
(+)-Camphor-mediated kinetic resolution of allylalanes: a strategy towards enantio-enriched cyclohex-2-en-1-ylalane†
Michaël
Coffinet
,
Fabien
Massicot
,
Jomy
Joseph
,
Jean-Bernard
Behr
,
Florian
Jaroschik
and
Jean-Luc
Vasse
*
Institut de Chimie Moléculaire, CNRS (UMR 7312) and Université de Reims, 51687 Reims Cedex 2, France. E-mail: jean-luc.vasse@univ-reims.fr
First published on 14th November 2016
Abstract
An efficient (+)-camphor-mediated kinetic resolution of racemic cyclohex-2-en-1-ylalane is described. This approach provides an enantiomerically enriched form of the alane, in situ available for synthetic uses. Applied to the allylation of aldehydes, this protocol leads to the corresponding homoallylalcohols in a highly enantioselective manner.
Configurationally stable organometallic compounds are traditionally difficult to generate and to handle and are confined to a limited number of metals. In this context, the enantioselective generation of a chiral nucleophilic organometallic species is a very fascinating and challenging approach in asymmetric synthesis.1 Among synthetically useful organometallic reagents, allylmetals are of particular interest due to their singular reactivity towards carbonyl compounds and to the high synthetic potential of the resulting adducts. However allylmetals have to be distinguished depending on their configurational stability.2 While allylboranes, allylboronic esters3 and allylsilanes4 are configurationally stable and can be stereo-specifically transferred to a prochiral electrophile, allylzincs are prone to metallotropism and thus constitute ideal candidates to promote a dynamic kinetic resolution (Fig. 1).5
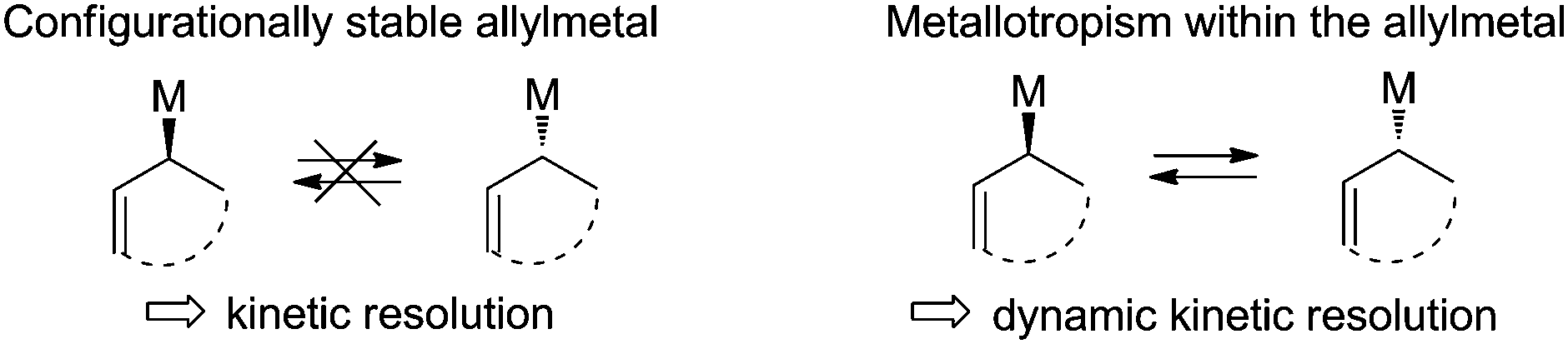 | ||
| Fig. 1 Classification of allylmetals. | ||
In the boron series, enantio-enriched allylboronic esters, based on a camphor6 or a tartaric acid7 template, have been described. More recently, Aggarwal's group developed a highly attractive enantioselective generation of chiral boronic esters8 which can be applied to the synthesis of enantiopure allylboronates.3b,9 In addition to this elegant approach, catalytic enantioselective preparations of allylmetals were also reported, including, hydrosilylation,10 hydroboration,11 silaboration12 of conjugated dienes and transition-metal catalyzed allylic addition.13 Nevertheless, among the allylmetals conventionally used in organic synthesis, the chiral integrity of allylalanes has never been experienced so far.
Recently, we reported a titanium-catalyzed hydroalumination of cyclic conjugated dienes which provided allylalanes in a straightforward manner.14 Their reaction with chiral imines occurred with low diastereoselectivity, in contrast to their allylzinc analogues for which an efficient dynamic kinetic resolution arose, affording homoallylic amines with high stereoselectivity.15 The poor stereoselectivity observed with allylalane could reflect the absence of a dynamic resolution between both enantiomers of the organoaluminium.
If allylalanes are really configurationally stable, a kinetic resolution of the racemic mixture could be envisioned to access an enantio-enriched allylalane.16 In this case, the discrimination of the couple of enantiomers may occur using a chiral electrophilic trap. In turn, the preserved enantiomer could next be allowed to react with a prochiral electrophile (Fig. 2).
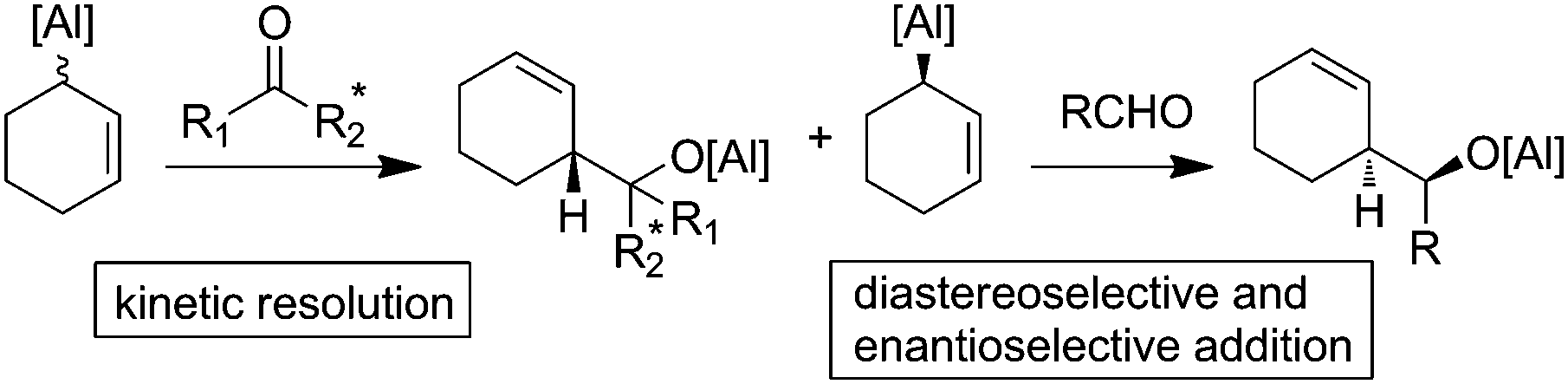 | ||
| Fig. 2 Towards a kinetic resolution of allylalanes. | ||
This study was initiated by identifying a carbonyl derivative able to kinetically discriminate the two enantiomers of the allylmetal. For that purpose, a series of chiral ketones was tested towards a racemic mixture of allylalane. Whereas low conversion was observed when using (+)-fenchone, alcohols derived from (−)-menthone and (+)-camphor were obtained quantitatively as a ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two diastereoisomers (Table 1, entries 2 and 3). To ascertain their relative stereochemistry, 1a and 1b were hydrogenated (H2, Pd/C) (Table 1) to give 1′a and 1′b respectively. Both were obtained as a sole isomer, indicating that the initial diastereomers are syn and anti isomers resulting from a nearly complete facial discrimination of the carbonyl group (endo addition) (Table 1). It was thus postulated that a divergent evolution within the couple of enantiomers of the allylmetal occurred, one leading to the syn adduct, the other to the anti.
:1 mixture of two diastereoisomers (Table 1, entries 2 and 3). To ascertain their relative stereochemistry, 1a and 1b were hydrogenated (H2, Pd/C) (Table 1) to give 1′a and 1′b respectively. Both were obtained as a sole isomer, indicating that the initial diastereomers are syn and anti isomers resulting from a nearly complete facial discrimination of the carbonyl group (endo addition) (Table 1). It was thus postulated that a divergent evolution within the couple of enantiomers of the allylmetal occurred, one leading to the syn adduct, the other to the anti.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | n | Ketone | T (°C) | Equiv. | 1,2a (yield, %) | dr syn:anti |
| a Isolated yields. b Complex mixture of products along with starting material was obtained. | ||||||
| 1 | 1 | (+)-Fenchone | rt | 2 | <10% | ndb |
| 2 | 1 | (−)-Menthone | rt | 2 | 1a (76) | 1.8:1 |
| 3 | 1 | (+)-Camphor | rt | 2 | 1b (62) | 2:1 |
| 4 | 1 | (+)-Camphor | rt | 3 | 1b (60) | 20:1 |
| 5 | 1 | (±)-Camphor | rt | 2 | 1b (80) | 20:1 |
| 6 | 1 | (+)-Camphor | −20 | 2 | 1b (82) | 20:1 |
| 7 | 1 | (−)-Menthone | −20 | 2 | 1a (69) | 2.1:1 |
| 8 | 0 | (+)-Camphor | −20 | 3 | 2b (81) | 4:1 |
| 9 | 0 | (±)-Camphor | −20 | 2 | 2b (71) | 7.8:1 |
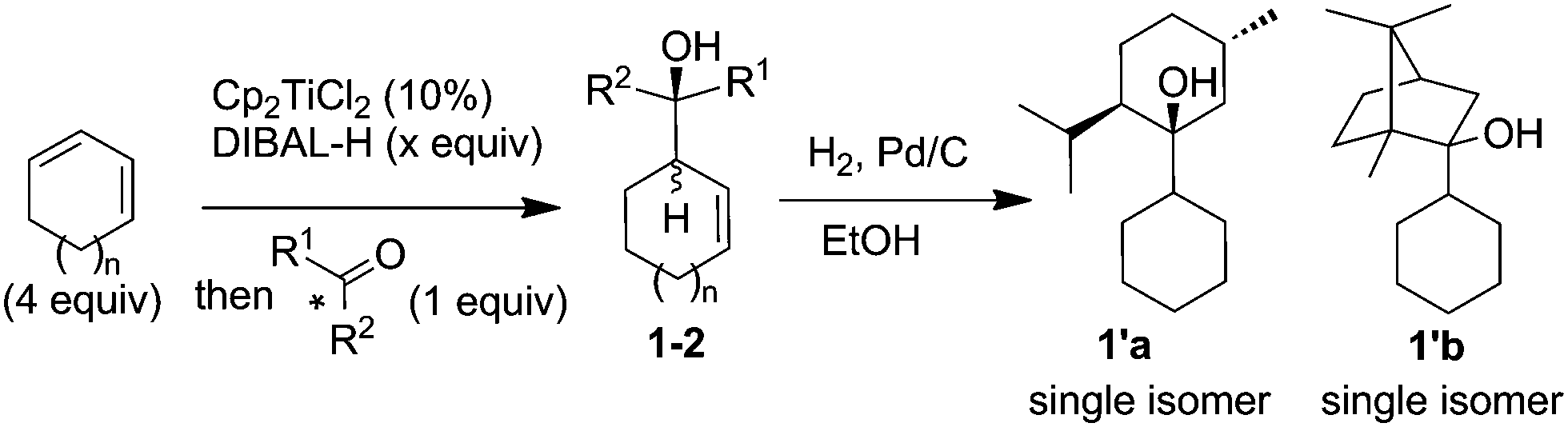
To estimate whether a kinetic resolution could also operate, additional experiments were carried out. Firstly, an excess of allylalane (3 equiv.) was reacted with (+)-camphor, resulting in a remarkable increase of the selectivity (dr > 20:1, Table 1, entry 4). Secondly, the allylalane was allowed to react with rac-camphor. In that case, the same diastereoisomer was obtained (dr > 20:1, entry 5). The absolute configuration of the major isomer 1b-syn, obtained using (+)-camphor, was unambiguously assigned through X-ray diffraction studies of its derivative 1′′b (Scheme 1).
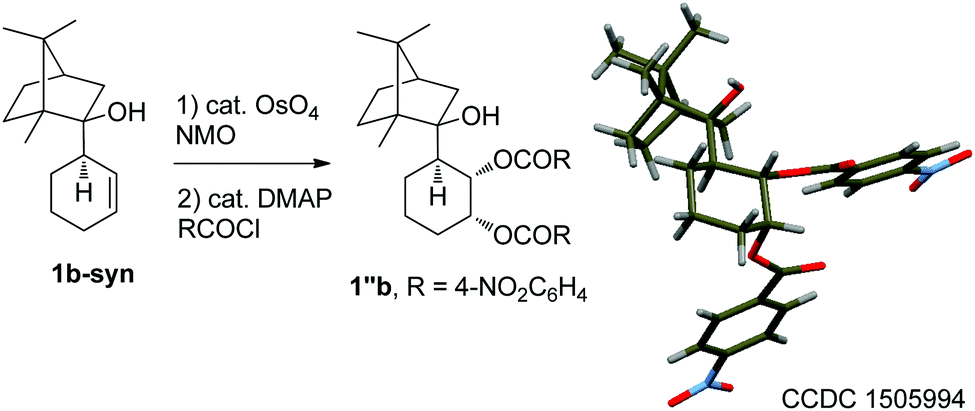 | ||
| Scheme 1 Characterisation of 1b-syn. | ||
These results indicate that a chiral recognition occurred. However, a competition persists between the two enantiomeric forms of the alane at rt, unless an excess of alane is used.
Ideally, for an optimal kinetic resolution, the total consumption of one enantiomer must be ensured using two equivalents of the racemic mixture with respect to the enantiomerically pure selector. Under these conditions, It was found that at −20 °C, the kinetic resolution was efficient using (+)-camphor as the chiral selector (entry 6), only one single isomer of 1b being observable according to NMR analysis accuracy. The rate difference between the two competing reactions might be significantly high at −20 °C to limit the consumption of one enantiomer. In contrast, the selectivity remained low with (−)-menthone (entry 7).
Finally, similar sequences were applied to cyclopentadiene, however lower selectivities were observed (entries 8 and 9).
The optimized conditions stated above should allow the seclusion of one single enantiomer of the allylalane in the reaction mixture. Thus, the whole sequence was tested by adding benzaldehyde at the last stage. In the case of cyclohexadiene, the corresponding homoallylalcohol was obtained with an enantiomeric excess of 87% by operating at −20 °C, which was slightly enhanced at −30 °C (Table 2, entries 1 and 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | n | RL | Rs | 3,4b (yield, %) | drc | erd |
| a Carried out at −20 °C. b Isolated yields. c Calculated from the crude reaction mixture. d Determined by HPLC on chiral phase. e 1.4 equiv. of (+)-camphor were used. f Could not be unambiguously determined from the crude reaction mixture, and was estimated by combining all product-containing fractions collected during the purification. g (−)-Camphor was used as the chiral selector. | ||||||
| 1a | 1 | Ph | H | 3a (65) | 98:2 |
93.5:6.5 |
| 2 | 1 | Ph | H | 3a (85) | 98:2 |
94.5:5.5 |
| 3 | 0 | Ph | H | 4a (83) | >98:2 |
70:30 |
| 4e | 0 | Ph | H | 4a (71) | >98:2 |
74:26 |
| 5 | 1 | 4-Br-C6H4 | H | 3b (89) | 96:4 |
92:8 |
| 6 | 1 | 3,4-Cl2-C6H3 | H | 3c (55) | 94:6 |
93.5:5.5 |
| 7 | 1 | 6-Br-piperonyl | H | 3d (82) | 97:3 |
92:8 |
| 8 | 1 | 4-MeCO2-C6H4 | H | 3e (75) | 93:7 |
93:7 |
| 9 | 1 | 3-Pyridyl | H | 3f (50) | 95:5 |
93.5:6.5 |
| 10 | 1 | 3-Furyl | H | 3g (83) | >98:2 |
93.5:6.5 |
| 11 | 1 | 2-Indolyl | H | 3h (64) | >98:2 |
93:7 |
| 12 | 1 | (E)-Styryl | H | 3i (80) | >98:2 |
93.5:6.5 |
| 13 | 1 | CH(Ph)2 | H | 3j (58) | >98:2 |
90.5:9.5 |
| 14 | 1 | TBSO–(CH2)3 | H | 3k (66) | >98:2 |
94.5:5.5 |
| 15 | 1 |
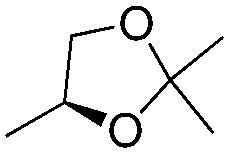
|
H | 3l (52) | nd | drf = 93:7 |
| 16g | 1 |
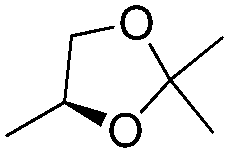
|
H | 3m (50) | nd | drf = 6:94 |
| 17 | 1 | 2-Me-C6H4 | Me | 3n (58) | >98:2 |
93:7 |
| 18 | 1 | α-Tetralone | 3o (53) | >98:2 |
93:7 |
|
| 19 | 1 | Ph | C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2OTr CCH2OTr |
3p (55) | 88:12 |
93:7 |
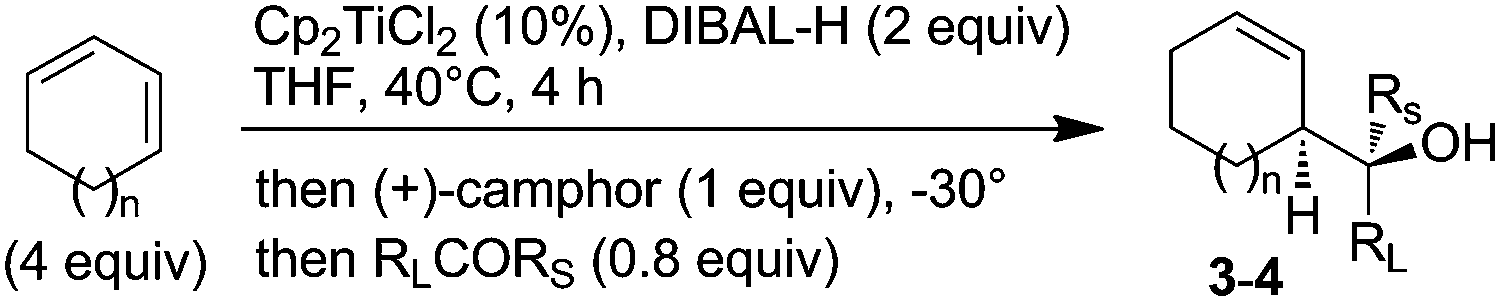
The same sequence was applied at −30 °C to cyclopentadiene, however poor enantioselectivity was obtained (entry 3), even by employing an excess of chiral selector (entry 4), as it could be anticipated from the initial results (Table 1, entries 8 and 9).
A series of aldehydes was next tested under the better conditions (e.g. −30 °C), affording homoallylic alcohols containing aromatic (entries 5–8), heteroaromatics (entries 9–11), vinylic (entry 12) or alkyl groups (entries 13 and 14) with good enantiomeric ratios (up to 94.5:5.5). The absolute configuration of homoallylic alcohols 3 was assigned by analogy with previously reported 3a and 3i.
Additionally, when enantiopure glyceraldehyde acetonide was used as the electrophile, it is noteworthy that the configuration of the newly formed stereogenic centers is subordinated to the sole configuration of the allylalane. Typically, a diastereomeric ratio of greater than 90:10 was obtained using either (+) or (−)-camphor as the chiral selector with an opposite stereoselection (entries 15 and 16), as previously reported with enantiomerically pure allyltin complexes.17
Finally, the methodology can also be applied to sterically dissymmetrical ketones (entries 17–19).
To rationalize the observed selectivities, several aspects should be taken in consideration. Firstly, the nucleophilic addition exclusively occurs at the endo face of (+)-camphor, irrespective of the allylalane configuration, which is in agreement with previously reported endo-selective allylation of camphor-derivatives.18 The predominant formation of 1b-syn is thus likely to result from the reaction of the (S)-enantiomer of the allylalane with (+)-camphor, and assumed to proceed through a Traxler–Zimmerman-like transition state, where the bulkier fragment of camphor is located in a pseudo equatorial position (Fig. 3).19 Competitively, the reaction of the (R)-enantiomer with (+)-camphor might produce 1b-anti, presumably through a different mechanistic pattern.
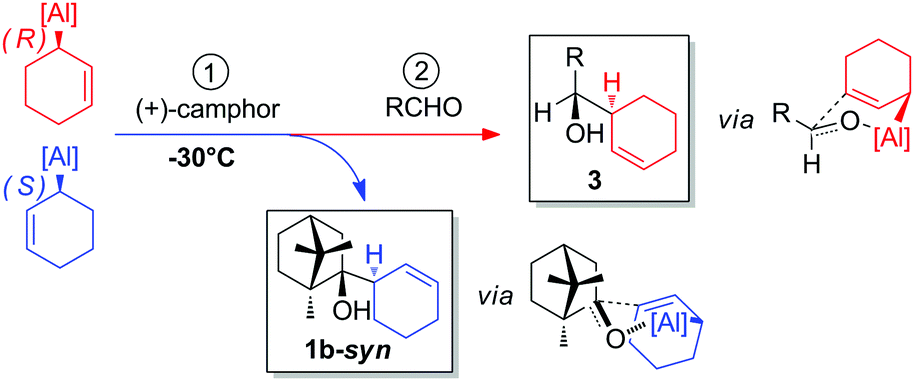 | ||
| Fig. 3 Mechanism proposal. | ||
Secondly, the reaction involving the (R)-enantiomer appears to be kinetically disfavored over that of the (S)-enantiomer at −30 °C, resulting in an efficient resolution in the case of the cyclohex-2-en-1-ylalanre.
Thirdly, once the resolution was effective, typically after 2 h at −30 °C, the remaining (R)-enantiomer was subsequently allowed to react with the aldehyde to give the pseudo-enantiomer of the camphor-adduct via a similar chair-like transition state (Fig. 3).
Interestingly, the procedure could be further valorized by converting the camphor-derived homoallylalcohol 1b-syn, side product of the resolution process. Indeed, an effective chirality transfer20 was observed when applying the palladium-mediated retroallylation/cross-coupling reaction developed by Oshima.21 Thus, the access to enantio-enriched 3-arylcyclohex-1-ene 5a–c could be achieved from 1b-syn by using arylbromide in the presence of Pd(OAc)2, P(Tol)3 and Cs2CO3 in toluene. Moreover, (+)-camphor was obtained as the co-product and could thus be recycled (Scheme 2).
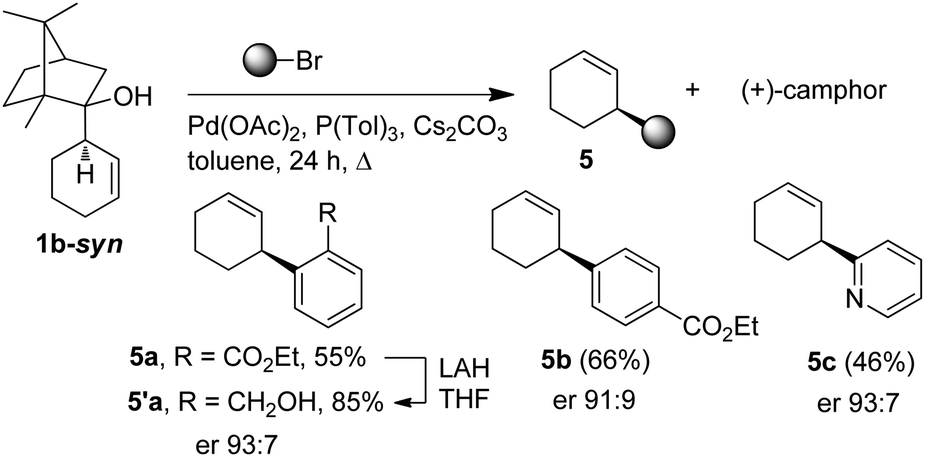 | ||
| Scheme 2 Valorisation of side product 1b-syn. | ||
In summary, an efficient kinetic resolution of the two enantiomers of cyclic allylalanes using readily available (+)-camphor as the chiral selector is described. This approach enables to in situ provide the alane in the enantiomerically enriched form, which diastereoselectively reacts with carbonyl compounds, affording homoallylalcohols with enantiomeric ratios up to 94.5:5.5. In parallel, the cyclohex-3-enylisoborneol, generated during the resolution step, could be converted into 3-arylcyclohexenes with er up to 93:7. Optimization of the chiral selector structure and generalization of the method to others allylalanes are currently under investigations.
MNRT and CNRS for financial support, Sylvie Lanthony, Sylviane Chevreux and Dominique Harakat for technical assistance are gratefully acknowledged.
Notes and references
- For recent examples of enantioselective C–Si bond-forming reactions see: (a) E. Hartmann and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 6195 CrossRef CAS PubMed; (b) E. Hartmann and M. Oestreich, Org. Lett., 2012, 14, 2406 CrossRef CAS PubMed; (c) K.-s. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 2898 CrossRef CAS PubMed; (d) I. Ibrahem, S. Santoro, F. Himo and A. Córdova, Adv. Synth. Catal., 2011, 353, 245 CrossRef CAS; (e) V. Pace, J. P. Rae, H. Y. Harb and D. J. Procter, Chem. Commun., 2013, 49, 5150 RSC; (f) J. Plotzitzka and C. Kleeberg, Organometallics, 2014, 33, 6915 CrossRef CAS ; for recent examples of enantioselective C–B bond forming reactions see: ; (g) J. M. O’Brien, K.-s. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10630 CrossRef PubMed; (h) R. Corberán, N. W. Mszar and A. H. Hoveyda, Angew. Chem., Int. Ed., 2011, 50, 7079 CrossRef PubMed; (i) F. Meng, H. Jang and A. H. Hoveyda, Chem. – Eur. J., 2013, 19, 3204 CrossRef CAS PubMed; (j) N. Matsuda, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2013, 135, 4934 CrossRef CAS PubMed; (k) H. Jang, B. Jung and A. H. Hoveyda, Org. Lett., 2014, 16, 4658 CrossRef CAS PubMed ; for examples of enantioselective C–Sn bond formation reaction see: ; (l) T. Jia, P. Cao, D. Wang, Y. Lou and J. Liao, Chem. – Eur. J., 2015, 21, 4918 CrossRef CAS PubMed; (m) M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2004, 126, 3688 CrossRef CAS PubMed.
- Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207 CrossRef CAS.
- (a) F. Peng and D. G. Hall, J. Am. Chem. Soc., 2007, 129, 3070 CrossRef CAS PubMed; (b) J. L.-Y. Chen, H. K. Scott, M. J. Hesse, C. L. Willis and V. K. Aggarwal, J. Am. Chem. Soc., 2013, 135, 5316 CrossRef CAS PubMed; (c) H. Ito, S. Kunii and M. Sawamura, Nat. Chem., 2010, 2, 972 CrossRef CAS PubMed.
- (a) D. Li, T. Tanaka, H. Ohmiya and M. Sawamura, Org. Lett., 2010, 12, 3344 CrossRef CAS PubMed; (b) Y. Yoshimura, M. Ohta, T. Imahori, T. Imamichi and H. Takahata, Org. Lett., 2008, 16, 3449 CrossRef PubMed; (c) H. S. Park, J. Wook Han, R. Shintani and T. Hayashi, Tetrahedron: Asymmetry, 2013, 24, 418 CrossRef CAS; (d) J. Wook Han and T. Hayashi, Tetrahedron: Asymmetry, 2002, 13, 325 CrossRef; (e) J.-M. Adam, L. de Fays, M. Laguerre and L. Ghosez, Tetrahedron, 2004, 60, 7325 CrossRef CAS.
- (a) J. Bejjani, C. Botuha, F. Chemla, F. Ferreira, S. Magnus and A. Perez-Luna, Organometallics, 2012, 31, 4876 CrossRef CAS; (b) C. J. Cordier, R. J. Lundgren and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 10946 CrossRef CAS PubMed; (c) N. P. Mulholland, G. Pattenden and I. A. S. Walters, Org. Biomol. Chem., 2008, 6, 2782 RSC; (d) T. Ling, V. R. Macherla, R. R. Manam, K. A. Mc Arthur and B. C. M. Potts, Org. Lett., 2007, 9, 2289 CrossRef CAS PubMed; (e) L. R. Reddy, B. Hu, M. Prashad and K. Prasad, Org. Lett., 2008, 10, 3109 CrossRef CAS PubMed.
- (a) F. R. Struth and C. H. Hirschhäuser, Eur. J. Org. Chem., 2016, 958 CrossRef CAS; (b) M. Chen and W. R. Roush, Org. Lett., 2010, 12, 2706 CrossRef CAS PubMed.
- J. Pietruszka, N. Schöne, W. Frey and L. Grundl, Chem. – Eur. J., 2008, 14, 5178 CrossRef CAS PubMed.
- (a) J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778 CrossRef CAS PubMed; (b) V. Bagutski, R. M. French and V. K. Aggarwal, Angew. Chem., Int. Ed., 2010, 49, 5142 CrossRef CAS PubMed; (c) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174 CrossRef CAS PubMed; (d) S. Balieu, G. E. Hallett, M. Burns, T. Bootwicha, J. Studley and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 4398 CrossRef CAS PubMed; (e) S. Roesner, D. J. Blair and V. K. Aggarwal, Chem. Sci., 2015, 6, 3718 RSC.
- (a) R. P. Sonawane, V. Jheengut, C. Rabalakos, R. Larouche-Gauthier, H. K. Scott and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 3760 CrossRef CAS PubMed; (b) P. J. Unworth, D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 9846 CrossRef PubMed; (c) J. L.-Y. Chen and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 10992 CrossRef CAS PubMed; (d) M. Althaus, A. Mahmood, J. R. Suárez, S. P. Thomas and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 4025 CrossRef CAS PubMed.
- (a) K.-s. Lee, H. Wu, F. Haeffner and A. H. Hoveyda, Organometallics, 2012, 31, 7823 CrossRef CAS PubMed; (b) J. W. Han and T. Hayashi, Tetrahedron: Asymmetry, 2010, 21, 2193 CrossRef CAS; (c) J. W. Han, N. Tokunaga and T. Hayashi, Helv. Chim. Acta, 2002, 85, 3848 CrossRef CAS; (d) T. Hayashi, K. Kabeta, T. Yamamoto, K. Tamao and M. Kumada, Tetrahedron Lett., 1983, 24, 5661 CrossRef CAS; (e) T. Hayashi, J. W. Han, A. Takeda, J. Tang, K. Nohmi, K. Mukaide, H. Tsuji and Y. Uozumi, Adv. Synth. Catal., 2001, 343, 279 CrossRef CAS.
- (a) H. C. Brown, K. S. Bhat and P. K. Jadhav, J. Am. Chem. Soc., 1985, 107, 2564 CrossRef CAS; (b) H. C. Brown, K. S. Bhat and P. K. Jadhav, J. Chem. Soc., Perkin Trans. 1, 1991, 2633 RSC; (c) Y. Sasaki, C. Zhong, M. Sawamura and H. Ito, J. Am. Chem. Soc., 2010, 132, 1226 CrossRef CAS PubMed; (d) E. González, L. Muñoz-Hernández, E. Alicea, B. Singaram, G. W. Kabalka and J. A. Soderquist, Org. Lett., 2015, 17, 4368 CrossRef PubMed.
- (a) M. Gerdin and C. Moberg, Adv. Synth. Catal., 2005, 347, 749 CrossRef CAS; (b) M. Gerdin, M. Penhoat, R. Zalubovskis, C. Pétermann and C. Moberg, J. Organomet. Chem., 2008, 693, 3519 CrossRef CAS.
- (a) L. Carosi and D. G. Hall, Angew. Chem., Int. Ed., 2007, 46, 5913 CrossRef CAS PubMed; (b) Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 3160 CrossRef CAS PubMed; (c) J. K. Park and D. T. McQuade, Synthesis, 2012, 1485 CAS; (d) M. A. Kacprzynski, T. L. May, S. A. Kazane and A. H. Hoveyda, Angew. Chem., Int. Ed., 2007, 46, 4554 CrossRef CAS PubMed; (e) M. Takeda, R. Shintani and T. Hayashi, J. Org. Chem., 2013, 78, 5007 CrossRef CAS PubMed; (f) L. B. Delvos, D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 4650 CrossRef CAS PubMed.
- J. Joseph, F. Jaroschik, K. V. Radhakrishnan, D. Harakat, J.-L. Vasse and J. Szymoniak, Chem. – Eur. J., 2014, 20, 5433 CrossRef CAS PubMed.
- M. Coffinet, S. Lamy, F. Jaroschik and J.-L. Vasse, Org. Biomol. Chem., 2016, 14, 69 CAS.
- The access to enantio-enriched allyltin derivatives by kinetic resolution was previously reported by reacting a racemic mixture of allyltin with enantiopure cyclohexylidene glyceraldehyde: F. Fliegel, I. Beaudet and J.-P. Quintard, J. Organomet. Chem., 2001, 624, 383 CrossRef CAS.
- (a) J. A. Marshall, Chem. Rev., 1996, 96, 31 CrossRef CAS PubMed; (b) J. A. Marshall, J. Org. Chem., 2007, 72, 8153 CrossRef CAS PubMed.
- (a) B. Schmidt and L. Staude, J. Organomet. Chem., 2006, 691, 5218 CrossRef CAS; (b) D. J. Dixon, R. A. J. Horan and N. J. T. Monck, Org. Lett., 2004, 6, 4423 CrossRef CAS PubMed; (c) S. Sezer, Y. Gümrükçü, E. Sahin and C. Tanyeli, Tetrahedron: Asymmetry, 2008, 19, 2705 CrossRef CAS; (d) C.-L. K. Lee, C.-H. A. Lee, K.-T. Tan and T.-P. Loh, Org. Lett., 2004, 6, 1281 CrossRef CAS PubMed; (e) W. H. Bunnelle, M. A. Rafferty and S. L. Hodges, J. Org. Chem., 1987, 52, 1603 CrossRef CAS.
- (a) Z. Peng, T. D. Blümke, P. Mayer and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 8516–8519 CrossRef CAS PubMed; (b) H. Ren, G. Dunet, P. Mayer and P. Knochel, J. Am. Chem. Soc., 2007, 129, 5376 CrossRef CAS PubMed.
- R. Wakabayashi, D. Fujino, S. Hayashi, H. Yorimitsu and K. Oshima, J. Org. Chem., 2010, 75, 4337 CrossRef CAS PubMed.
- (a) S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2006, 128, 2210 CrossRef CAS PubMed; (b) M. Iwasaki, S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2007, 129, 4463 CrossRef CAS PubMed; (c) S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2007, 129, 12650 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of new compounds, HPLC copies. CCDC 1505994 (1b-syn). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08649g |
| This journal is © The Royal Society of Chemistry 2017 |