the samples are stable for months). In stark contrast, CsPbI3–OA had already partially transformed from the cubic to orthorhombic phase within one day. Not surprisingly, by day 7 the sample had completely transformed into the orthorhombic phase. Notably, the peak widths in Fig. 3b progressively decrease over time, indicating that CsPbI3–OA is exhibiting grain growth throughout the phase transformation process.23
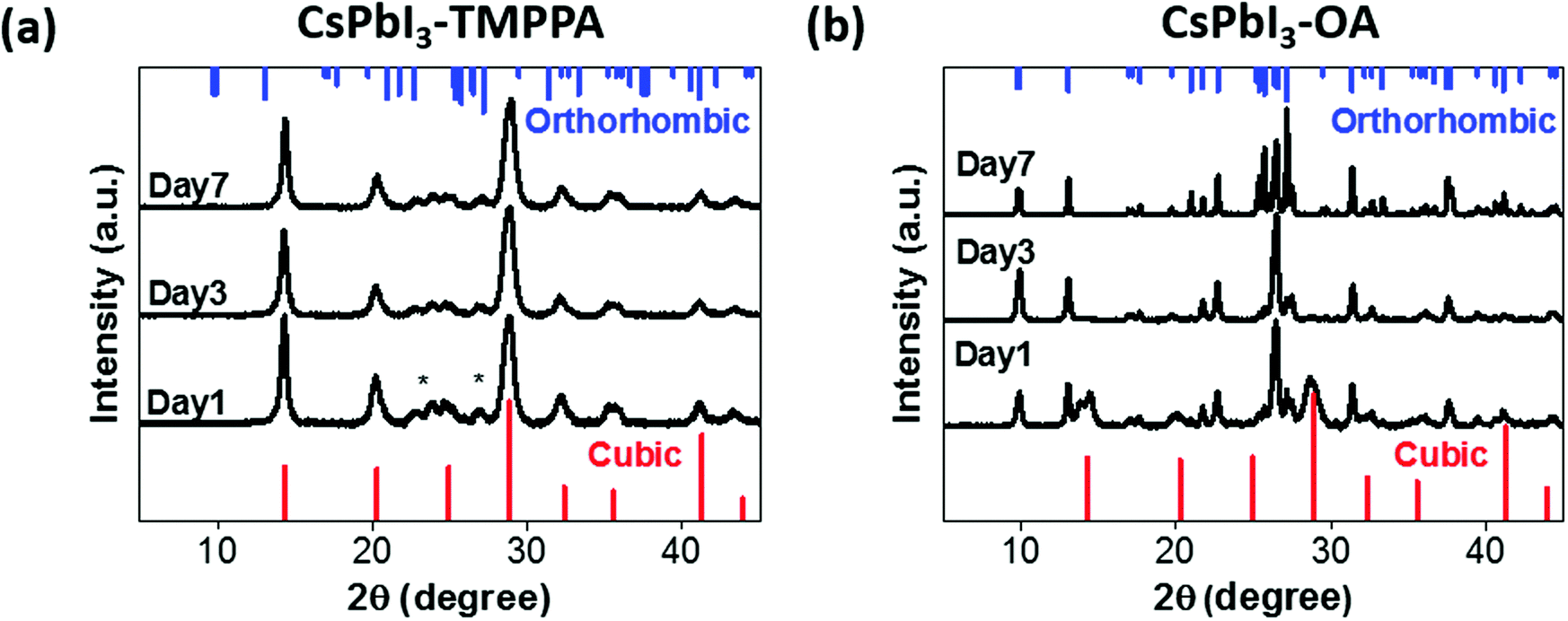 | ||
| Fig. 3 XRD patterns of (a) CsPbI3–TMPPA and (b) CsPbI3–OA. Standard cubic phase is shown in red (bottom), orthorhombic phase in blue (top). (*) unidentified peaks. | ||
Transmission electron microscopy (TEM) was performed to verify the structural characteristics of the as-prepared and phase transformed CsPbI3 samples. Nanocrystals of α-CsPbI3–TMPPA and α-CsPbI3–OA exhibited a cubic shape (Fig. 4a–d), and were measured to have an edge length of approximately 12 nm and 10 nm, respectively (Fig. S4, ESI†). Consistent with the (100) plane of the α-phase, these samples possess inter-planar distances of 0.62 nm (Fig. 4b and d).13,14 The formation of δ-CsPbI3 was only observed in CsPbI3–OA (Fig. 4e and f), as confirmed by the presence of an inter-planar distance of 0.91 nm, which corresponds to the (011) plane.14 These crystals no longer retain their cubic morphology and instead transform into nanorods and sheets that are hundreds of nanometers in size. Notably, small cubes of ∼10 nm width were also found in these samples, likely due to the presence of α-CsPbI3 NCs that have not yet fully transformed. From these observations, it is reasonable to posit that the α-to-δ phase transformation involves NCs aggregating and then structurally re-arranging to form the rod- and sheet-shaped crystals.
 | ||
| Fig. 4 TEM and HRTEM images of α-CsPbI3–TMPPA (a and b), α-CsPbI3–OA (c and d) and δ-CsPbI3–OA (e and f). | ||
To explore the potential mechanism for TMPPA stabilising α-CsPbI3, we examined solutions of washed NCs using Nuclear Magnetic Resonance (NMR) spectroscopy. As TMPPA is the only significant phosphorous-containing component of the reaction,2431P NMR measurements provide a clear insight into its chemical state, in contrast to 1H and 13C NMR measurements, where the convolution of signals from the OLA ligand and residual ODE and tert-butanol21 makes the unambiguous identification of TMPPA impossible (Fig. S5 and S6, ESI†).
The 31P NMR spectrum of a concentrated solution of once-washed CsPbI3–TMPPA NCs revealed that the signal attributed to TMPPA had significantly shifted and broadened compared to the reference spectra of TMPPA (Fig. S7 and S8, ESI†). While this can be a sign of strong bonding to a NC surface,25,26 the 31P NMR signal of TMPPA is also known to shift and broaden in the presence of a base, due to the formation of ion pairs in solution.24 To differentiate between these possibilities, a measurement of a solution in which TMPPA was reacted with an excess of OLA to form an oleylammonium–TMPPA− ion pair resulted in a spectrum that contained a peak with the same position and width as that observed for the CsPbI3 NCs (Fig. S9 and S10, ESI†). Furthermore, a control measurement of a Pb–TMPPA complex showed the TMPPA signal is in an unambiguously different position compared to the CsPbI3–TMPPA NC solution (Fig. S11, ESI†). These results confirmed that TMPPA was not binding to Pb on the surface of the nanocrystal.
Diffusion coefficients, as measured by 2D diffusion-ordered spectroscopy (DOSY), were also used to determine whether TMPPA was interacting with surface of the CsPbI3 NCs.21 The diffusion coefficients of TMPPA and the TMPPA species present in a concentrated solution of CsPbI3 NCs were both measured to be ∼520 μm2 s−1. These diffusion coefficients are approximately one order of magnitude larger than that expected for a species binding to the surface of NCs of a size synthesised in this study.21 Notably, while it was expected that the solution containing the ion pair would give a lower diffusion coefficient compared to the free TMPPA species, dialkyl phosphinic acids are known to form hydrogen bond dimers in apolar solvents,27 which would give a species with a comparable solvodynamic radius. Overall, these results indicated that in a once-washed solution of CsPbI3 NCs, TMPPA exists in an ion pair with oleylammonium and does not bind to the NC surface.
To test this hypothesis, a solution of CsPbI3 NCs was washed a second time in an attempt to remove all TMPPA species. Remarkably, an extended 31P NMR measurement (∼5 h) of a twice-washed solution of phase stable α-CsPbI3 NCs only yielded extremely weak signals due to P containing species (Fig. S12, ESI†). In comparison, a broad peak at 5.5 ppm, which has been attributed to OLA species dynamically interacting with the NC surface, could be detected in the same solution with a standard 1H NMR measurement (Fig. S5, ESI†).21 This indicates that after two washes, TMPPA was not present in the solution in any significant quantity and that it does not play a role in stabilising the phase of α-CsPbI3 by coordinating to the NC surface.
This finding is in contrast to syntheses utilising OA, in which oleylammonium-oleate and OA are co-passivants with oleylammonium bromide and OLA for CsPbBr3 NCs.21,22 We hypothesise that the α-to-δ phase transformation in NCs is associated with this surface chemistry. This is supported by the addition of OA or oleylammonium-oleate to an α-CsPbI3–TMPPA solution, which resulted in a rapid transformation to the δ-phase (Fig. S13 and S14, ESI†). Conversely, the addition of TMPPA to α-CsPbI3–OA solution resulted in a slower α-to-δ transformation, although it did not prevent it (Fig. S15 and S16, ESI†).
In conclusion, we have developed a new synthesis for high-quality CsPbI3 nanocrystals that harnesses an alkyl phosphinic acid as a replacement for conventionally used oleic acid. Importantly, the NCs retain their cubic perovskite phase in solution, which has been a significant challenge to date. We have found that, unlike for oleic acid, the phosphinic acid and its ion-pairs do not bind to the NC surface. This ensures that in our synthesis oleylammonium iodide and/or oleylamine are the predominant surface ligands. Based on this, we suggest that the phase instability in this system is directly associated with the destabilisation of this surface chemistry. Further research is required to understand precisely why oleic acid and/or its ion pair drive this instability and what factors prevent the alkyl phosphinic acid used in this study from binding to the α-CsPbI3 nanocrystal surface.
Notes and references
- A. Swarnkar, R. Chulliyil, V. K. Ravi, M. Irfanullah, A. Chowdhury and A. Nag, Angew. Chem., Int. Ed., 2015, 54, 15424–15428 CrossRef CAS PubMed.
- Q. A. Akkerman, V. D’Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- K. Wu, G. Liang, Q. Shang, Y. Ren, D. Kong and T. Lian, J. Am. Chem. Soc., 2015, 137, 12792–12795 CrossRef CAS PubMed.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed.
- J.-S. Lee, P. Ramasamy, D.-H. Lim, B. Kim, S.-H. Lee and M.-S. Lee, Chem. Commun., 2015, 52, 3–6 Search PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- D. Zhang, S. W. Eaton, Y. Yu, L. Dou and P. Yang, J. Am. Chem. Soc., 2015, 137, 9230–9233 CrossRef CAS PubMed.
- I. Lignos, S. Stavrakis, G. Nedelcu, L. Protesescu, A. J. Demello and M. V. Kovalenko, Nano Lett., 2016, 16, 1869–1877 CrossRef CAS PubMed.
- F. Palazon, Q. A. Akkerman, M. Prato and L. Manna, ACS Nano, 2016, 10, 1224–1230 CrossRef CAS PubMed.
- C. C. Lin, A. Meijerink and R. S. Liu, J. Phys. Chem. Lett., 2016, 7, 495–503 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grä Tzel, Energy Environ. Sci., 2016, 9, 1989–1997 CAS.
- C. K. Møller, Nature, 1958, 182, 1436 CrossRef.
- D. M. Trots and S. V. Myagkota, J. Phys. Chem. Solids, 2008, 69, 2520–2526 CrossRef CAS.
- G. E. Eperon, G. M. Paterno, R. J. Sutton, A. Zampetti, A. A. Haghighirad, F. Cacialli and H. J. Snaith, J. Mater. Chem. A, 2015, 3, 19688–19695 CAS.
- S. Dastidar, D. A. Egger, L. Z. Tan, S. B. Cromer, A. D. Dillon, S. Liu, L. Kronik, A. M. Rappe and A. T. Fafarman, Nano Lett., 2016, 16, 3563–3570 CrossRef CAS PubMed.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS PubMed.
- Y. Sun, Y. Chen, L. Tian, Y. Yu, X. Kong, J. Zhao and H. Zhang, Nanotechnology, 2007, 18, 275609 CrossRef.
- K. Nose, Y. Soma, T. Omata and S. Otsuka-Yao-Matsuo, Chem. Mater., 2009, 21, 2607–2613 CrossRef CAS.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943–7954 CrossRef CAS PubMed.
- A. Monshi, World J. Nano Sci. Eng., 2012, 2, 154–160 CrossRef.
- D. Cholico-Gonzalez, M. Avila-Rodriguez, G. Cote and A. Chagnes, J. Mol. Liq., 2013, 187, 165–170 CrossRef CAS.
- M. Kuno, J. K. Lee, B. O. Dabbousi, F. V. Mikulec and M. G. Bawendi, J. Chem. Phys., 1997, 106, 9869–9882 CrossRef CAS.
- J. S. Owen, J. Park, P.-E. Trudeau and A. P. Alivisatos, J. Am. Chem. Soc., 2008, 130, 12279–12281 CrossRef CAS PubMed.
- J. A. Walmsley, J. Phys. Chem., 1984, 88, 1226–1231 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental information, NMR spectra, and additional experimental results. See DOI: 10.1039/c6cc08282c |
| This journal is © The Royal Society of Chemistry 2017 |