
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg+ coordination polymers of a chiral thiol ligand bearing an AIE fluorophore†
Dan-Dan
Tao
,
Qian
Wang
,
Xiao-Sheng
Yan
,
Na
Chen
,
Zhao
Li
and
Yun-Bao
Jiang
*
Department of Chemistry, College of Chemistry and Chemical Engineering, MOE Key Laboratory of Spectrochemical Analysis and Instrumentation, and iChEM, Xiamen University, Xiamen 361005, China. E-mail: ybjiang@xmu.edu.cn
First published on 30th November 2016
Abstract
We report that in the Ag+ coordination polymers of a chiral thiol ligand containing an AIE fluorophore, tetraphenylethene (TPE), the TPE chromophores experience H-type aggregation, and yet a substantial enhancement of the fluorescence is observed, though to a lesser extent than that in the aggregates of the thiol ligand itself, which undergoes J-type aggregation. We show that this is not due to the difference in the freedom of the rotation of the fluorophore in the two types of aggregate, but is due to a small increase in the radiation rate constant in the coordination polymers while the much higher radiationless rate constant remains more or less unchanged.
The phenomenon of AIE (aggregation induced emission)1–3 has now become well recognized since its discovery in 2001 and has since found tremendously significant applications.4–8 The emission of an AIE fluorophore (also termed an AIEgen) in the aggregate state is more enhanced than when it exists in the monomer form. This is a basic observation that has been made with AIE, and has been attributed to the restriction of intramolecular movement. In the majority of cases, AIEgen molecules undergo self-assembly when the solvent composition is tuned or when the solvatophilicity of the AIEgen is varied. For example, the leading AIEgen, TPE (tetraphenylethylene), aggregates in water-rich ethanol–water binary mixtures. Other forms of aggregation, in particular the assembling of AIEgens in the presence of, for example, a covalent polymeric species, have not drawn enough attention, despite being reported in many cases in which the AIEgens might in principle randomly bind to the polymeric backbone. We noted that, despite the generally similar overall observation of an enhancement in the emissions of both kinds of aggregates, the extent of the enhancement differs quite a lot, being at 102 and 101 orders of magnitude, respectively. It thus becomes clear that a detailed photophysical mechanistic examination is necessary for the comprehensive understanding of the mechanism of AIE,9 given the importance of AIE in many areas, ranging from photophysical investigations to the design and synthesis of new AIEgens, and in sensing and imaging applications. We thus decided to explore the photophysical mechanism of less examined aggregates in the presence of an inducing species.
We envisioned that forming Ag+ coordination polymers of a thiol ligand10 that bears an AIEgen could be an approach to investigate this important issue, since the Ag+⋯Ag+ interactions11,12 that hold together the Ag-SR units in a manner like in a necklace (Scheme 1) could ensure the expected aggregation of the AIEgens. We thus designed a chiral thiol ligand employing a cysteine residue that was modified into the amide derivative of TPE (L- and D-1, Scheme 1), bearing in mind that the chirality of the cysteine residue offered an additional way of looking into the structure of the coordination polymers.
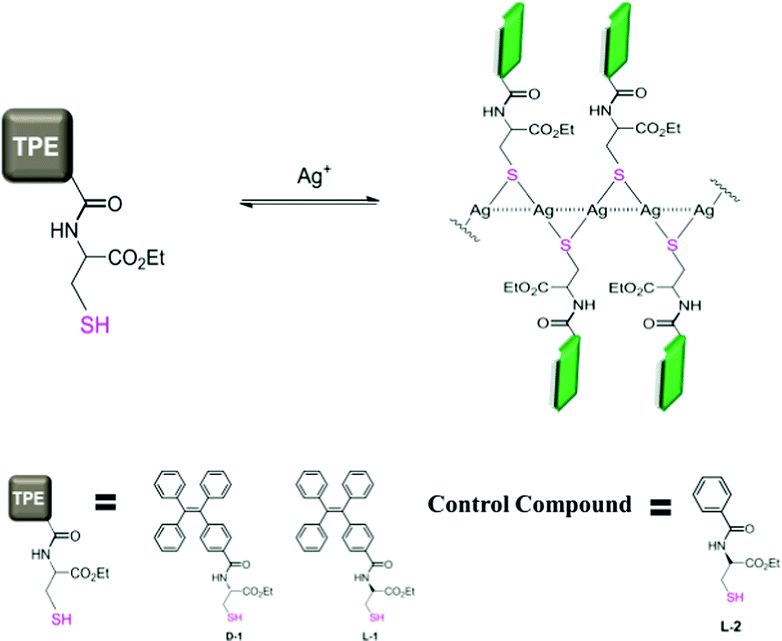 | ||
| Scheme 1 Formation of the coordination polymers of Ag+ and a chiral thiol ligand containing an AIEgen allows the induced aggregation of AIEgens. Dashed lines indicate the Ag+⋯Ag+ interactions. The colours of the TPE fluorophore are indicative of the brightness of the emission. Details of the syntheses of L-/D-1 and L-2 are provided in the ESI.† | ||
The formation of coordination polymers of Ag+ with L-1 in EtOH, in which 1 is well dissolved and hence exists in its monomer form, was first monitored by absorption. Fig. 1 shows that, with increasing Ag+ concentration, the absorption at 325 nm of 1, assigned to the TPE chromophore, is blue-shifted to 290 nm and enhanced. A plot of the absorbance at 290 nm versus the concentration of Ag+ (Fig. 1b), together with a Job plot (see Fig. S1, ESI†), suggests a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry between Ag+ and the thiol ligand 1. Variation in the profiles was found to be similar for L- and D-1 at other concentrations (Fig. S2–S5, ESI†). To confirm that the 290 nm band corresponded to the blue-shifting of the band from 325 nm, we synthesized the control compound L-2 (Scheme 1) in which the TPE chromophore in L-1 was replaced by a phenyl group. In the absorption titration trace of L-2 with Ag+, no sharp peak at 290 nm was observed (Fig. S6, ESI†), indicating that the absorption found at 290 nm in the absorption spectra of the L-1–Ag+ aggregates was related to the TPE chromophore that was originally located at 325 nm. Dynamic light scattering (DLS) data (Fig. S7, ESI†) showed that the average hydrodynamic diameter of 1 in EtOH was ca. 1 nm, in agreement with its monomer form, while that of 1 in the presence of Ag+ was around 100 nm. This suggests that the species formed in the solutions of 1 and Ag+ are polymers of the 1:1 Ag+-SR repeating unit. Similar observations were made at other concentrations of 1 in the presence of Ag+ (Fig. S8–S11, ESI†). Furthermore, 1H NMR titrations indicated that the resonance of –SH disappeared gradually upon the addition of Ag+, together with broadening of the signals of the protons of the –CH2 group that links the –SH unit to the TPE chromophore and appearance of well-resolved signals for the aromatic protons (Fig. S12, ESI†), confirming the formation of an Ag–S bond and, subsequently, of the coordination polymers.
:1 stoichiometry between Ag+ and the thiol ligand 1. Variation in the profiles was found to be similar for L- and D-1 at other concentrations (Fig. S2–S5, ESI†). To confirm that the 290 nm band corresponded to the blue-shifting of the band from 325 nm, we synthesized the control compound L-2 (Scheme 1) in which the TPE chromophore in L-1 was replaced by a phenyl group. In the absorption titration trace of L-2 with Ag+, no sharp peak at 290 nm was observed (Fig. S6, ESI†), indicating that the absorption found at 290 nm in the absorption spectra of the L-1–Ag+ aggregates was related to the TPE chromophore that was originally located at 325 nm. Dynamic light scattering (DLS) data (Fig. S7, ESI†) showed that the average hydrodynamic diameter of 1 in EtOH was ca. 1 nm, in agreement with its monomer form, while that of 1 in the presence of Ag+ was around 100 nm. This suggests that the species formed in the solutions of 1 and Ag+ are polymers of the 1:1 Ag+-SR repeating unit. Similar observations were made at other concentrations of 1 in the presence of Ag+ (Fig. S8–S11, ESI†). Furthermore, 1H NMR titrations indicated that the resonance of –SH disappeared gradually upon the addition of Ag+, together with broadening of the signals of the protons of the –CH2 group that links the –SH unit to the TPE chromophore and appearance of well-resolved signals for the aromatic protons (Fig. S12, ESI†), confirming the formation of an Ag–S bond and, subsequently, of the coordination polymers.
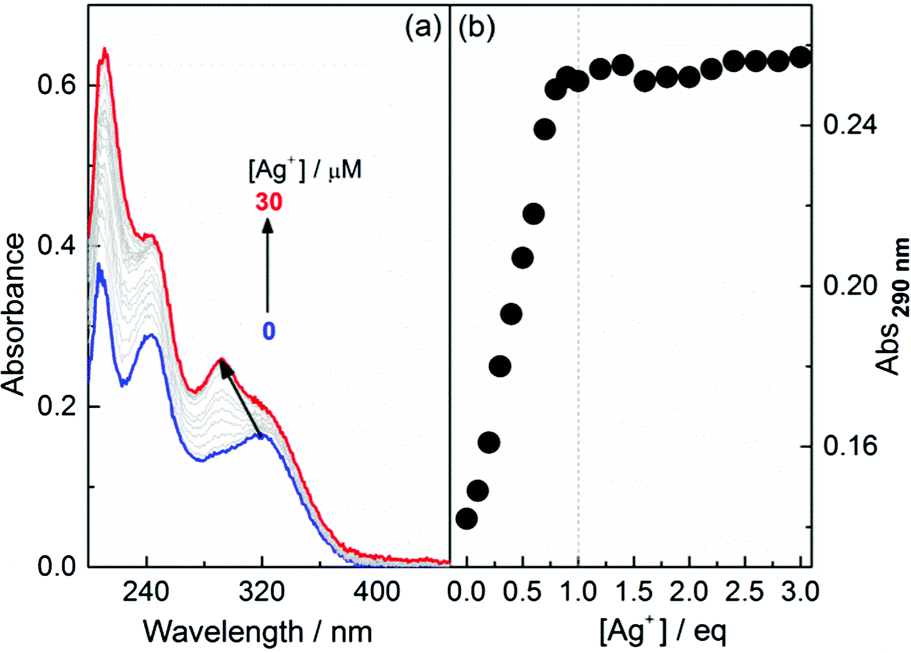 | ||
| Fig. 1 (a) Absorption spectra of L-1 in EtOH in the presence of Ag+ and (b) plots of the absorbance at 290 nm against the concentration of Ag+. [L-1] = 10 μM. | ||
A dilute ethanol solution of 1 was found to be practically nonfluorescent when excited at 325 nm (Fig. 2, and see Fig. S13–S16 for L-1 at other concentrations, ESI†), which suggests that 1 exists in its monomeric form in EtOH. Upon addition of Ag+, however, a dramatic enhancement in the emission at 465 nm was observed, until 1 eq. of Ag+ was introduced and the intensity leveled off, achieving a 20-fold enhancement. This observation confirms that the formation of Ag+–thiol coordination polymers effectively triggers AIE. The apparent binding stoichiometry of Ag+ with 1 was again shown to be 1:1 (Fig. 2b). The ratio of the enhancement is in the same order of magnitude as that observed with AIEgens when attaching to a polymeric backbone13 or when aggregation is induced by the presence of small molecules.4,14,15 This enhancement is, however, less substantial than that observed in the corresponding self-assembled aggregates that could amount to an enhancement of 102 orders of magnitude.5,16 For example, the emission of L-1 in water–ethanol binary solvents showed an up to 300-fold enhancement (Fig. S17, ESI†). This observation is significant for the comprehensive understanding of the AIE effect.
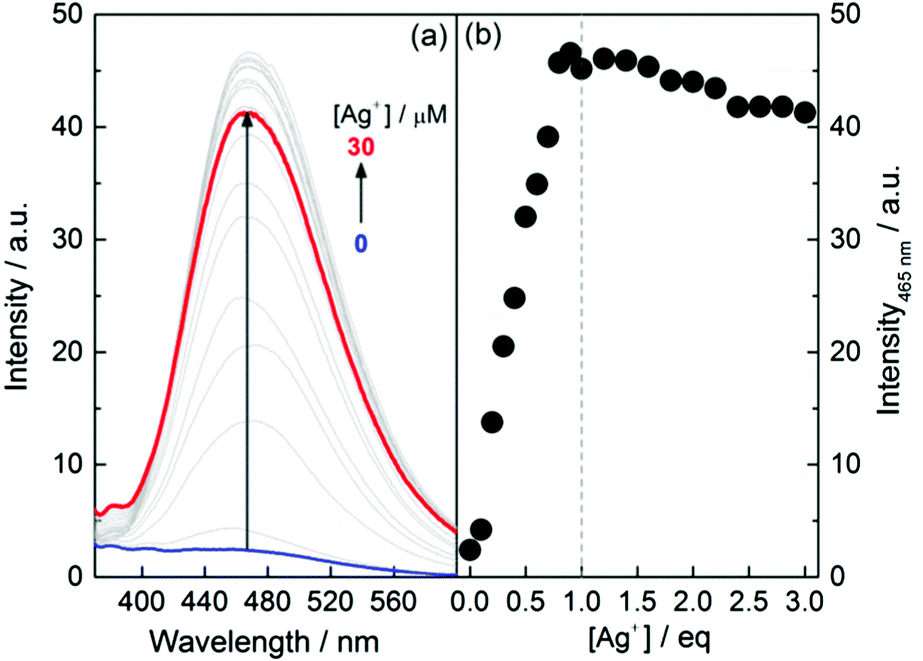 | ||
| Fig. 2 (a) Fluorescence spectra of L-1 in EtOH in the presence of Ag+ and (b) the plot of intensity at 465 nm versus the concentration of Ag+. [L-1] = 10 μM, λEx = 325 nm. | ||
To understand this huge difference in the extent of the enhancement of emission, we first monitored the CD spectra of L-/D-1 in ethanol in the presence of Ag+ or water. The formation of Ag+–L-1 or Ag+–D-1 coordination polymers in ethanol results in a chirality transfer from the cysteine residue to the TPE chromophore, with CD signals at 290 nm and 320 nm, and to the in situ created chromophore related to the Ag+⋯Ag+ interaction, exhibiting a CD signal at 340 nm (Fig. S18a, ESI†). The mirror-image CD profiles observed from the Ag+–L-1 and Ag+–D-1 solutions confirmed the origin of the chirality to be from the cysteine residue (for the CD profiles of the Ag+ coordination polymers of L-/D-1 at other concentrations, see Fig. S19–S21, ESI†). The CD signals developed from the solutions of L-1 and D-1 in ethanol with increasing water content also showed the transfer of chirality from the cysteine residue to the TPE chromophore in the aggregate forms, as suggested by the CD signal at the TPE absorption maximum of 325 nm, and supported by the mirror-image CD profiles (Fig. S18b, ESI†). Note that the profiles of the CD spectra of L-/D-1 in ethanol in the presence of Ag+ and those of L-/D-1 in water-rich ethanol solutions differ quite a lot, reflecting the difference in the aggregation of the TPE chromophores. This can also be seen in the morphologies of the two aggregates, being fibrils and spherical dots, respectively (Fig. S22, ESI†).
We next measured the photophysical parameters of the two systems, i.e. the fluorescence lifetime, quantum yield, and polarization. An increase in the polarization (P) of 1 in ethanol with increasing Ag+ concentration was observed, leveling off after 1 equivalent of Ag+ was added (Fig. 3a). The polarization of the Ag+/1 polymers in ethanol, ca. 0.20, however, was lower than that of the aggregates of 1 in water-rich ethanol solutions (ca. 0.37, Fig. 3b). Following the Perrin equation,17 taking together the data of their lifetime (Fig. S23, ESI†) and size (Fig. S11, ESI†), we calculated the fundamental polarization, P0, of the Ag+/1 coordination polymers in ethanol to be 0.20, while that of the aggregates of 1 in water-rich ethanol solutions were calculated to be 0.38 (Fig. S24 and S25, ESI†). Both values were close to the respective values measured for the Ag+/1 coordination polymers (0.20, Fig. 3a) and the aggregates of 1 (0.37, Fig. 3b). This means that, in both the Ag+/1 coordination polymers in ethanol and the aggregates of 1 in water-rich ethanol solutions, the rotational diffusion of the TPE fluorophore of 1 in the excited state is limited so that no additional depolarization can occur. This is important since it implies that the difference in the enhancement of the fluorescence in the Ag+/1 coordination polymers and the aggregates of 1 is not due to the different rotational freedom of the fluorophore.
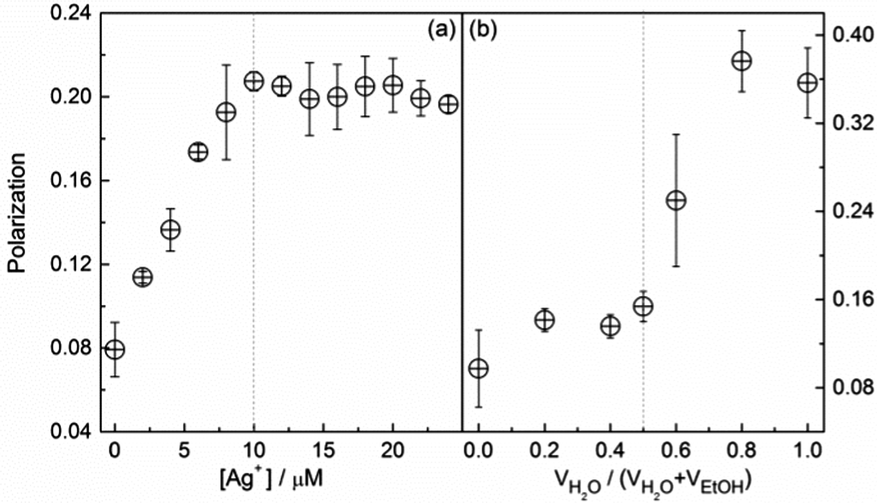 | ||
| Fig. 3 Fluorescence polarization of L-1 in EtOH versus (a) the concentration of Ag+ and (b) the water volume fraction. [L-1] = [Ag+] = 10 μM; λEx = 325 nm, λEm = 465 nm. | ||
We thus calculated the radiation and radiationless rate constants, kr and krl, using the measured data of the lifetimes (Fig. S23, ESI†) and quantum yields (Tables S1 and S2, ESI†). We noted that the fluorescence lifetime of 1 in ethanol decreases, from ca. 6.0 ns for the free 1 to 2.8 ns for the Ag+/1 coordination polymers (Fig. S23a, ESI†). Meanwhile the lifetime of 1, which is ca. 6.6 ns in water-deficient solutions in which it exists in its free state, drops to ca. 4.5 ns when it aggregates in water-rich solutions (Fig. S23b, ESI†). Such a small difference in the lifetimes of the coordination polymers and the aggregates could not explain the huge differences in the fluorescence enhancement. Indeed, we found that, in the case of the Ag+/1 coordination polymers, krl (108 s−1 order of magnitude) is much higher than kr (105–106 s−1), but it increases by only 2 times while kr increases by up to 375 times in the presence of 1 eq. or more of Ag+ (Fig. 4a). Note that, in the Ag+/1 coordination polymers, the krl remains high and unchanged when they are formed at a concentration of 1 eq. of Ag+ (Fig. 4a). With aggregates of 1 in water-rich solutions, however, the kr of the aggregates of 1 in water is 1.6 × 108 s−1, which is ca. 104 times that of free 1, while krl (4.0 × 107 s−1) remains more or less unchanged, until in pure water it drops to two thirds of that of free 1 (Fig. 4b).
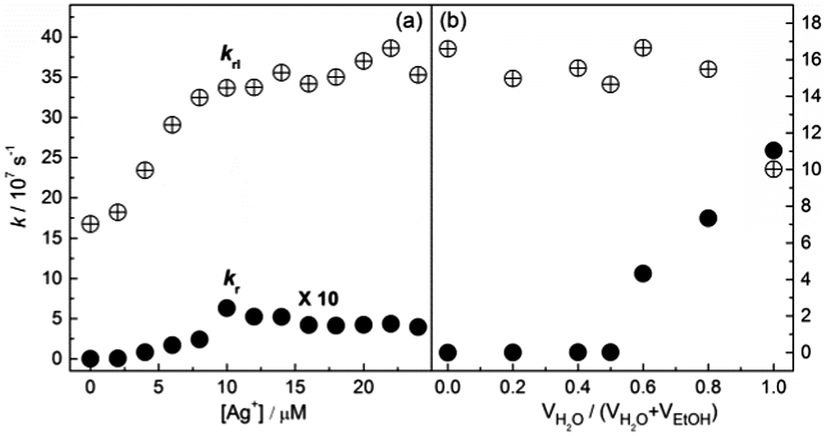 | ||
| Fig. 4 Radiation and radiationless rate constants, kr and krl, of L-1 in EtOH as a function of (a) the concentration of Ag+ and (b) the volume fraction of water. kr = Φ/τ, krl = (1 − Φ)/τ, in which Φ is the quantum yield and τ is the lifetime. Detailed data of Φ can be found in Tables S1 and S2 in the ESI.† | ||
It is worth noting that the formation of the 1-Ag+ coordination polymers results in a blue shift of the absorption of 1, whereas aggregation of 1 in water-rich ethanol solutions leads to a red-shift (Fig. S18, ESI†). This means that in the Ag+ coordination polymers the TPE chromophores of 1 undergo an H-type aggregation, while in the aggregates of 1 in the water-rich ethanol solutions the TPE chromophores are stacked in a J-type arrangement. This explains the extremely low kr value of the Ag+/1 coordination polymers (Fig. 4a), since H-aggregates are known to be nonfluorescent because of their highly efficient internal conversion.18 Yet, we observed a substantial fluorescence enhancement, demonstrating that the much stronger AIE effect is in competition with the quenching effect led by the H-aggregation, as indicated by the more substantial increase in kr than in krl (Fig. 4a).
Interestingly, this difference in the type of aggregation also leads to dramatically different CD profiles depending on the enantiomeric excess (ee) of the enantiomers of 1 that form the coordination polymers with Ag+ in ethanol or the aggregates in water-rich ethanol (Fig. S26, ESI†). In the case of the Ag+ coordination polymers, a weak chiral amplification was observed with a “Z”-shaped curve19 (Fig. S26c, ESI†), whereas an “opposite-Z” shaped curve was found for the aggregates of L-/D-1 in 9:1 H2O–EtOH (v/v) (Fig. S26d, ESI†) that has been very rarely observed.20 Note that for the Ag+/L-1 coordination polymers the first Cotton effect is positive (with contribution from a signal at ca. 350 nm which is related to the Ag+⋯Ag+ interaction, Fig. S26a, ESI†), while that of the aggregates of L-1 is negative (Fig. S26b, ESI†), again reflecting the different profiles of the aggregation of the TPE chromophores.
In summary, we have detailed AIE in an alternative manner looking at induced aggregation in the presence of Ag+, which led to supramolecular structures drastically different from the aggregates of the same AIEgens. In the Ag+ coordination polymers of a thiol ligand that contained an AIEgen, the AIEgens underwent H-type aggregation that would have otherwise led to fluorescence quenching, as in classic H-aggregates, but the Ag+ coordination polymers were fluorescent, with a quantum yield of 0.011 in ethanol. This observation clearly shows the power of AIE in competing with highly efficient internal conversion led by exciton-coupling in H-aggregates, and resulting in a net substantial enhancement of the fluorescence. In the two kinds of aggregates studied (the aggregates of the thiol ligand in water-rich ethanol solutions and the induced aggregates of the coordination polymers of the thiol ligand with Ag+), despite huge differences in the extent of the enhancement of the emission, the rotational freedom of the fluorophore TPE in both was limited so that no additional depolarization was observed, while the radiationless rate constants remained high and unchanged. Our findings would be of significance for the comprehensive mechanistic understanding of AIE and, more importantly, for designing new AIEgens, in particular in the context of induced aggregates, for example, to improve fluorescence enhancement by preventing efficient exciton-coupling.
We greatly appreciate the support of this work by the NSF of China (grants 21275121, 21435003, 91427304 and J1310024), and the Program for Changjiang Scholars and Innovative Research Teams in Universities, administrated by the MOE of China (grant IRT13036).
Notes and references
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- Y. Liu, C. Deng, L. Tang, A. Qin, R. Hu, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2011, 133, 660–663 CrossRef CAS PubMed.
- Y. Ma, Y. Zeng, H. Liang, C.-L. Ho, Q. Zhao, W. Huang and W.-Y. Wong, J. Mater. Chem. C, 2015, 3, 11850–11856 RSC.
- C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 62–65 CrossRef CAS PubMed.
- J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS PubMed.
- M. Zhang, G. Feng, Z. Song, Y.-P. Zhou, H.-Y. Chao, D. Yuan, T. T. Y. Tan, Z. Guo, Z. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241–7244 CrossRef CAS PubMed.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2016 DOI:10.1038/NCHEM.2612.
- (a) J.-S. Shen, D.-H. Li, Q.-G. Cai and Y.-B. Jiang, J. Mater. Chem., 2009, 19, 6219–6224 RSC; (b) J.-S. Shen, D.-H. Li, M.-B. Zhang, J. Zhou, H. Zhang and Y.-B. Jiang, Langmuir, 2011, 27, 481–486 CrossRef CAS PubMed; (c) D.-H. Li, J.-S. Shen, N. Chen, Y.-B. Ruan and Y.-B. Jiang, Chem. Commun., 2011, 47, 5900–5902 RSC; (d) Q. Zhang, Y. Hong, N. Chen, D.-D. Tao, Z. Li and Y.-B. Jiang, Chem. Commun., 2015, 51, 8017–8019 RSC; (e) Q. Wang, Z. Li, D.-D. Tao, Q. Zhang, P. Zhang, D.-P. Guo and Y.-B. Jiang, Chem. Commun., 2016, 52, 12929–12939 RSC.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- I. G. Dance, L. J. Fitzpatrick, A. D. Rae and M. L. Scudder, Inorg. Chem., 1983, 22, 3785–3788 CrossRef CAS.
- X. Lou, C. W. T. Leung, C. Dong, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, RSC Adv., 2014, 4, 33307–33311 RSC.
- L. Liu, G. Zhang, J. Xiang, D. Zhang and D. Zhu, Org. Lett., 2008, 10, 4581–4584 CrossRef CAS PubMed.
- H. Tong, Y. Hong, Y. Dong, M. Haussler, J. W. Y. Lam, Z. Li, Z. Guo, Z. Guo and B. Z. Tang, Chem. Commun., 2006, 3705–3707 RSC.
- W. Z. Yuan, H. Zhao, X. Y. Shen, F. Mahtab, J. W. Y. Lam, J. Z. Sun and B. Z. Tang, Macromolecules, 2009, 42, 9400–9411 CrossRef CAS.
- M. Vincent, B. De Foresta, J. Gallay and A. Alfsen, Biochemistry, 1982, 21, 708–716 CrossRef CAS PubMed.
- U. Rösch, S. Yao, R. Wortmann and F. Würthner, Angew. Chem., Int. Ed., 2006, 45, 7026–7030 CrossRef PubMed.
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS PubMed.
- X. Wu, X.-X. Chen, B.-N. Song, Y.-J. Huang, Z. Li, Z. Chen, T. D. James and Y.-B. Jiang, Chem. – Eur. J., 2014, 20, 11793–11799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses and 1H and 13C NMR spectra of L-/D-1, DLS data, spectral titration traces and fluorescence quantum yields. See DOI: 10.1039/c6cc08596b |
| This journal is © The Royal Society of Chemistry 2017 |