
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of ramariolide natural products and discovery of their targets in mycobacteria†
Johannes
Lehmann
a,
Johannes
Richers
b,
Alexander
Pöthig
b and
Stephan A.
Sieber
*a
aDepartment of Chemistry, Center for Integrated Protein Science Munich (CIPSM), Technische Universität München, Lichtenbergstraße 4, 85747 Garching, Germany. E-mail: stephan.sieber@tum.de
bCatalysis Research Center (CRC), Technische Universität München, Ernst-Otto-Fischer Straße 1, 85747 Garching, Germany
First published on 11th November 2016
Abstract
Ramariolides A–D are natural products with antibacterial activity. To exploit their cellular mechanism, we here devise the first total synthesis and prepare a photoprobe for target identification. Antibacterial testing against several pathogenic strains including Mycobacterium tuberculosis revealed the highest potency for ramariolide A. Chemical proteomics unraveled binding to essential proteins for amino acid anabolism.
Treatment of bacterial infections by classical antibiotics is severely impaired due to the development of multiple resistant strains.1,2 In particular tuberculosis caused by Mycobacterium tuberculosis, responsible for millions of deaths worldwide,3 represents a major challenge for drug development due to its largely impermeable membrane barrier.4 Thus, even antibiotic-sensitive strains require the application of several different antibiotics including combinations of synthetic pharmaceuticals such as isoniazid as well as natural products such as rifampicin.5,6 Natural products have always been a rich source of antibiotic discovery addressing a wealth of different targets.7 Although the discovery of new natural product antibiotics is declining,8 their detailed study still represents a major inspiration for the exploration of unprecedented pathways and the identification of druggable targets. In 2012 Anderson and co-workers reported the isolation of ramariolides A–D (Fig. 1) from the coral mushroom Ramaria cystidiphora and highlighted their potent antibacterial activity.9 All ramariolide compounds belong to the group of butenolides with ramariolide A and B exhibiting a unique spirocyclic acetal functionality. Despite their promising bioactivities and interesting structural features no total synthesis and no investigation of their in-depth mode of action have been reported up to now.
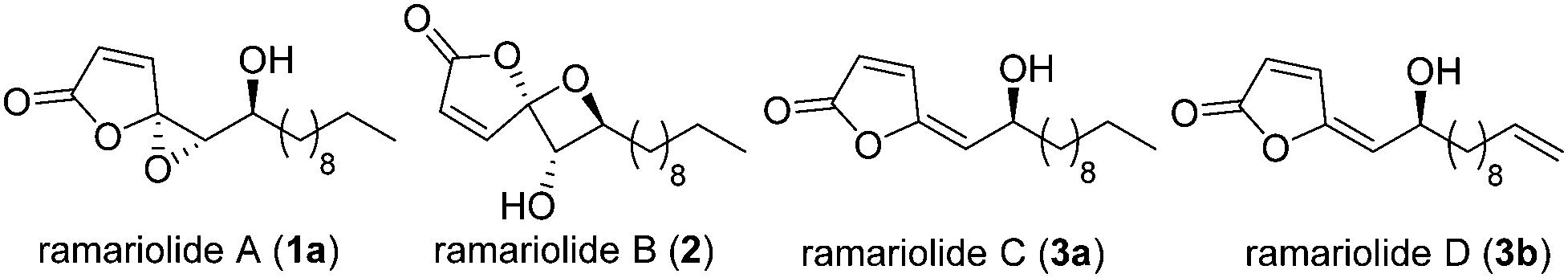 | ||
| Fig. 1 Molecular structures of ramariolides A–D. | ||
Here, we present the first total synthesis of all four natural products as well as their structural analogs for target identification. Activity-based protein profiling (ABPP) in the mycobacterial model strain M. smegmatis mc2 155 revealed several protein binders allowing us to decipher the antimycobacterial mode of action.
Structural examination of the individual natural products ramariolide A (1a), B (2), C (3a) and D (3b) revealed long chain fatty acids (4a, 4b) as commercially available precursors (Scheme 1). These were converted into α-bromo ketones in excellent yields (5a, 5b) in three consecutive steps.10,11 Transformation to the corresponding phosphonium-ylides was quantitative12 and enabled olefination using maleic anhydride yielding the butenolide moiety.13 However, the reaction was challenged by partial homo-olefination as well as polymerization caused by the acceptor-substituted ylide. Although dilution could not prevent this side reaction completely, the butenolides were obtained in good yields favoring the desired (E)-product (6a–c) over the (Z)-isomer (7a–c) in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.14 Subsequent reduction15 revealed racemic ramariolides C (3a) and D (3b) both matching published spectral data.9 Ramariolide A was obtained by epoxidation of the allylic alcohol using Sharpless conditions favoring the anti-diastereomer in a 87:13 ratio.16,17 Use of (+)-tartrate ester gave rise to the enantioenriched natural product in 55% ee. As the optical rotation of the synthetic product differed from reported values, we utilized chiral europium salt-assisted NMR titration to determine absolute values (Fig. S1, ESI†).18
:1 ratio.14 Subsequent reduction15 revealed racemic ramariolides C (3a) and D (3b) both matching published spectral data.9 Ramariolide A was obtained by epoxidation of the allylic alcohol using Sharpless conditions favoring the anti-diastereomer in a 87:13 ratio.16,17 Use of (+)-tartrate ester gave rise to the enantioenriched natural product in 55% ee. As the optical rotation of the synthetic product differed from reported values, we utilized chiral europium salt-assisted NMR titration to determine absolute values (Fig. S1, ESI†).18
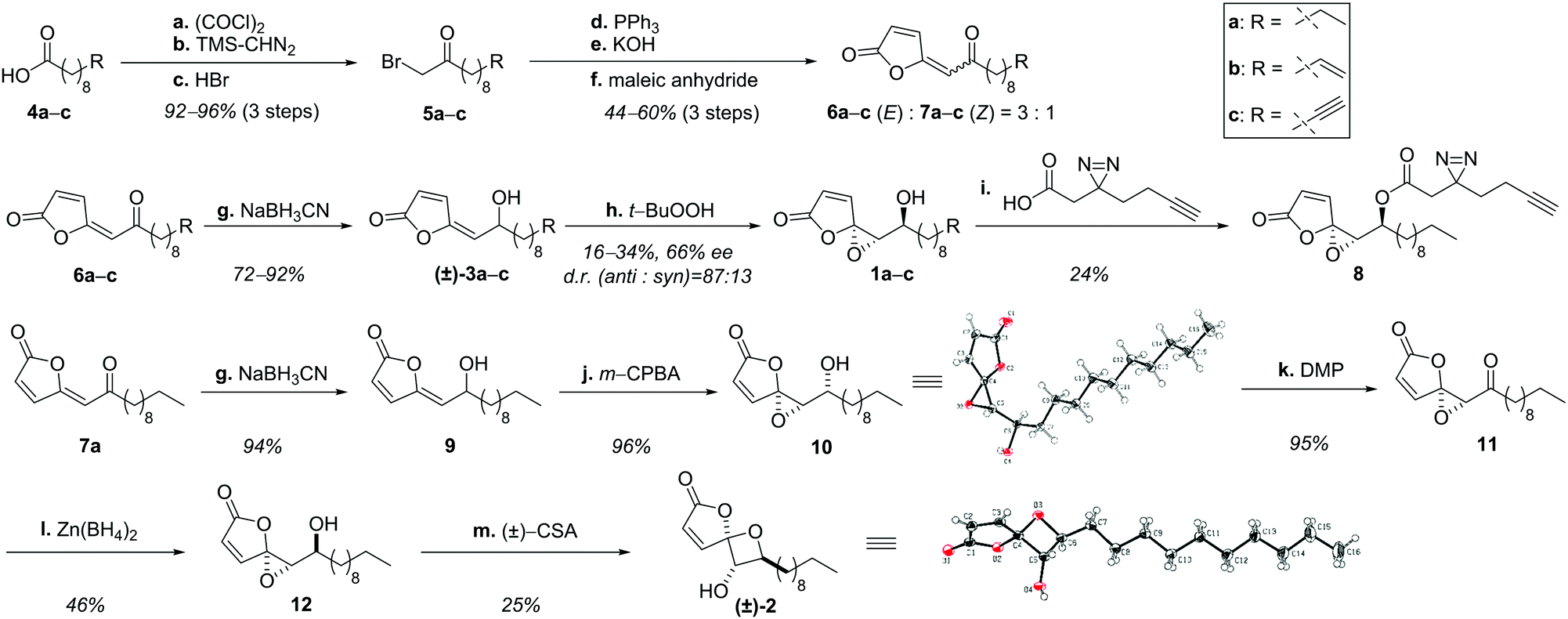 | ||
| Scheme 1 Consecutive synthesis of ramariolides A–D. (a) (COCl)2, DMF, CH2Cl2, rt, 3 h; (b) TMS-CHN2, ACN, 0 °C, 16 h; (c) HBr, Et2O, 60 °C, 3 h; (d) PPh3, CHCl3, rt, 16 h; (e) KOH, MeOH:H2O = 4:1, rt, 3 h; (f) maleic anhydride, PhMe, 110 °C; (g) NaBH3CN, THF, rt, 1 h then add HCl for 1 h; (h) Ti(Oi-Pr)4, (+)-DIPT, t-BuO2H, −32 °C, 16 h; (i) (COCl)2, DMF, CH2Cl2, rt, 2 h, then add 1a and pyridine, 2 h; (j) m-CPBA, CH2Cl2, 3 h; (k) DMP, CH2Cl2, rt, 2 h; (l) Zn(BH4)2, THF, 0 °C, 2 h, then add HCl; (m) (±)-CSA, CH2Cl2, rt, 3 h. DMF = N,N-dimethylformamide, TMS = trimethylsilyl-, THF = tetrahydrofuran, DIPT = diisopropyl tartrate, m-CPBA = meta-chloroperoxybenzoic acid, DMP = Dess–Martin periodinane, CSA = camphorsulfonic acid. | ||
Next, we pursued conversion of ramariolide A to B by reacetalisation of the epoxide to an oxetane moiety applying either alkaline or Brønsted/Lewis acidic conditions.19 Since only the formation of the epimeric spirocycle of ramariolide B (15) was observed, we rationalized that the inverted stereochemistry of the Z-configured intermediate 7a could be crucial to accomplish the synthesis of the desired isomer. Remarkably, analogous reduction and epoxidation resulted in exclusive formation of the syn-configured α-hydroxy epoxide 10, which was structurally verified by X-ray crystallography. All standard procedures for inversion of the alcohol function were unsuccessful. Instead, a sequence of oxidation and subsequent stereoselective reduction utilizing zinc-(II)-borohydride yielded the correctly configured substrate 12.20
After screening a variety of reaction conditions, the use of camphorsulfonic acid finally led to formation of 2,21 which was confirmed by spectral data as well as X-ray structural analysis. Interestingly, the previously isolated spirocyclic epimer 15 was validated as a side product by NOE experiments (Fig. S2, ESI†), supporting an acyclic intermediate (14) (Fig. S3, ESI†).
All natural products were tested against a series of pathogenic bacterial strains to determine their minimal inhibitory concentration (MIC) of bacterial growth (Table S1, ESI†). Interestingly, all ramariolides were active against diverse Gram-positive strains with ramariolide A exhibiting the most pronounced potency against Staphylococcus aureus USA300 (30 μM) as well as mycobacteria including Mycobacterium tuberculosis H37Rv (25 μM). Since antimycobacterial compounds are rare, we were interested in unraveling their molecular targets responsible for growth inhibition. Therefore, a ramariolide-based probe exhibiting a terminal alkyne was designed, which allowed for modification with functionalized azides via click chemistry after target binding.22,23 Inspired by the terminal olefin-containing ramariolide D, we utilized this blueprint for designing an alkyne analog.24 The synthesis followed the established procedure yielding probe 1c without the need for alkyne protection. In contrast to control substances 1a and 1b, terminal alkynylated compound 1c did not maintain antimicrobial activity suggesting that the altered bond geometry negatively interferes with target binding (Table S1, ESI†).
Validation of the compound structure for alternative modification sites revealed the hydroxy functionality of ramariolide A as a synthetically accessible position for the introduction of an alkyne tag. Although ramariolide A contains protein-reactive epoxide as well as Michael acceptor moieties, the mode of target binding could be driven by (non-covalent) reversible interactions. The natural product was thus also provided with a diazirine UV-crosslinker located next to the alkyne moiety.25 Diazirines covalently attach to amino acids in physical proximity to the binding pocket upon UV irradiation thus enabling mass spectrometric (MS) target identification under protein denaturing conditions.26 Corresponding probe 8 was accessible by esterification of the secondary alcohol (Scheme 1). Satisfyingly, the modified structure retained antimycobacterial activity, an important prerequisite for following ABPP studies.
Mycobacterial cells were grown to mid-log phase, harvested and incubated with DMSO or probe 8 in PBS for 1.5 h. UV-irradiation for 10 min at 365 nm and 4 °C was performed to account for covalent as well as reversible target binding. Cells were lysed at 4 °C by sonication and soluble as well as insoluble fractions were separated by centrifugation. Protein fractions were clicked to biotin azide, enriched on avidin beads and released by tryptic digest (Fig. S4, ESI†).27 Cleaved peptides were modified by dimethyl labeling using light, medium and heavy isotope reagents.28 A label switch was performed throughout the biological replicates and differentially labeled samples were pooled prior to LC–MS/MS analysis. Identified proteins were ranked in corresponding volcano plots as a (log2−) ratio of 8 to DMSO treatment (Fig. 2A) against statistical significance (−log10p-value). Proteins enriched by a factor >2 with a p-value of 0.05 or below were considered hits (Table S2, ESI†). Evaluation of the soluble protein fraction revealed five hits as essential proteins for mycobacterial growth, while none were detected in the insoluble fraction (Fig. S5, ESI†).29 These hits comprise the 30S ribosomal proteins S4 (RpsD) and S5 (RpsE), the caseinolytic protease subunit X (ClpX) and the metabolic enzymes aspartate kinase (Ask) and homoserine dehydrogenase (Hsd). RpsD and RpsE are small ribosome-associated proteins, which are highly abundant soluble proteins found in prokaryotic cells. Previous proteomics experiments with E. coli also identified these proteins as possible contaminants during affinity enrichment.30 Therefore, these proteins were not considered in more detail. The molecular chaperon ClpX is well-studied and known to assist the ClpP subunit in protein degradation.31–33 Analysis of overexpressed mycobacterial protein did not reveal binding with probe 8 (Fig. S6, ESI†). Finally, Ask and Hsd catalyze subsequent steps in amino acid anabolism and have been shown to be essential for microbacterial growth.34,35 It is thus intriguing to speculate that ramariolide A might compete with natural substrates for enzyme binding by blocking their active site pockets.
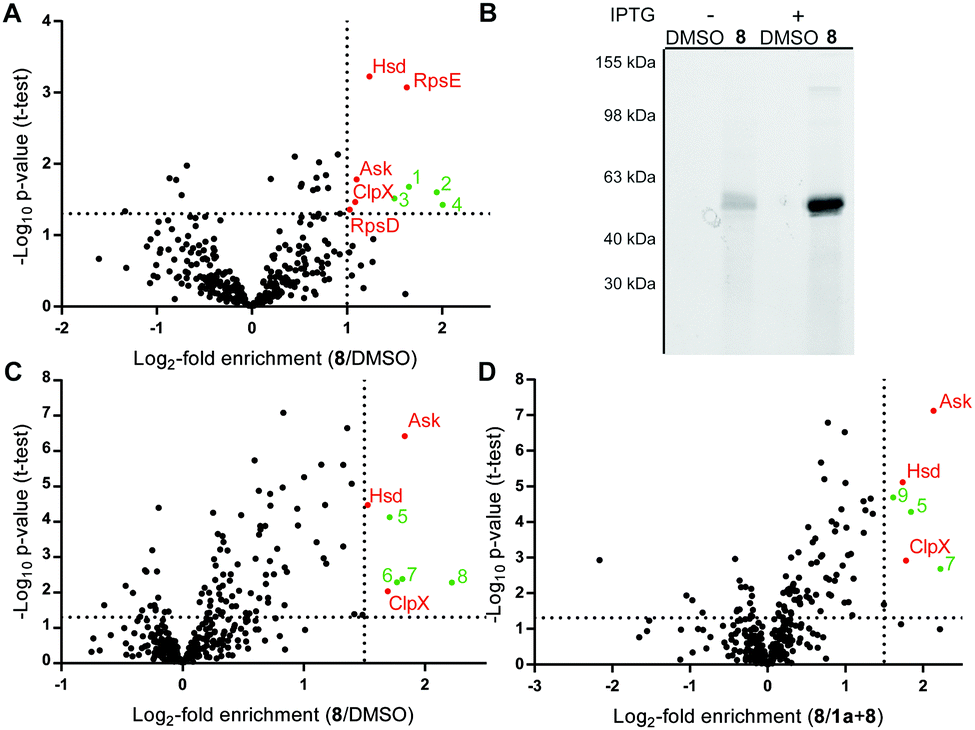 | ||
| Fig. 2 (A) Scatter plot of gel-free ABPP dimethyl labeling experiments with 30 μM 8vs. DMSO in M. smegmatis mc2155 soluble fraction after UV-irradiation (criteria: enrichment ≥2 and p-value ≤0.05). (B) Fluorescence scan of gel-based ABPP experiments with recombinant expressed mycobacterial Ask labeled with 8 in E. coli. “+” indicates induction of protein expression by addition of IPTG; “−“ represents absence of inducer. (C) Scatter plot of gel-free ABPP dimethyl labeling experiment with 30 μM 8vs. DMSO in M. smegmatis mc2155 soluble fraction without UV-irradiation. (criteria: enrichment ≥3 and p-value ≤0.05) (D) Scatter plot of M. smegmatis mc2155 soluble fraction preincubated with 100 μM 1a followed by treatment with 30 μM 8 (criteria: enrichment ≥3 and p-value ≤0.05). Red dots indicate proteins essential for bacterial growth. Green dots depict non-essential protein hits. DMSO = dimethylsulfoxide, IPTG = isopropyl-β-D-thiogalactopyranosid. | ||
In order to validate this hypothesis we cloned mycobacterial ask in E. coli and conducted gel-based ABPP experiments. Addition of 8 to Ask-overexpressing cells followed by rhodamine-azide click chemistry resulted in a strong fluorescent band of predicted molecular size (Fig. 2B) upon induction of protein expression, indicating a covalent mode of binding. Furthermore gel based labeling of purified protein was successful in contrast to heat disrupted control samples.
These results suggest a specific interaction of ramariolide A with a natively folded protein (Fig. S8, ESI†). Based on the covalent binding mode we repeated gel-free ABPP using probe 8 against DMSO (Fig. 2C) as well as competition with unmodified ramariolide A (Fig. 2D) without UV crosslinking. Importantly, Ask was consistently among the most significantly enriched proteins in both experiments. Although these findings highly imply interference of ramariolide natural products with amino acid anabolism in mycobacteria, binding to other target enzymes cannot be excluded and is under further evaluation.
In summary we provided a first total synthesis of the family of ramariolides and verified their antimicrobial activity against Gram-positive strains. Molecular targets in mycobacteria were identified with customized probes. Essential members of the bacterial amino acid metabolism were found as interactors and validated by in situ and in vitro experiments providing strategies for future drug development.
S. A. S. acknowledges funding by the Deutsche Forschungs-gemeinschaft, FOR1406 and the ERC starting grant (250924-antibacterials) (to S. A. S.). The Studenstiftung des deutschen Volkes is acknowledged for funding support of JL. We gratefully thank Imke Plitzko for analysis of NOE NMR results, Christian Bartelmus for HRMS measurements and Vadim Korotkov for proof reading of the manuscript as well as helpful suggestions. The Harvard School of Public Health is gratefully acknowledged for use of their facilities to conduct studies on Mycobacterium tuberculosis H37Rv.
Notes and references
- B. R. Laible, S. D. J. Med., 2014, 67, 30 Search PubMed.
- J. O'Neill, AMR Rev., 2016 Search PubMed , http://amr-review.org/Publications.
- WHO, 2016, 20.
- M. Sani, E. N. Houben, J. Geurtsen, J. Pierson, K. de Punder, M. van Zon, B. Wever, S. R. Piersma, C. R. Jimenez, M. Daffe, B. J. Appelmelk, W. Bitter, N. van der Wel and P. J. Peters, PLoS Pathog., 2010, 6, e1000794 Search PubMed.
- W. McDermott, C. Muschenheim, C. Clark, D. F. Elmendorf, Jr. and W. U. Cawthon, Trans. Assoc. Am. Physicians, 1952, 65, 191 CAS.
- V. Nitti, Helv. Med. Acta, 1965, 32, 603 CAS.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670 CrossRef CAS PubMed.
- D. G. Brown, T. Lister and T. L. May-Dracka, Bioorg. Med. Chem. Lett., 2014, 24, 413 CrossRef CAS PubMed.
- R. M. Centko, S. Ramon-Garcia, T. Taylor, B. O. Patrick, C. J. Thompson, V. P. Miao and R. J. Andersen, J. Nat. Prod., 2012, 75, 2178 CrossRef CAS PubMed.
- H. Ren and W. D. Wulff, Org. Lett., 2010, 12, 4908 CrossRef CAS PubMed.
- L. Wang, C. Cherian, S. K. Desmoulin, L. Polin, Y. Deng, J. Wu, Z. Hou, K. White, J. Kushner, L. H. Matherly and A. Gangjee, J. Med. Chem., 2010, 53, 1306 CrossRef CAS PubMed.
- D. M. Bradley, R. Mapitse, N. M. Thomson and C. J. Hayes, J. Org. Chem., 2002, 67, 7613 CrossRef CAS PubMed.
- D. W. Knight and G. Pattenden, J. Chem. Soc., Perkin Trans. 1, 1979, 62 RSC.
- J. S. Fowler and S. Seltzer, J. Org. Chem., 1970, 35, 3529 CrossRef CAS.
- G. Struve and S. Seltzer, J. Org. Chem., 1982, 47, 2109–2113 CrossRef CAS.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974 CrossRef CAS.
- N. S. Kim, J. R. Choi and J. K. Cha, J. Org. Chem., 1993, 58, 7096 CrossRef CAS.
- W. H. Pirkle and P. L. Rinaldi, J. Org. Chem., 1979, 44, 1025 CrossRef CAS.
- G. B. Payne, J. Org. Chem., 1962, 27, 3819 CrossRef CAS.
- R. Cambie, N. Renner, P. Rutledge and P. Woodgate, Aust. J. Chem., 1991, 44, 61 CrossRef CAS.
- J. Chen, P. Gao, F. Yu, Y. Yang, S. Zhu and H. Zhai, Angew. Chem., Int. Ed. Engl., 2012, 51, 5897 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 2002, 41, 2596 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- J. Lehmann, M. H. Wright and S. A. Sieber, Chemistry, 2016, 22, 4666 CrossRef CAS PubMed.
- Z. Li, P. Hao, L. Li, C. Y. Tan, X. Cheng, G. Y. Chen, S. K. Sze, H. M. Shen and S. Q. Yao, Angew. Chem., Int. Ed. Engl., 2013, 52, 8551 CrossRef CAS PubMed.
- R. A. G. Smith and J. R. Knowles, J. Am. Chem. Soc., 1973, 95, 5072 CrossRef CAS PubMed.
- M. H. Wright and S. A. Sieber, Nat. Prod. Rep., 2016, 33, 681 RSC.
- P. J. Boersema, R. Raijmakers, S. Lemeer, S. Mohammed and A. J. Heck, Nat. Protoc., 2009, 4, 484 CrossRef CAS PubMed.
- C. M. Sassetti, D. H. Boyd and E. J. Rubin, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12712 CrossRef CAS PubMed.
- D. Mellacheruvu, Z. Wright, A. L. Couzens, J. P. Lambert, N. A. St-Denis, T. Li, Y. V. Miteva, S. Hauri, M. E. Sardiu, T. Y. Low, V. A. Halim, R. D. Bagshaw, N. C. Hubner, A. Al-Hakim, A. Bouchard, D. Faubert, D. Fermin, W. H. Dunham, M. Goudreault, Z. Y. Lin, B. G. Badillo, T. Pawson, D. Durocher, B. Coulombe, R. Aebersold, G. Superti-Furga, J. Colinge, A. J. Heck, H. Choi, M. Gstaiger, S. Mohammed, I. M. Cristea, K. L. Bennett, M. P. Washburn, B. Raught, R. M. Ewing, A. C. Gingras and A. I. Nesvizhskii, Nat. Methods, 2013, 10, 730 CrossRef CAS PubMed.
- J. Leodolter, J. Warweg and E. Weber-Ban, PLoS One, 2015, 10, e0125345 Search PubMed.
- K. R. Schmitz and R. T. Sauer, Mol. Microbiol., 2014, 93, 617 CrossRef CAS PubMed.
- D. Frees, K. Sorensen and H. Ingmer, Infect. Immun., 2005, 73, 8100 CrossRef CAS PubMed.
- J. D. Cirillo, T. R. Weisbrod, L. Pascopella, B. R. Bloom and W. R. Jacobs, Jr., Mol. Microbiol., 1994, 11, 629 CrossRef CAS PubMed.
- V. Sritharan, P. R. Wheeler and C. Ratledge, Eur. J. Biochem., 1989, 180, 587 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1508942 and 1508943. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08365j |
| This journal is © The Royal Society of Chemistry 2017 |