
Chemically synthesized and cross-linked PDMS as versatile alignment medium for organic compounds†
Yulia E.
Moskalenko‡§
,
Viktor
Bagutski§
and
Christina M.
Thiele
 *
*
Clemens-Schöpf-Institut für Organische Chemie und Biochemie, Technische Unitersität Darmstadt, Alarich-Weiss-Strasse 16, 64287 Darmstadt, Germany. E-mail: cthiele@thielelab.de
First published on 23rd November 2016
Abstract
A useful procedure for the preparation of chemically synthesized and cross-linked polydimethylsiloxane (PDMS) gels is presented, which does not require β-irradiation for cross-linking. NMR spectra of high quality are obtained, such that even mixtures of compounds exhibiting similar NMR spectra like interconverting stereoisomers can be investigated in the residual dipolar coupling (RDC) approach of organic structure determination.
The determination of conformation and configuration of organic compounds usually involves the measurement of 3J couplings1 and the nuclear Overhauser effect (NOE).2 Residual dipolar couplings (RDCs) become increasingly important in the structure elucidation process due to their complementary information content.3 While 3J couplings and NOEs can be measured in isotropic solution, the compound in question needs to be oriented with respect to the magnetic field to get access to RDCs. Anisotropy in the orientation of the compound is induced via suitable alignment media, which should be compatible with organic solvents and solutes, should be homogeneous and ideally also scalable. For organic compounds there are two main classes of alignment media: namely lyotropic liquid crystals and anisotropically swollen cross-linked polymer gels.3 The former have the advantage of a quick sample preparation; scalability, however, is limited.4,5 For the latter it is known that a scaling of the order induced in the compound can be performed either via different cross-linking degrees (before sample preparation)6 or even on the readily prepared gel using a gel-stretching device.7,8 Swelling of gels in the NMR sample tube can be a time-consuming process, though. A series of cross-linked polymer gels already exist. Apart from poly(methyl methacrylate) (PMMA) gels, from which initiators and cross-linkers can be removed via reversible compression and release,8 and thermally cross-linked polystyrene9, polymer gels normally contain some impurities due to their fabrication process (residual monomer, radical initiator, cross-linker). These can lead to solute incompatibilities or residual signals in the NMR spectrum. In this respect PDMS gels stand out as they have so far been made by irradiation with β-rays.10 Furthermore, the residual signal of PDMS appears in a region in the spectrum (around 0 ppm in 1H, 13C and 29Si) where it does not overlap with signals of commonly studied compounds.
In this communication, we present a procedure for the preparation of PDMS sticks requiring neither β-irradiation nor radical initiators during polymerisation and/or cross-linking. It is shown that the gels obtained are suitable as alignment medium even for sensitive compounds. High-quality NMR spectra are obtained, which allow investigating mixtures of compounds exhibiting similar NMR spectra such as stereoisomers.
The beauty of the synthetic procedure (see Scheme 1) lies in the use of one single starting material octamethylcyclotetrasiloxane (D4) for polymer, initiator and cross-linker.11,12
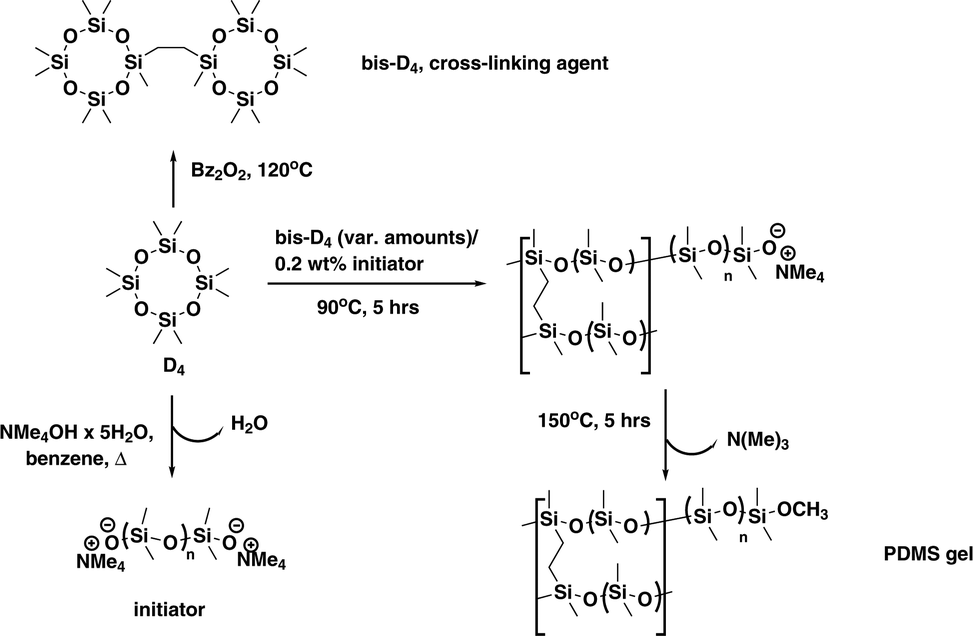 | ||
| Scheme 1 Synthetic procedure for the preparation of PDMS gels: starting from octamethylcyclotetrasiloxane (D4), the cross-linking agent bis(heptamethylcyclotetrasiloxanyl)-ethane (bis-D4) as well as the initiator bis(tetramethylammonium)oligodimethylsiloxanediolate are prepared. | ||
Thus no non-silicone cross-linkers are used alleviating problems of unwanted rearrangements as well as parasitic signals of compounds related to the manufacturing process of the gel. The PDMS gel with living anionic ends would allow for exploitation of self-healing properties,11 which can in principle be used for direct preparation of the gel inside the NMR sample tube. This has not been exploited here, though.
An annealing step at 150 °C is used to “decatalyse” the living polymer (removal of NMe3),11,13 making the polymer gel stable and thus suitable for its use as alignment medium. While dibenzoyl peroxide (Bz2O2) is used in the preparation of the cross-linking agent bis(heptamethylcyclotetrasiloxanyl)-ethane (bis-D4), this can be removed by various purification steps (see ESI†) before the actual gel is produced. We envisaged that using this living anionic polymerisation would allow for the preparation of a homogeneous and contamination-free alignment medium with improved spatial regularity ensured by the essentially equilibrative character of the polymerisation process.
For the preparation of the gels an array of molds easily prepared from commercially available PFTE tubing (3 or 3.2 mm inner diameter) was filled with degassed polymerising mixture of varying cross-linker content and subjected to polymerisation at 90 °C (see ESI†) over 5 h to afford the PDMS-sticks of an appropriate quality after thermal decatalysing¶ (see Scheme 1). The sticks obtained via this procedure were introduced into NMR tubes and swollen in CDCl3. Quadrupolar deuterium splitting ΔνQ of CDCl3 is taken as indicator of anisotropy.
The swelling process of the gel sample over time and the assessment of its homogeneity can conveniently be performed by 2H imaging14, a technique that allows for obtaining 2H spectra as a function of z-position (see Fig. 1).
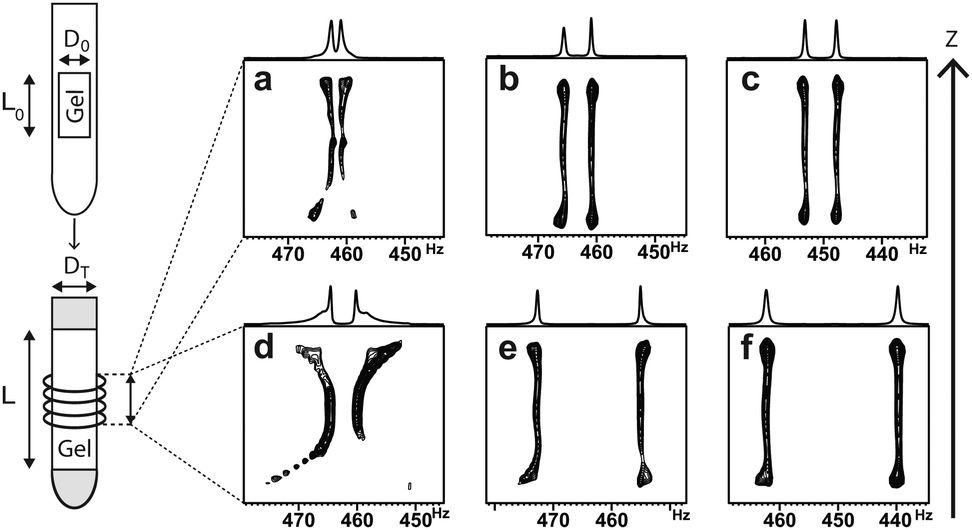 | ||
| Fig. 1 2H NMR images of PDMS gels at 300 K after swelling in CDCl3 (5 mm OD NMR tubes) over 18 h (a and d), 4 days (b and e) and 8 days (c and f). Gels contained 0.5 mol% of bis-D4 (top row: a–c) and 1.3 mol% of bis-D4.(bottom row: d–f). | ||
As solvent is added to the top and to the bottom of the NMR tube in the sample preparation process (see ESI†), these regions display the largest quadrupolar splittings in the beginning (see Fig. 1a and d) indicating the largest degrees of swelling. Within one week (see Fig. 1c and f) samples reach sufficient axial homogeneity for RDC investigations. Only a slight increase of ΔνQ (<5% per day) is observed afterwards indicating an asymptotical behaviour of swelling near the saturation point. The fully equilibrated gels exhibit ΔνQ in a range between 7 and 48 Hz as it is shown in Table 1. As can be seen from the values in Table 1 and is known for other gels6,15 the degree of swelling can be influenced by the choice of the diameters of the mold, the inner diameter of the NMR tube (entries 1–3 vs. 4 and 5) and the cross-linker density. Here, beyond 2 mol% of cross-linker no increase in splitting is observed anymore.
While we did not do the detailed investigation here, it is to be expected that the different quadrupolar splittings of the solvent translate into different degrees of orientation and thus into different sizes of RDCs for the solutes to be investigated.
A clear advantage of our gel preparation protocol is that the parameters required for the characterization of the physical state of the gels are well known, allowing for the modeling of ΔνQ, the measured quadrupolar splitting of the gel solvent. As further discussed in the theory section of ESI,† one has ΔνQ = Δν0QεϕS, where S is the order parameter for monomer orientations, ϕ is the chain monomer volume fraction and Δν0Q = 168 kHz16 is the quadrupolar splitting of perfectly aligned CDCl3 molecules. The efficiency factor ε accounts for the transfer of orientation between the partially oriented chain monomers and the solvent molecules during a solvent–monomer encounter; it is an intrinsic property of each given solvent–monomer pair. The careful measurement of gel and tube dimensions at all stages of our experiments and the knowledge of the cross-linking molar ratios allows computing ϕ and S from standard polymer–gel theory. We found (see ESI†) good agreement between theory and experiments and determine the efficiency for this particular gel–solvent pair ε = 6.5 ± 0.5 × 10−3, roughly at one part per 150. This in turn provides for the quantitative prediction of ΔνQ values for any tube stretching experiments performed for the polymer/solvent pair PDMS/CDCl3. Predictions for other polymer solvent systems would simply require performing similar sets of experiments to extract the corresponding orientation efficiency factors ε. We believe that this will allow for the tailor-made synthesis of PDMS-sticks in the future.
To highlight the suitability of the synthesized PDMS gel for the acquisition of high quality RDC data sets we have chosen the ubiquitous natural sesquiterpene β-(−)-caryophyllene (BCP). It is deemed a challenging object for structure elucidation due to its known complex conformational composition exhibiting more than one signal set (Fig. 2).17
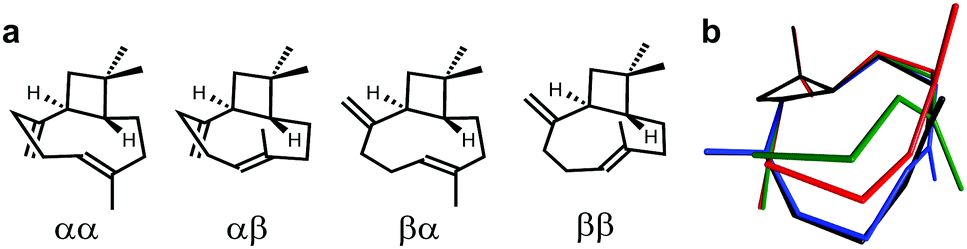 | ||
| Fig. 2 (a) β-(−)-Caryophyllene conformers (descriptors convention: α – “down”, β – “up”, first: methylene group, second: alkenyl-CH3) and (b) their cumulative 3D representation (αα – green, αβ – red, βα – blue and ββ – black). Putative conformer αβ is not present in the mixture as it is energetically disfavoured.17 | ||
The composition of the solution equilibrium of conformations has been determined NMR-spectroscopically at low temperature (120 K) previously,17 but no assignment of diastereotopic protons has been published so far. Thus BCP is the ideal candidate to demonstrate the suitability of chemically synthesized PDMS gel for the analysis of complex mixtures.
For the RDC measurements we thus equilibrated a PDMS gel with 1.3 mol% cross-linker density in a BCP/CDCl3 (5 μL mL−1) solution. The spectra were of excellent quality (see Fig. 3).
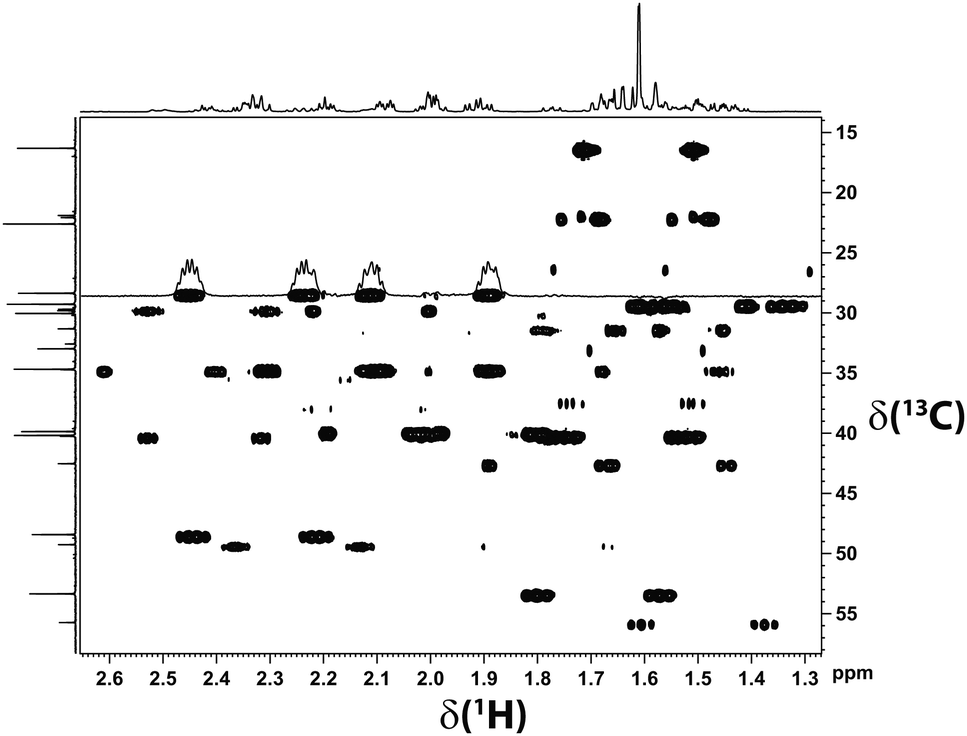 | ||
| Fig. 3 Section of the CLIP-HSQC spectrum18 of β-(−)-caryophyllene recorded in a chemically synthesized PDMS gel equilibrated in CDCl3. The high quality of the spectrum allows for the precise extraction of 1DC–H. Note that two signal sets of different proportion are visible. The quality of the acquired data is very high; even the fine structure of the multiplets is visible. | ||
As can be seen in Fig. 3, two signal sets – one of higher and one of lower intensity – are obtained. At low temperatures the minor signal set corresponds to the ββ-conformer, whereas the remaining signal set was assigned to interconverting αα- and βα-conformers.17
Accordingly, two sets of RDCs were obtained for the two signal sets; values varied between −1 and +4 Hz at 279 K (using the J + 2D convention for T) (see ESI†). Thus we fitted the RDCs belonging to the signal set of lower intensity to the DFT calculated geometry of the ββ-conformer using our in-house developed software RDC@hotfcht.19,20 As can be seen in Fig. 4a an excellent fit of RDCs of the minor signal set to the coordinates of the ββ-conformer was obtained. Fitting of these data to the (DFT derived) coordinates of the αα- or βα-conformers yielded a significantly poorer agreement (as indicated by a larger Q-factor) (see ESI†) confirming the assignment of the minor signal set to the ββ-conformer.
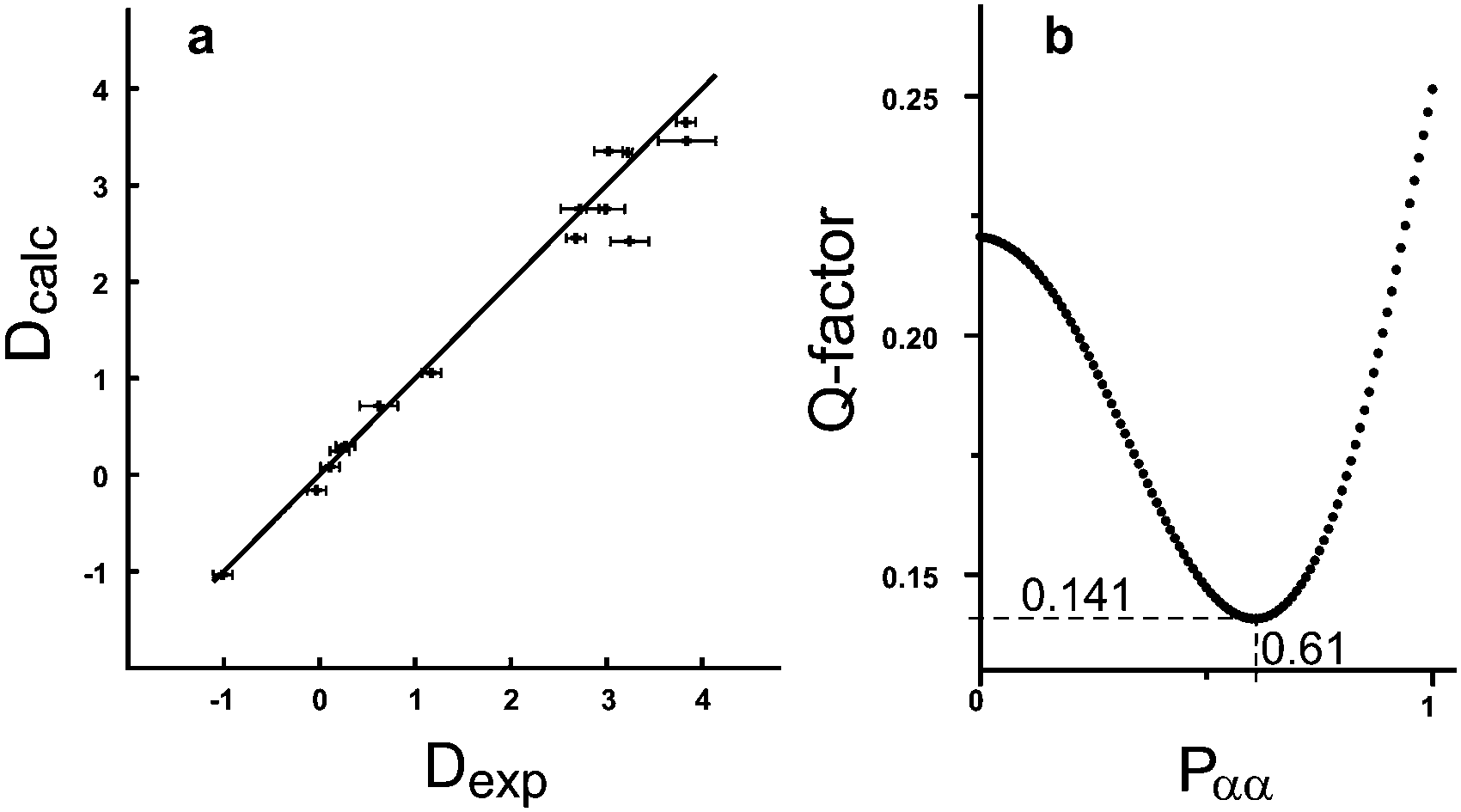 | ||
| Fig. 4 (a) Correlation plot of 15 RDCs measured for the minor signal set to the coordinates of the ββ-conformer of BCP (Q = 0.074); (b) population scan (1% steps) for the 17 RDCs of the major signal set to the coordinates of the αα- and βα-conformers. The population with the best agreement with experimental data is Pαα = 0.61 (Q = 0.141). | ||
For the major signal set 17 RDCs were obtained. As it is expected that this signal set does not belong to a single conformer, but is due to interconverting conformers, flexibility needs to be taken into account in the fitting procedure.20 Fitting the RDCs in a multi-conformer single-tensor analysis to the coordinates of the αα- and βα-conformer (1% step size in the population scan) leads to an excellent agreement with experimental data at αα![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :βα = 61:39 (see Fig. 4b). Thus this composition is assumed to be the one present in solution (in the anisotropic sample at 279 K). This outcome turns out to be in excellent agreement with the previously published results of both – an experimental (63% of αα at 120 K)17 and a computational study (67.8% of αα)21. Additionally, we provide the so far missing diastereotopic assignment of all protons (see ESI†).
:βα = 61:39 (see Fig. 4b). Thus this composition is assumed to be the one present in solution (in the anisotropic sample at 279 K). This outcome turns out to be in excellent agreement with the previously published results of both – an experimental (63% of αα at 120 K)17 and a computational study (67.8% of αα)21. Additionally, we provide the so far missing diastereotopic assignment of all protons (see ESI†).
In terms of its practical application as alignment medium it is important to note that PDMS is not limited to CDCl3 as solvent, but also swells in THF10,22 and even at lower temperatures (253 K was chosen here). As might be expected, swelling time is increased with decreasing temperature (see ESI†).
The applicability at lower temperatures greatly extends the scope towards covering air- and heat-sensitive (non-basic, see below) organometallic compounds. We have used this feature in the investigation of structure and dynamics of η1-coordinated Pd-intermediates.22 As can be seen in Fig. 5 the spectral quality is excellent despite the two species A and B being present simultaneously.
 | ||
| Fig. 5 CLIP-HSQC spectrum18 of diastereomeric Pd-complexes A and B22 in chemically cross-linked PDMS (1.25 mol% cross-linker density) swollen in THF-d8 (swelling performed at 253 K, measurement performed at 293 K). The phosphoramidite ligand indicated is MonoPhos™. | ||
Thus chemically synthesized PDMS is a versatile alignment medium, which owing to the excellent spectral quality allows for investigating mixtures of rather similar compounds. An application to the structural analysis of a peptide will be presented separately.23
It is worth mentioning here that in terms of solute and solvent compatibility the inherent sensitivity of PDMS towards Brønsted bases needs to be taken into account.24 Contrary to our expectation neither the Pd-complexes described above nor the trimethylamine liberated in the decatalysing step cause any problems in anhydrous solvents. In a wet solvent, however, slow decomposition of the PDMS-gel may happen. Slow decomposition was also observed in the case of an attempted RDC measurements of (+)-isopinocampheol (IPC) in chloroform (see ESI†). Interestingly, the same tendency for decomposition in the presence of (+)-IPC has been also displayed by a sample of PDMS produced by β-irradiation (gift from the Luy group). Thus this incompatibility is not a result from the synthetic procedure.
On a positive side, this feature can readily be exploited to develop a facile procedure for the removal of used PDMS from the NMR tubes. It was found that the simple addition of THF slightly basified with a few drops of methanolic NaOH completely liquefies the gel. This allows for the easy recovery of the expensive “Young's” NMR-tubes, which are a prerequisite for NMR measurements of highly air- and moisture sensitive compounds.
In this communication, we have presented a valuable procedure for the chemical synthesis of PDMS gels for the use as alignment medium in NMR-spectroscopy. The high regularity obtained by the synthetic procedure enables the acquisition of spectra of excellent quality, such that even mixtures of compounds exhibiting similar NMR spectra can be investigated. This is exemplified here for the sesquiterpene β-(–)-caryophyllene (BCP) and for a mixture of η1-coordinated Pd-intermediates. The known incompatibility of PDMS with Brønsted bases is exploited for introducing a cleaning procedure for expensive NMR tubes.
Furthermore, the known cross-linker content and careful monitoring of the swelling geometry allowed for the successful comparison between polymer gel theory and experiments, providing the calibration of the orientation efficiency factor for the PDMS/CDCl3 pair, which will enable the preparation of PDMS gel sticks with predefined orientational order.
The authors thank Burkhard Luy for providing PDMS sticks obtained by β-irradiation and for helpful discussions concerning solute compatibility. We thank Dr C. M. Marques (ICS CNRS, Stasbourg) for his help with the evaluation of gel anisotropy and prediction of solvent quadrupolar splittings. PD Dr S. Immel and Dr V. Schmidts provided help with the DFT calculations of the BCP conformers. This work was supported by the ERC (St. G. 257041 to C. M. T.).
Notes and references
- C. A. G. Haasnoot, F. A. A. M. de Leeuw and C. Altona, Tetrahedron, 1980, 36, 2783–2792 CrossRef CAS.
- F. A. L. Anet and A. J. R. Bourn, J. Am. Chem. Soc., 1965, 87, 5250–5251 CrossRef CAS; C. P. Butts, C. R. Jones and J. N. Harvey, Chem. Commun., 2011, 47, 1193–1195 RSC.
- Reviews: C. M. Thiele, Eur. J. Org. Chem., 2008, 5673–5685 CrossRef CAS; G. Kummerlöwe and B. Luy, Trends Anal. Chem., 2009, 28, 483–493 CrossRef; R. R. Gil, Angew. Chem., Int. Ed., 2011, 50, 7222–7224 CrossRef PubMed; V. Schmidts, Magn. Reson. Chem., 2016 DOI:10.1002/mrc.4543.
- A. Marx and C. Thiele, Chem. – Eur. J., 2009, 15, 254–260 CrossRef CAS PubMed.
- C. M. Thiele, Angew. Chem., Int. Ed., 2005, 44, 2787–2790 CrossRef CAS PubMed.
- B. Luy, K. Kobzar and H. Kessler, Angew. Chem., Int. Ed., 2004, 43, 1092–1094 CrossRef CAS PubMed; B. Luy, K. Kobzar, S. Knör, J. Furrer, D. Heckmann and H. Kessler, J. Am. Chem. Soc., 2005, 127, 6459–6465 CrossRef PubMed; R. R. Gil, C. Gayathri, N. V. Tsarevsky and K. Matyjaszewski, J. Org. Chem., 2008, 73, 840–848 CrossRef PubMed.
- R. Tycko, F. J. Blanco and Y. Ishii, J. Am. Chem. Soc., 2000, 122, 9340–9341 CrossRef CAS; P. W. Kuchel, B. E. Chapman, N. Müller, W. A. Bubb, D. J. Philp and A. M. Torres, J. Magn. Reson., 2006, 180, 256–265 CrossRef PubMed; G. Kummerlöwe, F. Halbach, B. Laufer and B. Luy, Open Spectrosc. J., 2008, 2, 29–33 CrossRef.
- C. Gayathri, N. V. Tsarevsky and R. R. Gil, Chem. – Eur. J., 2010, 16, 3622–3626 CrossRef CAS PubMed.
- A.-C. Pöppler, H. Keil, D. Stalke and M. John, Angew. Chem., Int. Ed., 2012, 51, 7843–7846 CrossRef PubMed.
- J. C. Freudenberger, P. Spiteller, R. Bauer, H. Kessler and B. Luy, J. Am. Chem. Soc., 2004, 126, 14690–14691 CrossRef CAS PubMed.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS PubMed.
- O. K. Johannson, US Pat., 2762827, 1956; S. W. Kantor, US Pat., 2785147, 1957; S. W. Kantor and R. C. Ostoff, US Pat., 2793222, 1957.
- A. R. Gilbert and S. W. Kantor, J. Polym. Sci., 1959, 11, 35–58 CrossRef.
- P. Trigo-Mouriño, C. Merle, M. R. M. Koos, B. Luy and R. R. Gil, Chem. – Eur. J., 2013, 19, 7013–7019 CrossRef PubMed.
- T. Montag and C. M. Thiele, Chem. – Eur. J., 2013, 19, 2271–2274 CrossRef CAS PubMed.
- B. Deloche, A. Dubault and D. Durand, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 1419–1421 CrossRef CAS.
- M. Hübner, B. Rissom and L. Fitjer, Helv. Chim. Acta, 1997, 80, 1972–1982 CrossRef.
- A. Enthart, J. C. Freudenberger, J. Furrer, H. Kessler and B. Luy, J. Magn. Reson., 2008, 192, 314–322 CrossRef CAS PubMed.
- V. Schmidts, PhD thesis, Technische Universität Darmstadt, 2013; R. Berger, C. Fischer and M. Klessinger, J. Phys. Chem. A, 1998, 102, 7157–7167 Search PubMed.
- C. M. Thiele, V. Schmidts, B. Böttcher, I. Louzao, R. Berger, A. Maliniak and B. Stevensson, Angew. Chem., Int. Ed., 2009, 48, 6708–6712 CrossRef CAS PubMed.
- M. Clericuzio, G. Alagona, C. Ghio and L. Toma, J. Org. Chem., 2000, 65, 6910–6916 CrossRef CAS PubMed.
- L.-G. Xie, V. Bagutski, D. Audisio, L. M. Wolf, V. Schmidts, K. Hoffmann, C. Wirtz, W. Thiel, C. M. Thiele and N. Maulide, Chem. Sci., 2015, 6, 5734–5739 RSC.
- M. Fredersdorf, M. Kurz, A. Bauer, M. O. Ebert, C. Rigling, L. Lannes and C. M. Thiele, manuscript in preparation.
- J. N. Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthesis procedure for PDMS sticks, RDC data on β-(−)-caryophyllene and IPC, investigation of PDMS decomposition, theory to predict quadrupolar splittings induced, representative spectra in PDMS gels. See DOI: 10.1039/c6cc08762k |
| ‡ Current address: Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, 05508-900 São Paulo-SP, Brazil. |
| § These authors have contributed equally to this work. |
| ¶ The polymerization process was terminated by heating at 150 °C for 5 h to accomplish thermal decomposition of the catalyst liberating a miniscule amount of trimethylamine. |
| This journal is © The Royal Society of Chemistry 2017 |