
Convenient route to tetraarylphosphonium polyelectrolytes via metal-catalysed P–C coupling polymerisation of aryl dihalides and diphenylphosphine†
Wang
Wan
a,
Xiaoyan
Yang
a and
Rhett C.
Smith
*ab
aDepartment of Chemistry, Clemson University, Clemson, SC 29634, USA
bCentre for Optical Materials Science and Engineering Technologies, Clemson University, Anderson, SC 29634, USA
First published on 8th December 2016
Abstract
A P–C bond-forming reaction has been applied to the convenient preparation of tetraarylphosphonium polyelectrolytes (TPELs) from aryl dihalides and diphenylphosphine. A TPEL having a thermal decomposition temperature of 460 °C that is also stable to heating at 65 °C in 6 M NaOH(aq) for at least 24 h has been prepared by this method.
Polyelectrolytes and polyelectrolyte complexes have attracted widespread recent interest as potentially sustainable and green-processable/recyclable materials.1–8 Phosphonium polyelectrolytes specifically have also been explored for delivery of material to cells and as ion-transporting membrane component of alkaline exchange fuel cells. Compared to alkylphosphonium polyelectrolytes, tetraarylphosphonium units ([PAr4]+) can be significantly more chemically stable, particularly to hydrolysis or nucleophilic attack.9–20 Furthermore, [PAr4]+ salts are the most thermally stable ionic liquids known,11 making these units excellent candidates for incorporation into polymers for high temperature applications.
The potential for TPELs to exhibit combined thermal and chemical stability led us to pursue simple transition metal-catalysed P–C bond-forming routes to tetraarylphosphonium polyelectrolytes (TPELs). In 2015, we reported the first such route based on established small molecule chemistry.10,11 Although this is successful, it requires the use of a bis(triarylphosphine) monomer (Scheme 1A). Such monomers generally require expensive organometallic reagents and time-consuming methodologies for their preparation, and there are few that are commercially available. Consequently, it is not practically feasible to rapidly and affordably prepare a wide range of TPELs by this method.
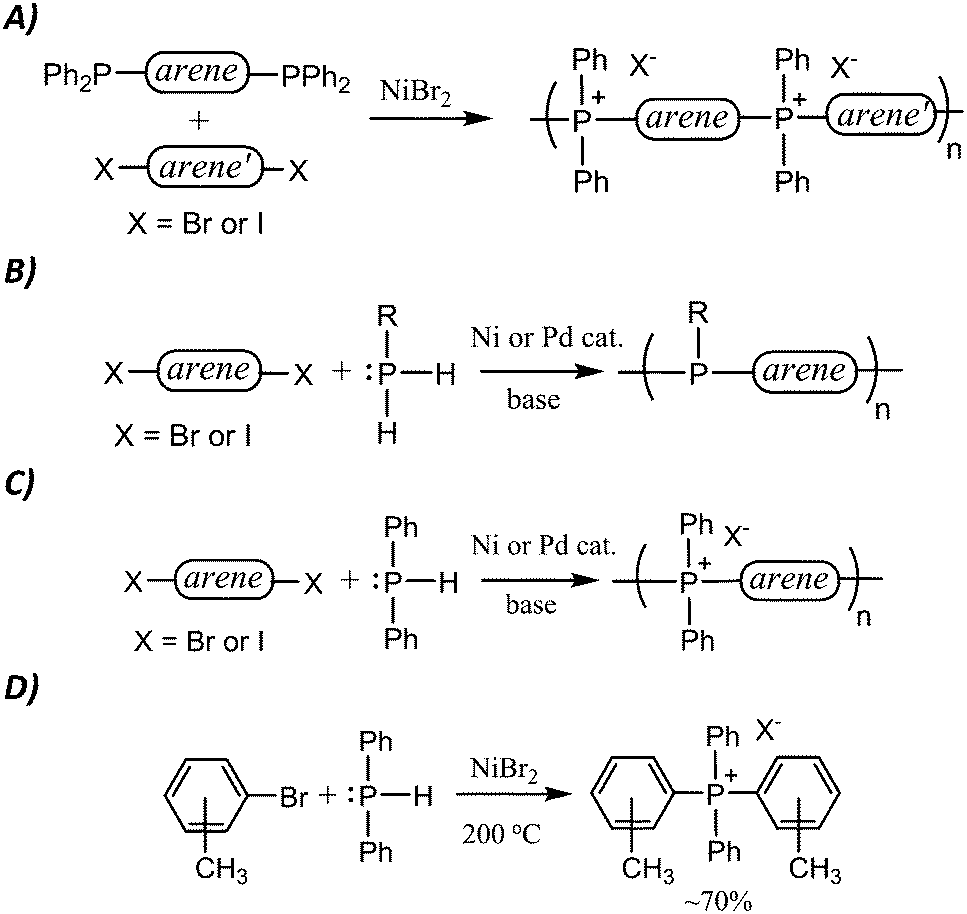 | ||
| Scheme 1 Established P–C coupling route to TPELs (A), and polyphosphines (B). The proposed new route to TPELs (C) employs chemistry similar to a little-explored synthesis (D) of small molecule [PAr4]+ salts from HPPh2. | ||
In our search for a more facile and affordable route to TPELs, we were inspired by a straightforward P–C coupling route to poly(triarylphosphine)s reported by Lucht in this journal (Scheme 1B).18 This route exploits commercial aryl- or alkylphosphines and commercial aryl dihalides as coupling partners to yield polyphosphine analogues of polyaniline. We reasoned that an analogous route to TPELs starting with commercial aryl dihalides and HPPh2 (Scheme 1C) might also be achievable.
In a 1980 report,21 Cristau et al. reported that HPPh2 could be converted to tetraarylphosphonium salts in ∼70% yield by NiBr2-catalyzed coupling with o-, m- or p-tolylbromide (Scheme 1D). In order to extend this coupling methodology to a polymerization route capable of reasonable degrees of polymerization, however, the yield had to be significantly improved. In the stepwise pathway (Scheme 2), deprotonation of [HPAr3]+ is required before the second P–C bond-forming step can occur, so we hypothesized that addition of a base might improve the reaction yield. As anticipated, simple addition of one equiv of diisopropylamine led to quantitative yields of the target [PAr4]+ salts under the reported conditions. We also found that, in addition to the NiBr2 catalyst used in the previous report, Pd2(dba)3 (dba = dibenzylideneacetone) was also an effective catalyst for the transformation shown in Scheme 1D. Having established appropriate conditions for high yield coupling in small molecular model reactions, polymerization of HPPh2 with several commercial dihalides was undertaken (Scheme 3). Properties of resultant polymers are summarized in Table 1.
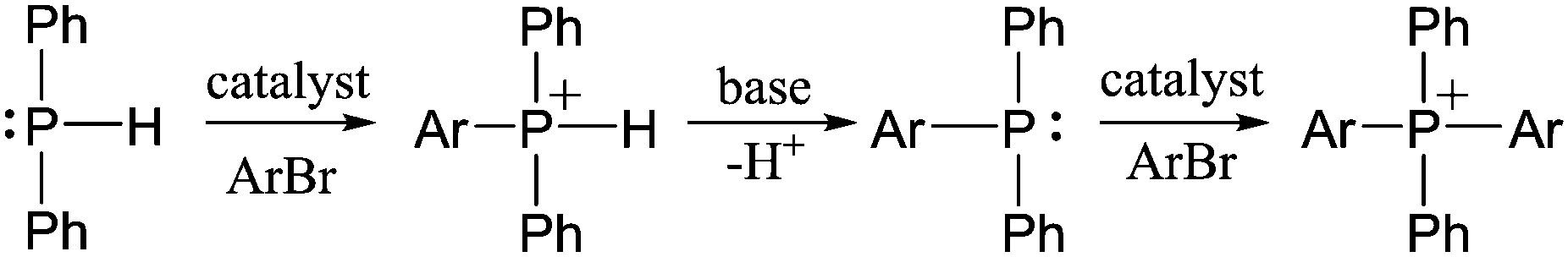 | ||
| Scheme 2 Pathway to formation of [Ar2PPh2]+ salts from HPPh2. | ||
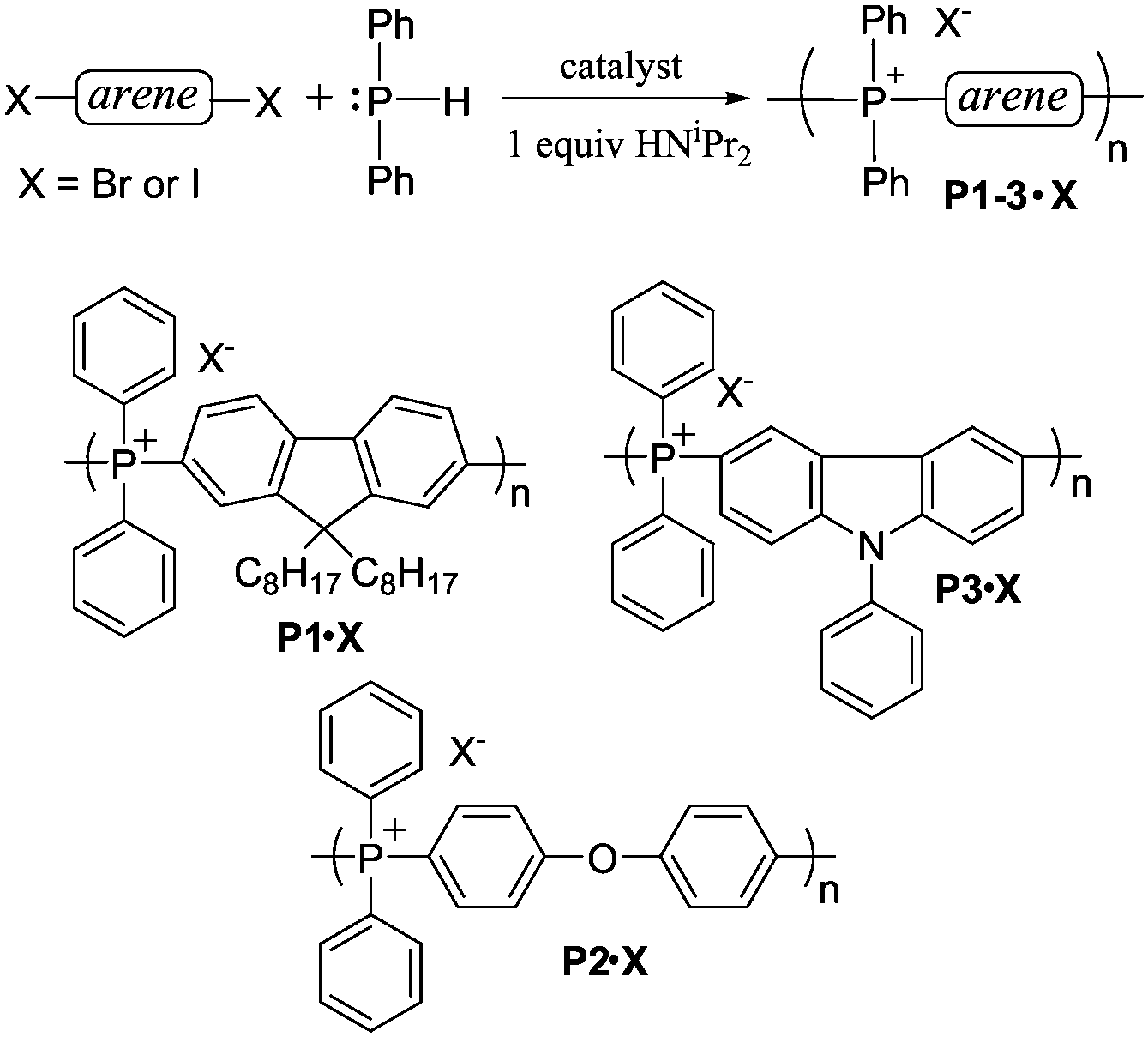 | ||
| Scheme 3 Synthesis of TPELs. | ||
| Polymer | Catalyst | M n (kDa) | X n | λ max (nm) | T d (°C) |
|---|---|---|---|---|---|
| a Number average molecular weight determined by NMR end group analysis. These Mn data are averages of at least two runs. b Degree of polymerization. c Wavelength of maximum absorption for the band attributable to the π–π* transition in the UV-vis spectrum in CH2Cl2. d Defined as the temperature at which 5% mass loss occurs under nitrogen as determined by thermogravimetric analysis. e Prepared via anion exchange with the corresponding poly(phosphonium bromide) made via NiBr2 catalysis (P1·NTf2 and P2·NTf2) and Pd2(dba)3 catalyst (P3·NTf2). | |||||
| P1·Br | NiBr2 | 11.1 | 17 | 331 | 344 |
| Pd2(dba)3 | 15.0 | 23 | 332 | 328 | |
| P2·Br | NiBr2 | 6.9 | 16 | 267 | 360 |
| Pd2(dba)3 | 13.0 | 30 | 268 | 371 | |
| P3·Br | Pd2(dba)3 | 13.2 | 26 | 288 | 414 |
| P1·NTf2 | NAe | 15.4 | 18 | 331 | 403 |
| P2·NTf2 | NAe | 10.8 | 17 | 267 | 440 |
| P3·NTf2 | NAe | 19.1 | 27 | 288 | 460 |
The NiBr2-catalyzed polymerization reactions were carried out at 180 °C, while Pd2(dba)3-catalyzed (dba = dibenzylideneacetone) reactions were carried out at 145 °C, and all reactions were initially carried out in ethylene glycol for 24 h. For P1, both aryl bromide and aryl iodide coupling partners were evaluated because aryl iodides generally give higher degrees of polymerization via C–C coupling reactions. At the elevated temperature employed for NiBr2-catalyzed reactions, however, the aryl iodides screened underwent some decomposition (accompanied by development of characteristic I2 colour in solution) and they were not useful coupling partners for polymerization under these conditions.
The degrees of polymerization (Xn) and number average molecular weights (Mn) for polymers produced from aryl dibromides was determined by analyzing the number of phosphine oxide end groups via NMR spectrometry, as summarized in Table 1. As with C–C coupling polymerizations, the degrees of polymerization (Xn) obtainable from Pd2(dba)3-catalyzed P–C coupling polymerization reactions were notably higher than those obtained by the NiBr2-catalyzed route (Table 1). These Xn values are also similar to or higher than corresponding values for reported alkylphosphonium ionomers prepared via nucleophilic substitution-driven condensation polymerization of diphosphines with alkyl or benzyl bromides (Xn generally ≤20).10,22–24
UV-vis absorption data (Table 1) confirm earlier indications10 that there is little or no π-conjugation through the phosphonium moieties along the polymer backbone, as the absorption spectra are similar to those of the monomers.
The thermal stability of [PAr4]+ units was one of the motivating factors for incorporating them into TPELs, so thermogravimetric analysis was employed to determine the decomposition temperatures (Td, Table 1) of P1–3. The bromide salts have reasonably good thermal stability (Td = 328–414 °C) and are observed to decompose via thermal loss of HBr. Small molecular [PAr4]+ ditriflamide ([NTf2]−, Tf = –SO2CF3) salts have significantly better thermal stability compared to their halide analogues,11 so P1–3·NTf2 analogues were prepared by anion exchange in an effort to increase their thermal stability. Elemental microanalysis of the P1–3·NTf2 indicated that ≥82.0% of bromide counteranions had been replaced with [NTf2]− counteranions in these materials. The thermal stabilities of P1–3·NTf2 was improved as anticipated, with Td values of >400 °C for all of the materials.
Chemical stability, particularly to alkaline conditions and nucleophilic attack, is another potentially useful property of TPELs. A film of each polymer P1–3·[NTf2] was thus submerged in NaOH(aq) for 24 h at room temperature or 65 °C. After exposure, each film was redissolved in CDCl3 and analysed by NMR spectrometry to determine the percentage of phosphonium units that had decomposed (Table 2). It was observed that FLoct (Chart 1, X− = Br− or [NTf2]−) undergoes complete decomposition via nucleophilic aromatic substitution (SNAr) reaction upon heating to 60 °C in 6 M NaOH(aq) for 24 h. The presence of electron donors, such as those present in P2 and P3, should slow down the SNAr pathway and afford greater stability. Films of P1–3·NTf2 treated at room temperature do not undergo any detectable decomposition. The percentage of decomposition at 65 °C with 6 M NaOH(aq) is inversely related to the electron donating capacity of the substituent para- to the phosphonium site (Table 2). Notably, P3·NTf2 undergoes very little if any decomposition under these conditions, making it the most chemically and thermally robust of the TPELs prepared.
| Polymer | [NaOH] | T (°C) | % decomp. (±5%) |
|---|---|---|---|
| P1·NTf2 | 1 M | 20 | 0 |
| 1 M | 65 | 7 | |
| 6 M | 65 | 26 | |
| P2·NTf2 | 1 M | 20 | 0 |
| 1 M | 65 | 7 | |
| 6 M | 65 | 7 | |
| P3·NTf2 | 1 M | 20 | 0 |
| 1 M | 65 | 0 | |
| 6 M | 65 | 0 | |
| FLoct | 6 M | 60 | 100 |
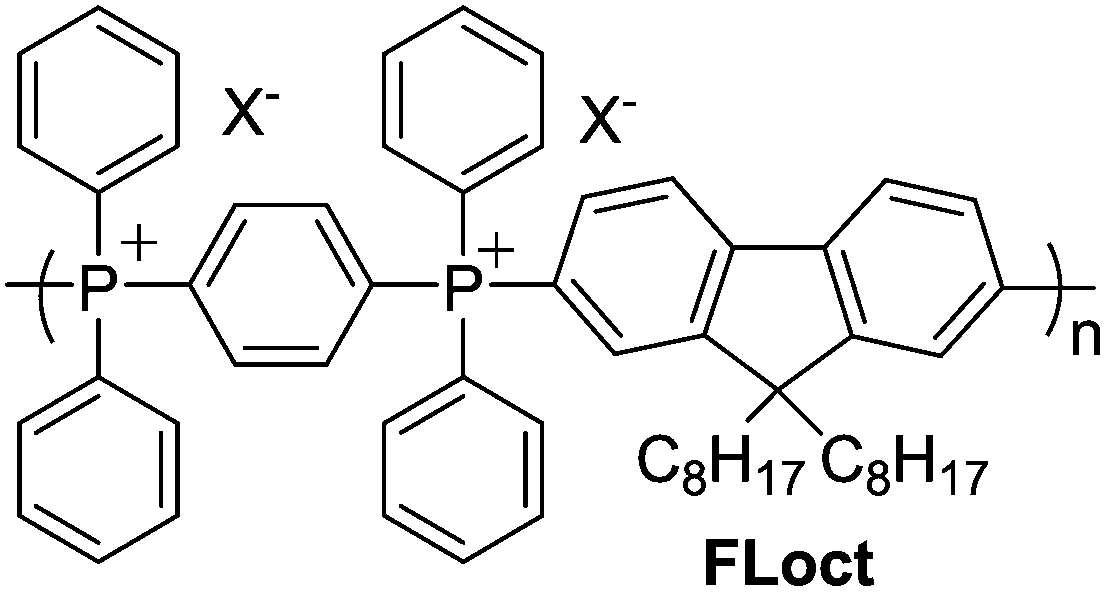 | ||
| Chart 1 Structure of FLoct, prepared via the method in Scheme 1A. | ||
A transition metal-catalysed P–C bond forming reaction was modified to allow the convenient preparation of tetraarylphosphonium polyelectrolytes (TPELs) from commercial aryl dihalides and diphenylphosphine. Both nickel and palladium catalysts are effective in C–P coupling reactions to yield polymers with degrees of polymerization up to about 30. Anion exchange of initial polymeric bromide salts allows ready access to ditriflamide salts having thermal stability of up to 460 °C. One TPEL prepared via this methodology is stable to heating at 65 °C in 6 M NaOH(aq) for at least 24 h, whereas alkylphosphonium polymers and even previously-reported TPELs undergo complete depolymerisation under these conditions. The improved stability suggests that P3·NTf2 and similar TPELs may hold promise for use in alkaline fuel cell applications. The synthetic protocol described herein promises to facilitate rapid access to a wide variety of thermally/chemically stability materials for a variety of applications.
Notes and references
- S. Chempath, J. M. Boncella, L. R. Pratt, N. Henson and B. S. Pivovar, J. Phys. Chem. C, 2010, 114, 11977–11983 CAS.
- J. B. Edson, C. S. Macomber, B. S. Pivovar and J. M. Boncella, J. Membr. Sci., 2012, 399, 49–59 CrossRef.
- S. Gu, R. Cai, T. Luo, Z. Chen, M. Sun, Y. Liu, G. He and Y. Yan, Angew. Chem., Int. Ed., 2009, 48, 6499–6502 CrossRef CAS PubMed.
- H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419–9426 CAS.
- H. Long and B. Pivovar, J. Phys. Chem. C, 2014, 118, 9880–9888 CAS.
- C. S. Macomber, J. M. Boncella, B. S. Pivovar and J. A. Rau, J. Therm. Anal. Calorim., 2008, 93, 225–229 CrossRef CAS.
- K. J. T. Noonan, K. M. Hugar, H. A. Kostalik, E. B. Lobkovsky, H. D. Abruna and G. W. Coates, J. Am. Chem. Soc., 2012, 134, 18161–18164 CrossRef CAS PubMed.
- J. E. Coughlin, A. Reisch, M. Z. Markarian and J. B. Schlenoff, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2416–2424 CrossRef CAS.
- S. J. Abraham and W. J. Criddle, J. Anal. Appl. Pyrolysis, 1985, 7, 337–349 CrossRef CAS.
- M. S. Bedford, X. Yang, K. M. Jolly, R. L. Binnicker, S. B. Cramer, C. E. Keen, C. J. Mairena, A. P. Patel, M. T. Rivenbark, Y. Galabura, I. Luzinov and R. C. Smith, Polym. Chem., 2015, 6, 900–908 RSC.
- C. G. Cassity, A. Mirjafari, N. Mobarrez, K. J. Strickland, R. A. O'Brien and J. H. Davis, Jr., Chem. Commun., 2013, 49, 7590–7592 RSC.
- S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112, 3179–3182 CAS.
- G. W. Fenton and C. K. Ingold, J. Chem. Soc., 1929, 2342–2357 RSC.
- G. L. Keldsen and W. E. McEwen, J. Am. Chem. Soc., 1978, 100, 7312–7317 CrossRef CAS.
- F. Y. Khalil, M. T. Hanna and M. ElBatouti, C. R. Acad. Sci., Ser. IIb: Mec., Phys., Chim., Astron., 1997, 325, 545–551 CAS.
- B. Siegel, J. Am. Chem. Soc., 1979, 101, 2265–2268 CrossRef CAS.
- M. Wada and S. Higashizaki, J. Chem. Soc., Chem. Commun., 1984, 482–483 RSC.
- B. L. Lucht and N. O. St Onge, Chem. Commun., 2000, 2097–2098 RSC.
- L. Horner, G. Mummenthey, H. Moser and P. Beck, Chem. Ber., 1966, 99, 2782–2788 CrossRef.
- L. Horner and H. Hoffmann, Chem. Ber., 1958, 91, 50–52 CrossRef CAS.
- H. J. Cristau, A. Chene and H. Christol, J. Organomet. Chem., 1980, 185, 283–295 CrossRef CAS.
- X. Yang, C. A. Conrad, W. Wan, M. S. Bedford, L. Hu, G. Chumanov and R. C. Smith, J. Mater. Chem. C, 2015, 3, 4537–4544 RSC.
- E. G. Tennyson, S. He, N. C. Osti, D. Perahia and R. C. Smith, J. Mater. Chem., 2010, 20, 7984–7989 RSC.
- C. A. Conrad, M. S. Bedford, A. A. Buelt, Y. Galabura, I. Luzinov and R. C. Smith, Polym. Int., 2015, 64, 1381–1388 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, elemental microanalysis data, UV-vis spectra and TGA traces. See DOI: 10.1039/c6cc08938k |
| This journal is © The Royal Society of Chemistry 2017 |