
Evolution of solid/aqueous interface in aqueous sodium-ion batteries†
Xiaowen
Zhan
and
Mona
Shirpour
*
Department of Chemical and Materials Engineering, University of Kentucky, Lexington, KY 40506, USA. E-mail: mona.shirpour@uky.edu
First published on 28th November 2016
Abstract
The microstructural and compositional evolution at the solid/aqueous solution interfaces are investigated to monitor the electrical properties of superionic conducting phosphates and the electrochemical failure of aqueous sodium-ion batteries.
The challenges associated with matching the intermittent solar- and wind-generated electricity and variable electricity demand pattern have motivated the development of energy storage systems. The systems suitable for storing renewable sources of energy need to be cost competitive, reliable, and safe in order to meet the needs of grid and microgrid applications. Each grid energy storage system has its own performance characteristics that makes it more suitable for certain grid services and less suitable for other grid applications. Battery systems (e.g., lithium-ion, sodium sulfur, lead acid, and flow batteries) are robust and their performance matches different grid requirements allowing the same storage system to provide multiple services. The low-cost lead-acid batteries are suitable for load leveling and grid stabilization, but have low energy density and a large environmental footprint. Sodium sulfur batteries exhibit high energy density, long discharge cycles and life, and fast response, but encounter liquid-handling difficulties at their operating temperature of about 300 °C. Flow batteries, with very long calendar and cycle lives, provide megawatt-scale storage capacity beyond the geographic constraints of hydroelectric facilities, but due to low energy density and complicated design, they are not the most suitable systems for sub-megawatt scale storage. Lithium- and sodium-ion batteries are best-suited for relatively short discharges (less than two hours) and do not handle deep discharges, so these batteries are more suited for power-management operations such as frequency regulation or as an uninterruptible power source.1,2 Sodium-ion batteries are an appealing lower-cost alternative to lithium-ion batteries because of the vast resources of sodium and the much lower materials costs.3,4 Besides the cost savings and abundance associated with the sodium precursors, aqueous sodium-ion batteries offer extra advantages of using much cheaper water-based electrolyte solutions, less costly manufacturing conditions, and safety, due to the absence of expensive and hazardous organic solutions.4–8 Therefore, aqueous sodium-ion batteries are promising candidates for (micro)grid storage applications that require low-cost energy storage systems with safety/low environmental issues because the units are large and always energized.1,2 In the aqueous (water-based) cells, both the anode and cathode should undergo redox processes approximately within the voltage stability region of water. The high-voltage water-in-salt aqueous electrolytes9 and sulfonate-containing electrolytes10 expand the electrochemical stability of water against oxygen evolution in aqueous lithium-ion cells thus enabling the use of higher potential cathode materials. On the other side, most of the anode materials for sodium-ion batteries, including carbon-based compounds, alloying metals, and titanates, have very low operating potentials vs. sodium, and fall in the region of hydrogen evolution (Fig. S1, ESI†). The NASICON (sodium (Na) Super Ionic CONductor)-type NaTi2(PO4)3 is perfectly located at the lower border of the water voltage stability region and operates at 2.1 V versus Na/Na+, thus provides access to the maximum possible energy available in this system. The aqueous sodium-ion batteries7,8,11–14 using NaTi2(PO4)3 anode and Na0.44MnO2 or Na2FePO4F22,23 cathode materials exhibit initial capacities close to the theoretical values; however, the cells cannot maintain their initial capacity upon extended cycling and their capacity especially when cycled slowly fades quickly. The capacity fade in aqueous lithium- and sodium- ion batteries has often been reported.6,15–21 The mechanism responsible for this capacity fading upon cycling has not yet been identified. The loss of electrical contact between the electrode particles,6 and the formation of amorphous layer between the electrode and carbon particles22 are suggested as possible causes for this capacity loss in batteries with NASICON-type electrodes. Several strategies are proposed for enhancement of performance; for example using polypyrrole coating19 and in situ formation of carbon at the precursor stage23 has helped the performance. Recently the performance and stability of the NaTi2(PO4)3/Na0.44MnO2 aqueous sodium-ion batteries is related to the electrolyte concentration.12 It is also reported that the removal of dissolved oxygen decreases the side reactions of LiTi2(PO4)3 anode in aqueous lithium-ion batteries.20 A very recent work24 shows that carbon coating on LiTi2(PO4)3 improves the capacity retention of aqueous cells to Coulombic efficiencies of >99% at 1C, 5C, and 10C, however the Columbic efficiency drops to ∼94% at C/5, and the carbon coating does not seem to be particularly effective at relatively slower charge–discharge rates. In this communication, phosphate–aqueous solution interface reactions are discussed as the cause of the electrochemical capacity fade and poor electrode utilization in aqueous sodium-ion batteries. It is demonstrated that amorphous surface layers inhibit the diffusion of sodium ions through the particles and are the main cause of capacity fade. We also show that the surface precipitates/phases clog the electrolyte transport pathways and are the possible origin of the poor electrode utilization.
Single-phase NaTi2(PO4)3 (herein after called NTP) was prepared by solid state method (Fig. S2 and S3, ESI†). A stoichiometric mixture of Na2CO3 (Sigma Aldrich, ≥99.5%), TiO2 (Aldrich, 99.7%), and 6NH4H2PO4 (Sigma Aldrich, ≥98%) was calcined in three steps (400 °C/6 h, 800 °C/24 h and 1000 °C/12 h in air) using intermediate milling and grinding. The nature of reactions on the surface of NTP particles after their exposure to water, as well as salt-containing aqueous solutions, under no electrochemical cycling was examined by a JEOL-2010F transmission electron microscopy (TEM) operated at 200 kV. The atomic nature and composition of the observed layer/species on the surface of NTP was studied using X-ray Photoelectron Spectroscopy (XPS; Thermo Scientific K-Alpha™+). For both characterizations, suspensions of NTP powders (0.9 g in 100 ml deionized water or 1 M Na2SO4 solution) were stirred in tightly closed containers on a hot plate at 80 °C for 10 days and then dried at 80 °C in an isothermal oven. The tests at 80 °C are chosen to accelerate the reactions without boiling the solvent. The cyclic voltammetry and the electrochemical impedance spectra were conducted (Biologic VMP3) using a three-electrode cell (Plate material evaluating cell from Bio-logic) with Pt foil as counter electrode and Ag/AgCl (3 M NaCl) reference electrode. The NTP anode electrode was prepared by casting a mixture of the NTP powders, acetylene black and polyvinylidene fluoride (PVDF) (in a weight ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25:5) on stainless steel mesh. The electrode area was 0.5 cm2, and the electrolyte was 1 M Na2SO4 aqueous solution.
:25:5) on stainless steel mesh. The electrode area was 0.5 cm2, and the electrolyte was 1 M Na2SO4 aqueous solution.
We did not observe any high-frequency semicircle in the initial electrochemical impedance spectra of the as-made cells (Fig. 1a), but a high-frequency semicircle in the range of 20–80 Ω cm−2 appeared in the impedance spectra of the cycled cells. The formation of a solid-electrolyte interphase (SEI) layer in non-aqueous (organic) lithium-ion batteries is a common observation and the high frequency semicircle is usually attributed to the diffusion of lithium ions through this SEI layer.25,26 On the other hand, the formation of a SEI layer in aqueous electrolytes has not been extensively confirmed. It is reported that SEI layer is not formed in the presence of aqueous electrolytes.9,27 Formation of an additional resistive component was also observed in the Nyquist and Bode plots of the NaTi2(PO4)3 electrodes in the absence of electrochemical intercalation and as a function of time (Fig. 1b).
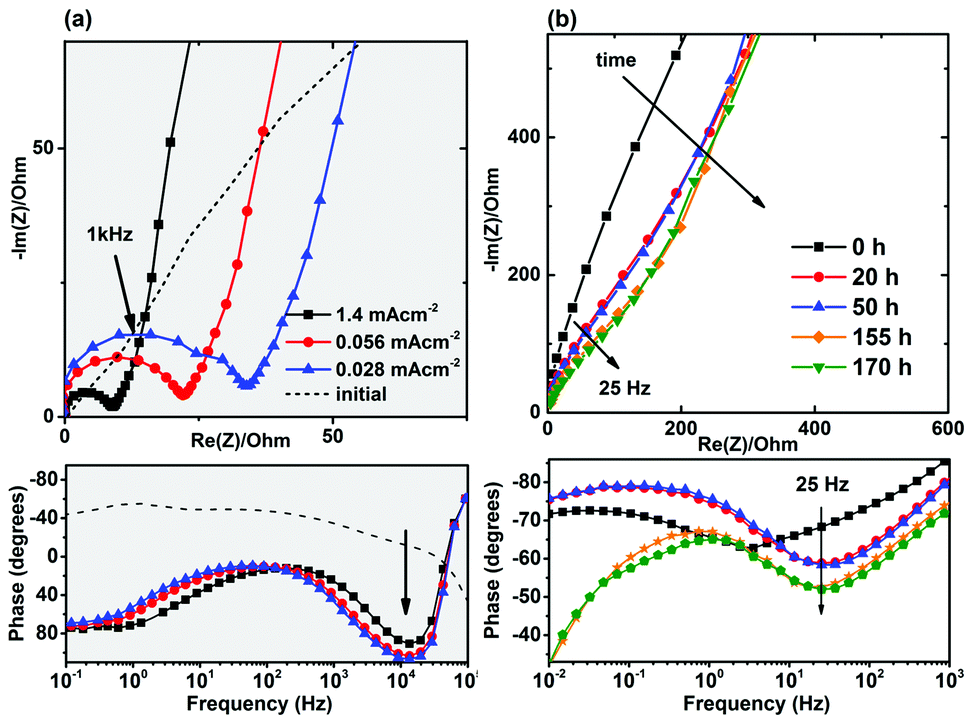 | ||
| Fig. 1 (a) Nyquist and Bode plots of NaTi2(PO4)3 electrode before and after cycling and at different current loadings of 1.4 mA cm−2 (=5C), 0.056 mA cm−2 (=C/5), and 0.028 mA cm−2 (=C/10). (b) Nyquist and Bode plots after immersing NaTi2(PO4)3 coated on stainless steel mesh (no electrochemical cycling). | ||
In addition, it should be noted that for the aqueous cells, reasonable initial capacities can be achieved only when the electrodes are intentionally made with high porosities and coated on stainless steel mesh instead of foil to provide access for the aqueous solution. This behavior, also reported by other groups and sometimes referred to as “poor electrode utilization”,12,19,23,28 is unexpected because the ionic conductivity of aqueous solutions is higher than that of organic electrolytes and better electrolyte access in aqueous cells is expected. This poor electrolyte penetration also directs us towards considering the occurrence of surface reactions.
The transmission electron microscopy images on NTP particles stirred in water (herein after called NTP-DIW; Fig. 2a and b) and in 1 M Na2SO4 solutions (herein after called NTP-NSO; Fig. 2c and d), show that (i) an amorphous surface layer (5–10 nm) forms on the surface of NTP particles after their exposure to water, as well as salt-containing aqueous solutions, and (ii) a secondary phase in the form of randomly distributed spherical caps (5–20 nm in diameter) forms on the surface of particles exposed to the salt-containing solution (1 M Na2SO4 solution; Fig. 2c). As marked in the figures, the interplannar spacings of the observed lattice fringes match well with certain d-spacing values obtained from XRD analysis of NTP powders. The synchrotron-based high resolution phase analysis (Fig. S3, ESI†) confirmed that NTP phase is stable in water and salt-containing solution and there is no evidence of major phase changes after the relatively aggressive exposure of these particles to aqueous environments.
 | ||
| Fig. 2 TEM images of NaTi2(PO4)3 particles (a and b) stirred in DI water and at room temperature (NTP-DIW sample), and (c and d) stirred in 1 M Na2SO4 solution and at room temperature (NTP-NSO sample). | ||
The XPS Ti2p scan of NTP-DIW (Fig. 3a) with one peak at 465.58 eV (Ti2p1/2) and one peak at 459.78 eV (Ti2p3/2) matches well with previously reported peaks of Ti4+ in NTP bonding environment.29 The other two peaks at 463. 08 eV (Ti2p1/2) and 458.58 eV (Ti2p3/2), are close to the characteristics of Ti–O–P linkage in Ti(HPO4)2 species.30 The O1s region (see Fig. 3b) is composed of three peaks at 530.98 eV, 532.88 eV and 536.28 eV, which correspond to O in NTP (P–O–Ti, P–O–Na, and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O),29 P–OH,29,31 and sodium Auger overlap with O1s, respectively. The O1s peak at ∼533 eV for P–OH bonding is assigned to HPO42− or H2PO4− groups.31,32 Thus, the amorphous layer observed on the surface of NTP powders exposed to deionized water consists of Ti(HPO4)2/Ti(H2PO4)4, both of which are known as amorphous titanium phosphates.33,34 The Ti2p scan of NTP-NSO is deconvoluted to three contributions presented in Fig. 3c. After exposure to Na2SO4 containing solution, the two characteristic peaks of NTP experience slight changes in their binging energy (Ti2p1/2 465.58 eV and Ti2p3/2 459.88 eV). The next pair of shoulders at 463.48 eV (Ti2p1/2) and 458.38 eV (Ti2p3/2) can be assigned to either Ti–O–P linkage in Ti(HPO4)2/Ti(H2PO4)4 or Ti–O–S in Ti(SO4)2,30 in which the former is ruled out considering the very small contribution of P–OH bonding in O1s shown in Fig. 3d. Thus, the cap shape precipitates observed in TEM images are amorphous form of titanium(IV) sulfate Ti(SO4)2, which was not detectable from the X-ray diffraction pattern.35 The peaks at 461.68 eV and 455.88 eV based on previous XPS studies belong to amorphous TiOS (456 eV and 462 eV).36 Compared to that of NTP-DIW, the major O1s peak of NTP-NSO shifts to a higher binding energy at 531.78 eV, probably due to the presence of S–O bonds; also, a stronger Na KLL Auger signal appears, which can be ascribed to extra sodium from Na2SO4.
O),29 P–OH,29,31 and sodium Auger overlap with O1s, respectively. The O1s peak at ∼533 eV for P–OH bonding is assigned to HPO42− or H2PO4− groups.31,32 Thus, the amorphous layer observed on the surface of NTP powders exposed to deionized water consists of Ti(HPO4)2/Ti(H2PO4)4, both of which are known as amorphous titanium phosphates.33,34 The Ti2p scan of NTP-NSO is deconvoluted to three contributions presented in Fig. 3c. After exposure to Na2SO4 containing solution, the two characteristic peaks of NTP experience slight changes in their binging energy (Ti2p1/2 465.58 eV and Ti2p3/2 459.88 eV). The next pair of shoulders at 463.48 eV (Ti2p1/2) and 458.38 eV (Ti2p3/2) can be assigned to either Ti–O–P linkage in Ti(HPO4)2/Ti(H2PO4)4 or Ti–O–S in Ti(SO4)2,30 in which the former is ruled out considering the very small contribution of P–OH bonding in O1s shown in Fig. 3d. Thus, the cap shape precipitates observed in TEM images are amorphous form of titanium(IV) sulfate Ti(SO4)2, which was not detectable from the X-ray diffraction pattern.35 The peaks at 461.68 eV and 455.88 eV based on previous XPS studies belong to amorphous TiOS (456 eV and 462 eV).36 Compared to that of NTP-DIW, the major O1s peak of NTP-NSO shifts to a higher binding energy at 531.78 eV, probably due to the presence of S–O bonds; also, a stronger Na KLL Auger signal appears, which can be ascribed to extra sodium from Na2SO4.
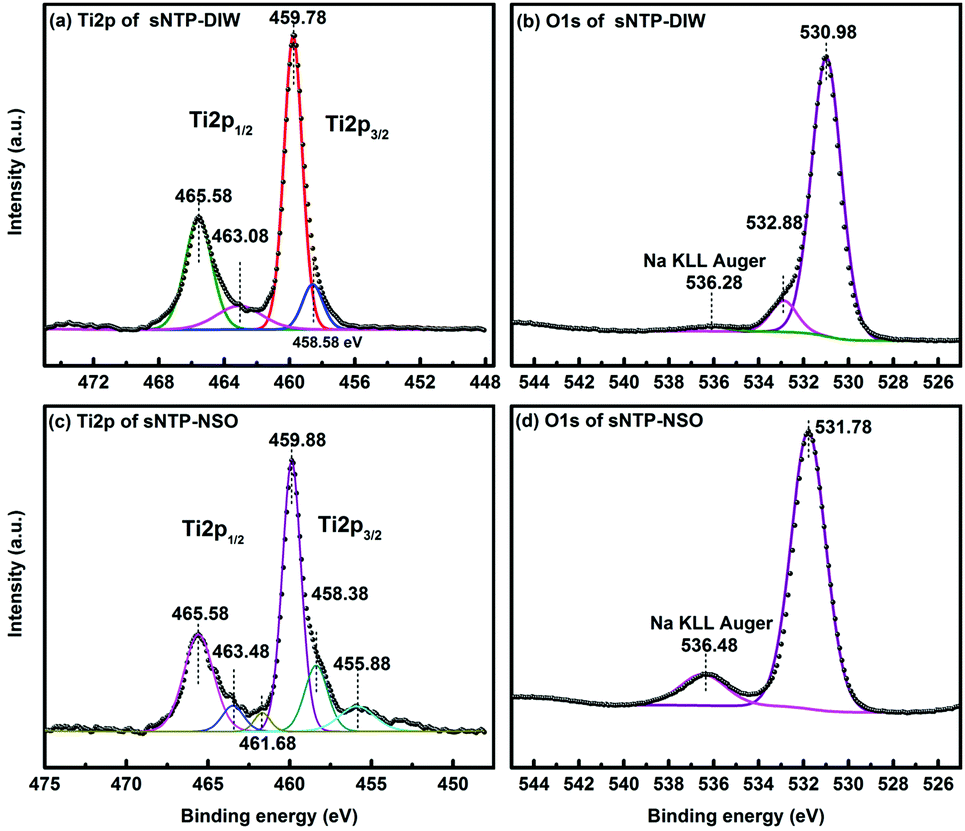 | ||
| Fig. 3 XPS spectra of the surface chemical composition: (a) Ti2p scan and (b) O1s scan of NTP-DIW sample, (c) Ti2p scan and (d) O1s scan of NTP-NSO sample. | ||
The initial increase in conductivity after adding NTP powder to deionized water (Fig. S4a, ESI†) corresponds to release of Ti4+ and Na+. ICP-OES analysis (Fig. S5, ESI†) confirmed the presence of excess sodium ions (33 mg L−1) and titanium ions (9.1 mg L−1) in the clear solution removed from top of the NTP particles. No remarkable change was observed when dispersing NTP in Na2SO4 solution that originally has a relatively high conductivity, and ICP-OES analysis showed release of small amount of Ti ions into the Na2SO4 solution (0.5 mg L−1). In deionized water, the initial increase in pH value (Fig. S4b, ESI†) is probably due to the hydrolysis of the released PO43− ions through these two reactions favored in relatively acidic environment: PO43− + H2O → HPO42− + OH− and/or HPO42− + H2O → H2PO4− + OH−. The following gradual drop in pH could be a result of hydrolysis of released Ti4+ ions. In Na2SO4 solution, the pH first decreases and then remains constant for both room temperature and 80 °C. The rise of H+ concentration can be explained as the hydrolysis of titanium sulfate formed on the surface of NTP. Ul'yanov suggested that the hydrolysis of Ti(SO4)2 mainly produces insoluble H2TiO3, and an increase in the amount of sulfides will hinder the hydrolysis of Ti(SO4)2.37 The Ti(SO4)2 is partly-soluble and its solubility increases in concentrated acidic environment when hydrolysis is well prevented.38 We conclude that in the salt containing solution the amorphous region appears as a result of Na+ and Ti4+ ions leave their lattice sites, and then insoluble Ti(SO4)2 and its hydrolysis products, i.e., H2TiO3, will precipitate on the NTP surface and suppress further Ti4+ release and hydrolysis. Based on the investigation of the Ti(SO4)2–H2SO4–NaOH–H2O system39 formation of Ti(SO4)2 or Na2SO4 precipitates strongly depends on the concentration of the anionic group, temperature, and basicity of the aqueous solutions. Further reactions during drying the suspension could introduce other species e.g., TiOS characterized by XPS.
Based on the findings of the charge-distributed multi-site complexation (CD-MUSIC) model on the surface adsorption/binding mechanisms on the surface of oxides,40 we suggest that the cation dissolution on the surface of NASICON is controlled by the anion type/concentration, pH, and temperature. More importantly, based on the well documented cases of Fe, Cr, Mn, and Ni dissolution in aqueous solutions,40 dissolution of transition-metal cations in the NASICON structure is a function of the oxidation state of the metal ions (Mn+). The observed increase in the amount of dissolved Ti (4.6 mg L−1) in Na2SO4 aqueous solution after the electrochemical cycling is therefore due to the reduction of Ti4+ to Ti3+ and increased solubility of titanium from surface (cycled between −1 V and 0 V for 20 cycles with a current density of 0.476 mA cm−2). This phenomena explains the accelerated formation of the resistive surface layer in the cycled NTP shown in Fig. 1a.
In summary, (i) dissolution of sodium and titanium cations and hydrolysis of surface groups result in the formation of amorphous transition-metal phosphate layers that are ionically and electronically insulating and lead to capacity fade, and (ii) in the presence of sulfate anions, insoluble titanium sulfate phases form on the surface of the particles and eliminate further surface hydrolysis, however, these precipitates are large enough to clog the pores and block the electrolyte access pathways to the NaTi2(PO4)3 particles embedded in the electrode. Further understanding of “phosphate–aqueous solution interface” reactions requires future investigations of the thermodynamic and kinetics behind the proposed reactions and the rate-determining steps to the formation of insoluble species in various environmental conditions.
Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The authors are immensely grateful to Dr. Marca Doeff (Lawrence Berkeley National Laboratory) for helpful discussions. The authors would like to thank the Department of Chemical and Materials Engineering at the University of Kentucky for its financial support of this work, and Dr. Jie Pan and Prof. Yang-Tse (YT) Cheng at the University of Kentucky for assisting with XPS measurements.
Notes and references
- A. A. Akhil, A. Georgianne Huff, B. Currier, B. C. Kaun, D. M. Rastler, S. Bingqing, A. Chen, D. L. Cotter, T. Bradshaw and W. D. Gauntlett, DOE/EPRI Electricity Storage Handbook, 2013 Search PubMed.
- I. Gyuk, M. Johnson, J. Vetrano, K. Lynn, W. Parks, R. Handa, L. Kannberg, S. Hearne, K. Waldrip and R. Braccio, Grid Energy Storage, U.S. Department of Energy, 2013 Search PubMed.
- B. L. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168–177 CrossRef CAS.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martinez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312–2337 CAS.
- H. Kim, J. Hong, K. Y. Park, H. Kim, S. W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- C. Wessells, R. A. Huggins and Y. Cui, J. Power Sources, 2011, 196, 2884–2888 CrossRef CAS.
- J. F. Whitacre, T. Wiley, S. Shanbhag, Y. Wenzhuo, A. Mohamed, S. E. Chun, E. Weber, D. Blackwood, E. Lynch-Bell, J. Gulakowski, C. Smith and D. Humphreys, J. Power Sources, 2012, 213, 255–264 CrossRef CAS.
- Z. Li, D. Young, K. Xiang, W. C. Carter and Y.-M. Chiang, Adv. Energy Mater., 2013, 3, 290–294 CrossRef CAS.
- L. M. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. L. Fan, C. Luo, C. S. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- K. Miyazaki, T. Shimada, S. Ito, Y. Yokoyama, T. Fukutsuka and T. Abe, Chem. Commun., 2016, 52, 4979–4982 RSC.
- M. Shirpour, X. Zhan and M. Doeff, Proceedings of the TechConnect World, 2015, 17–20.
- W. Wu, S. Shabhag, J. Chang, A. Rutt and J. F. Whitacre, J. Electrochem. Soc., 2015, 162, A803–A808 CrossRef CAS.
- B. L. Ellis, W. R. M. Makahnouk, W. N. Rowan-Weetaluktuk, D. H. Ryan and L. F. Nazar, Chem. Mater., 2010, 22, 1059–1070 CrossRef CAS.
- N. Recham, J.-N. Chotard, L. Dupont, K. Djellab, M. Armand and J.-M. Tarascon, J. Electrochem. Soc., 2009, 156, A993–A999 CrossRef CAS.
- Z. Li, D. Young, K. Xiang, W. C. Carter and Y. M. Chiang, Adv. Energy Mater., 2013, 3, 290–294 CrossRef CAS.
- X.-H. Liu, T. Saito, T. Doi, S. Okada and J.-i. Yamaki, J. Power Sources, 2009, 189, 706–710 CrossRef CAS.
- Y. Cui, Y. Hao, W. Bao, Y. Shi, Q. Zhuang and Y. Qiang, J. Electrochem. Soc., 2013, 160, A53–A59 CrossRef CAS.
- Y. Lin, W. Wang, J. Zhang and C. Dai, ECS Electrochem. Lett., 2014, 3, A105–A107 CrossRef CAS.
- A. I. Mohamed, N. J. Sansone, B. Kuei, N. R. Washburn and J. F. Whitacre, J. Electrochem. Soc., 2015, 162, A2201–A2207 CrossRef CAS.
- J. Y. Luo, W. J. Cui, P. He and Y. Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS PubMed.
- C. Wessells, F. La Mantia, H. Deshazer, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2011, 158, A352–A355 CrossRef CAS.
- A. I. Mohamed, ECS Meeting Abstracts, 2015, MA2015-01 167.
- W. Wu, J. Yan, A. Wise, A. Rutt and J. F. Whitacre, J. Electrochem. Soc., 2014, 161, A561–A567 CrossRef CAS.
- D. Sun, Y. Tang, K. He, Y. Ren, S. Liu and H. Wang, Sci. Rep., 2015, 5, 17452 CrossRef CAS PubMed.
- M. D. Levi and D. Aurbach, J. Phys. Chem. B, 1997, 101, 4630–4640 CrossRef CAS.
- E. Peled, D. Golodnitsky and G. Ardel, J. Electrochem. Soc., 1997, 144, L208–L210 CrossRef CAS.
- N. Alias and A. A. Mohamad, J. Power Sources, 2015, 274, 237–251 CrossRef CAS.
- W. Wu, A. Mohamed and J. F. Whitacre, J. Electrochem. Soc., 2013, 160, A497–A504 CrossRef CAS.
- J. Liu, J. Zhang, S. Cheng, Z. Liu and B. Han, Small, 2008, 4, 1976–1979 CrossRef CAS PubMed.
- K. J. A. Raj, R. Shanmugam, R. Mahalakshmi and B. Viswanathan, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2010, 49, 9–17 Search PubMed.
- P. W. Brown and B. Constantz, Hydroxyapatite and related materials, CRC press, 1994 Search PubMed.
- B. Demri and D. Muster, J. Mater. Process. Technol., 1995, 55, 311–314 CrossRef.
- K. Elghniji, M. Saad, M. Araissi, E. Elaloui and Y. Moussaoui, Mater. Sci.-Pol., 2014, 32, 617–625 CAS.
- C. Schmutz, P. Barboux, F. Ribot, F. Taulelle, M. Verdaguer and C. Fernandez-Lorenzo, J. Non-Cryst. Solids, 1994, 170, 250–262 CrossRef CAS.
- J. R. Sohn, E. H. Park and J. G. Kim, Studies in Surface Science and Catalysis, Elsevier, 2000, vol. 143, pp. 377–385 Search PubMed.
- D. Gonbeau, C. Guimon, G. Pfister-Guillouzo, A. Levasseur, G. Meunier and R. Dormoy, Surf. Sci., 1991, 254, 81–89 CrossRef CAS.
- A. Ul'yanov, Bull. Acad. Sci. USSR, 1960, 9, 553–559 CrossRef.
- R. Rich, Inorganic reactions in water, Springer Science & Business Media, 2007 Search PubMed.
- A. I. Ul'yanov, Russ. Chem. Bull., 1960, 9, 553–559 CrossRef.
- T. Hiemstra and W. H. Van Riemsdijk, J. Colloid Interface Sci., 1999, 210, 182–193 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemical potential window, SEM images, synchrotron-based HRXRD patterns and fitting, conductivity, pH, and ICP-OES measurements. See DOI: 10.1039/c6cc08901a |
| This journal is © The Royal Society of Chemistry 2017 |