
Water-induced helical supramolecular polymerization and gel formation of an alkylene-tethered perylene bisimide dyad†
Xu
Lin
a,
Hiroki
Kurata
a,
Deepak D.
Prabhu
a,
Mitsuaki
Yamauchi
a,
Tomonori
Ohba
 b and
Shiki
Yagai
*a
b and
Shiki
Yagai
*a
aDepartment of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan. E-mail: yagai@faculty.chiba-u.jp; Fax: +81-(0)43-290-3039; Tel: +81-(0)43-290-3368
bDepartment of Chemistry, Graduate School of Science, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
First published on 28th November 2016
Abstract
An alkylene-tethered perylene bisimide (PBI) dyad with hydrophilic substituents forms helical supramolecular polymers that can be visualized by AFM in THF–water mixtures. The supramolecular polymers also form thixotropic gel-like lyotropic mesophases in the mixtures.
Supramolecular polymers1 formed by stacking of π-conjugated electronic molecules are fascinating smart materials that can hybridize well-defined functionalities of small molecules and physical properties of polymers.2 Due to the steric and electronic effects of π-conjugated systems,3 the formation of such supramolecular polymers is often accompanied by helical architectures in different levels of hierarchy, which are particularly related to chiroptical properties4 and formation of gels.5 Recently, π-conjugated molecules that can supramolecularly polymerize in aqueous media in terms of their compatibility with biological as well as a variety of water-soluble organic and inorganic entities have been of particular interest to chemists.6 Most of the π-electronic supramolecular monomers that can aggregate in aqueous media are functionalized with specific hydrogen-bonding sites to enforce aggregation.7 The same situation holds true for perylene bisimide (PBI) dyes, which are arguably the most extensively studied functional π-conjugated molecules.8
Here we demonstrate that covalent dimerization of two PBI units through an alkylene tether is an effective molecular design for supramolecular monomers that can one-dimensionally aggregate in aqueous media. The concept underpinning our molecular design is based on a simple but rational idea to enforce intermolecular π–π stacking by covalent dimerization without introducing a hydrogen-bonding site. The effectiveness of this idea has been recently corroborated by our group using covalent PBI dimers (hereafter referred to as PBI dyads) with swallow-tailed hydrophobic chains in nonpolar media.9 An important feature in this molecular design is that the alkylene chain tethering two PBI units should be odd-numbered (n = 5, 7 and 9) so as to orient the two chromophores in “V-shape” geometry. In this study, we synthesized a new alkylene-tethered PBI dyad, 2, bearing hydrophilic swallow-tailed oligoethylene glycol chains10 (Fig. 1). Dyad 2 showed well-defined water-induced supramolecular polymerization and gelation in THF–water mixtures. It is striking that in the present system the helical handedness of supramolecular polymers could be unequivocally determined by AFM.
 | ||
| Fig. 1 Molecular structures of PBI dyad 2 and reference unimolecular PBI 1. | ||
Dyad 2 is moderately soluble in THF (cmax = 1 × 10−3 M). Concentration-dependent UV/Vis absorption measurements showed that 2 forms π–π stacked aggregates in pure THF (Fig. 2a). At c = 1 × 10−6 M, 2 displayed 0–0 (522 nm) and 0–1 (487 nm) vibronic transitions, and the intensity ratio of these transitions (ε0–0/ε0–1) is 1.1. This value is smaller than that of reference PBI 1 that is molecularly dissolved under the same condition (ε0–0/ε0–1 = 1.6, Fig. S1, ESI†), which demonstrates that 2 aggregates at this low concentration. Upon increasing the concentration, ε0–0/ε0–1 further decreased and the spectral change levelled off at 1 × 10−4 M to give ε0–0/ε0–1 of 0.6. The observed concentration dependence thus excludes the possibility of intramolecular folding via π–π stacking interaction.11 The plot of the fraction of aggregated molecules (αagg)12 as a function of concentration (cT) provided a sigmoidal curve, indicating that the process appears to be isodesmic as revealed by the relatively smooth slope of the plot.13 The curve could be fitted well with an isodesmic model, giving an association constant (K) of 1.0 × 106 M−1 (Fig. 2b).14 This K value is significantly higher than that of the unimolecular PBI derivative (K = 1.0 × 102 M−1).15
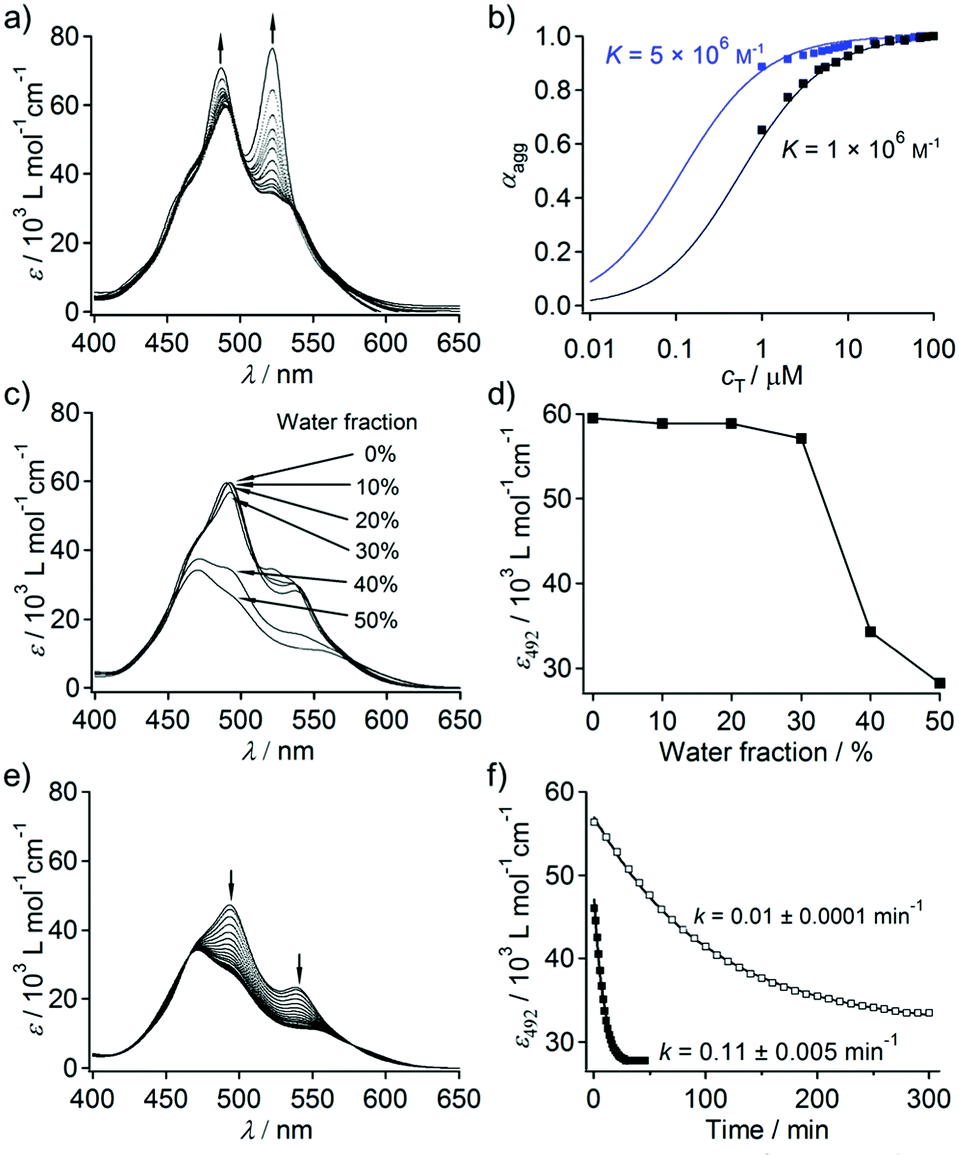 | ||
| Fig. 2 (a) Concentration-dependent UV/Vis spectra of 2 (c = 1 × 10−6 to 1 × 10−4 M) in THF at 20 °C. Arrows indicate the spectral changes with decreasing concentration. (b) Plot of the fraction of aggregated molecules (αagg) of 2 calculated from the absorption intensity at 522 nm as a function of cT as well as fitting according to the isodesmic model in pure THF (black squares and curve) and in THF/water (80/20) mixture (blue squares and curve). (c) UV/Vis spectra of 2 (c = 1 × 10−4 M) in THF/water mixtures with different water contents (0–50, v/v%). All spectra were measured after 6 hours of incubation. (d) Plots of molar extinction coefficient of 2 at 492 nm (ε492) versus different water contents. (e) Time-dependent (1–30 min) UV/Vis spectra of 2 (c = 1 × 10−4 M) in THF/water (50/50) mixture at 20 °C. Arrows indicate the spectral change with time. (f) Plot of ε492versus time for THF/water (50/50) (■) and THF/water (60/40) (□) solutions. k denotes the rate constant. The solid lines correspond to the theoretical fitting obtained from nonlinear regression analysis using a first-order kinetic model. | ||
When an appropriate volume of water is added to THF solutions of 2 so as to prepare THF/water solutions with the final concentration of 1 × 10−4 M, homogeneous solutions were obtained up to the water content of 50%. No significant UV/Vis spectral change was observed upon increasing the water content from 0 to 30% (Fig. 2c and d). However, concentration-dependent measurements (Fig. S3, ESI†) of THF/water (80/20) solutions revealed that even 20% of water can significantly enhance π–π stacking interactions, which was reflected in a 5 fold increase in the association constant (K) (blue curve, Fig. 2b). This was further confirmed by fluorescence measurements, wherein a significant quenching (ΦF = 0.33 in THF; 0.04 in THF/water (70/30)) along with a bathochromic shift of the excimer like fluorescence (λem–max = 634 nm in THF; 648 nm in THF/water (70/30)) was observed with increasing water content (Fig. S4, ESI†). Taking almost negligible absorption change into account, the observed fluorescence quenching suggests the formation of much more strongly exciton-coupled excimer species from the same ground-state aggregate structures through the enhanced solvophobic interaction.
Remarkably, large time-dependent absorption spectral changes were observed upon increasing the water content above 30%. For the THF/water (50/50) solution, a hypsochromic shift of 20 nm in the absorption maximum accompanied by a hypochromic effect was observed over 30 min (Fig. 2e). The equilibrated spectrum is characteristic of lamellarly aggregated PBIs with a lesser degree of rotational displacement in the π–π stacking.16 The time-dependent spectral transition can be attributed to the transformation of a kinetically trapped, less ordered stack of PBI chromophores into a thermodynamically stable stack. The spectral change follows a first-order kinetics with a rate constant k = 0.11 ± 0.005 min−1 (Fig. 2f). A much slower spectral transition was observed for the THF/water (60/40) solution (k = 0.01 ± 0.0001 min−1) with an equilibration time of 300 min (Fig. 2f and Fig. S5, ESI†). The time-dependent spectral transition implies that 2 forms kinetic aggregates upon adding water, which are slowly converted to thermodynamically stable aggregates.
The time evolution of the aggregates in the THF/water (50/50) mixture was investigated by AFM. Before adding water, a large number of short fibers (∼100 nm) were visualized (Fig. S6a, ESI†), and they had a height of ca. 0.6 nm (the inset in Fig. S6a, ESI†), which is comparable to the molecular length along the shorter molecular axis (ca. 0.7 nm). Upon addition of water, these short fibers were completely replaced by particulate aggregates after 1 min of incubation (Fig. S6b, ESI†). These particulates may correspond to the kinetic aggregates formed by interactions of hydrophilic oligoethylene glycol chains with water molecules, as no minimal change was observed in the absorption spectra. Incubation of 5 min afforded elongated fibers, which were visualized by AFM (Fig. S6c, ESI†), the population of which further increased and levelled off after 15 min. At this stage, very few particulate aggregates were still observed, which completely disappeared after 6 h of incubation. In contrast, such a time-evolved change in morphology was not observed for aggregates prepared from solution with lower water content (Fig. S7, ESI†). Remarkably, the supramolecular polymer fibers from the THF/water (50/50) mixture seemingly have helical morphology (Fig. S6d, ESI†), which motivated us to further study their morphology by AFM.
High-resolution AFM phase and height images (Fig. 3a and c) revealed that supramolecular fibers have an ordered helical structure with a pitch of ca. 5.0 nm and a pitch angle of ca. 120° (Fig. 3b).17 The helical nanofibers showed both right- (P) and left-handed (M) helical structures due to the absence of any internal and external chiral sources.18 However, the handedness of helicity in a given fiber is completely biased, which suggests cooperative helical stacking through water-induced supramolecular polymerization. The average thickness of the fibers is 2.8 ± 0.2 nm (Fig. 3d), which is significantly larger than that of the short fibers formed in THF. Because the width (ca. 15 nm) of the fibers obtained from AFM may include a tip-broadening effect, we analysed the fibers by transmission electron microscopy (TEM) and obtained a width of 5.0 ± 0.5 nm (Fig. S8, ESI†). This value is comparable to the molecular length along the long axis (ca. 5.0 nm). Based on these values, a helical one-dimensional stacking shown in Fig. 3b is proposed. The helical pitch of ca. 5.0 nm corresponds to the half turn (180°) of the dyad molecules. Assuming a distance of 0.3–0.5 nm between stacked dyad molecules, one helical pitch corresponds to approximately 10–16 molecules. Hence, the rotational angle (θ) between the neighboring dyad molecules is calculated to be in the range of 11–18°. This value is considerably smaller than that in the columnar aggregates of unimolecular PBI derivatives (θ = 55°).19 The smaller rotational displacement (i.e., larger overlap of π-systems) of the dyad is in line with its specific absorption spectra with the largely blue-shifted maxima in THF–water mixtures (vide supra). Such a helical feature was not observed for the fibers formed in the THF/water (60/40) mixture after prolonged aging after equilibration (Fig. S9a and c, ESI†). Although these non-helical fibers have lengths comparable to those of helical fibers formed in the THF/water (50/50) mixture, they are thinner than the helical fibers by 1 nm (Fig. S9d and e, ESI†). These findings demonstrate that fibers formed in the THF/water (60/40) mixture lack successive one-handed helical stacking, probably due to dynamic rotation of stacked dyad molecules (Fig. S9b, ESI†).20
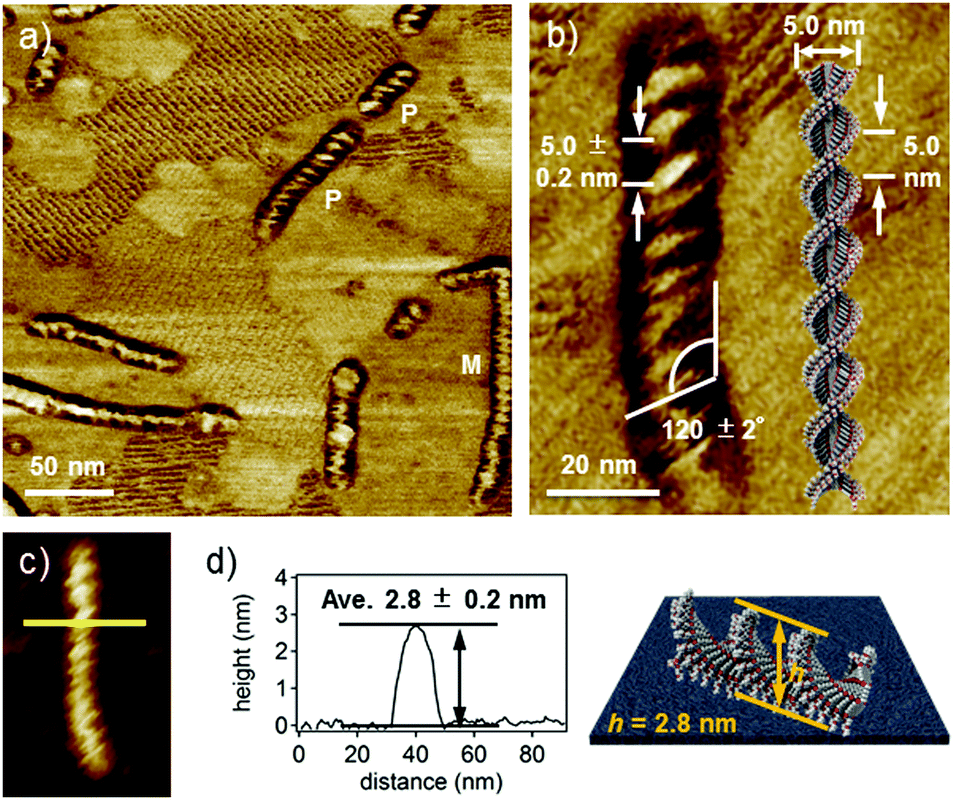 | ||
| Fig. 3 (a) AFM phase image of 2 (c = 1 × 10−4 M) spin-coated from THF/water (50/50) onto HOPG. P and M in (a) indicate right- and left-handed helical fibers. (b) Magnified image of a P-type fiber with the proposed aggregate structure. (c) AFM height image of the P-type fiber. (d) Cross-sectional analysis along the yellow line in (c) with schematic representation of the fiber on the substrate. | ||
When a hot THF solution of 2 (c = 2.0 × 10−3 M) was prepared by vigorous heating in a vial, an excess amount of 2 precipitated upon cooling to r.t. (Fig. 4a). The precipitates are composed of ill-defined fibrous structures, as shown by AFM (Fig. S10, ESI†). Interestingly, the addition of comparable amount of water to the precipitated solution resulted in a slow dissolution without heating (Fig. 4b), which eventually afforded a transparent gel-like liquid after several hours (Fig. 4c).21 The polarized optical microscopic image of the gel showed a birefringence (Fig. 4d), indicating the formation of lyotropic liquid crystals.22 AFM imaging of the dried gel showed an entangled network of parallelly bundled supramolecular polymer fibers, suggesting anisotropic organization of the fibers (Fig. S11, ESI†). The average inter-fiber distance was estimated to be 3.0 ± 0.2 nm (Fig. 4f), which was further confirmed by an explicit X-ray diffraction pattern that can be assigned to a hexagonal columnar packing with the lattice parameter a = 29.9 Å (Fig. 4e). The significant difference between the inter-fiber distance (3.0 nm) and the width of the modeled helix (ca. 5.0 nm) is ascribable to the interdigitation of branched oligoethylene glycol chains. To confirm this assumption, we added 10 equiv. of lithium triflate (CF3SO3Li) to a gel of 2 with an expectation that the lithium salt will chelate with oligoethylene glycol exteriors and thereby suppress the interdigitation. AFM images of the film prepared from the gel containing lithium salt displayed an increment of the inter-fiber distance to 4.5 ± 0.2 nm (Fig. 4g), which is much closer to the width of the modelled helix.
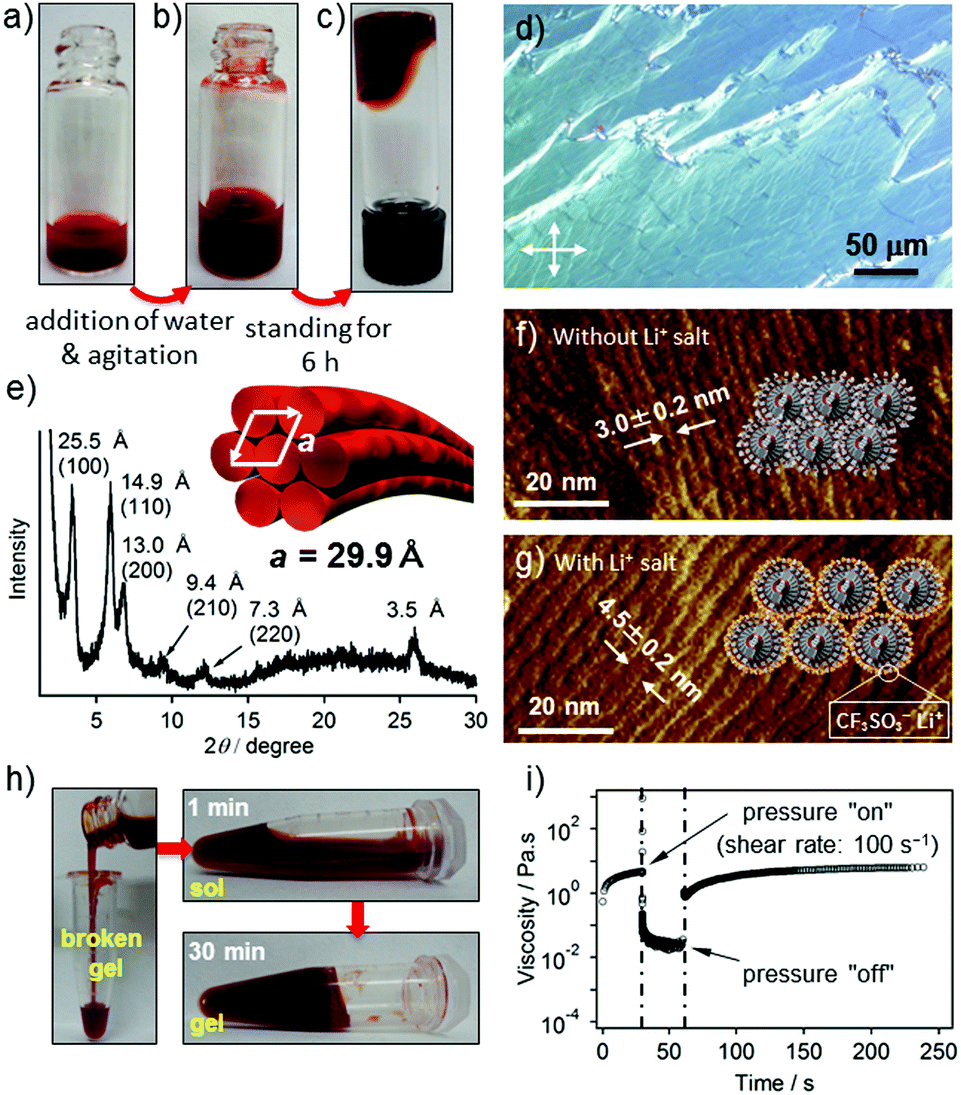 | ||
Fig. 4 Photographs of (a) precipitated solution of 2 in THF prepared by a heating–cooling procedure (c = 2.0 × 10−3 M, 0.5 mL), (b) THF/water mixed solution obtained after addition of 0.5 mL of water and (c) transparent gel-like liquid formed after 6 h. (d) Polarized optical microscopic image of dried gel of 2 sandwiched between two glass slides. (e) XRD diffraction pattern of the dried gel. (f and g) AFM error images of the dried gel (f) without Li+ salt and (g) with Li+ salt. (h) Photographs showing the thixotropic behavior of the THF/water (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) gel of 2 (c = 1.0 × 10−3 M). The gel broken by shaking recovered the original gel state in 30 min. (i) Step strain measurement (shear rate: 100 s−1) performed on the gel of 2 (c = 3.0 × 10−3 M) at 25 °C. :50) gel of 2 (c = 1.0 × 10−3 M). The gel broken by shaking recovered the original gel state in 30 min. (i) Step strain measurement (shear rate: 100 s−1) performed on the gel of 2 (c = 3.0 × 10−3 M) at 25 °C. | ||
The elastic response of the gel-like liquid formed by 2 in the THF–water mixture (50:50) was studied by rheological measurements. The values of dynamic moduli G′ and G′′ were independent over a wide range of oscillatory frequencies, indicating the solid like nature of the gel formed (Fig. S12a, ESI†). However, the value of storage modulus (G′) was only slightly higher than that of the loss modulus (G′′) measured with a strain amplitude of 10% (Fig. S12b, ESI†), suggesting that the gel is very weak.21a This can be explained by the bundling of the elementary fibers rather than three-dimensional entanglement: the former would reduce the number of junction points (Fig. S11, ESI†). Interestingly, the gel-like liquids exhibited thixotropic behavior. When the gel in a glass vial was disturbed by shaking to afford a homogeneous liquid, it remained homogeneous even after centrifugation (500 rpm) without any phase separation. Upon standing the liquid, the apparent viscosity gradually increased and eventually the original gel-like state was recovered within 30 min (Fig. 4h). The rheological measurement of the gel (c = 3.0 × 10−3 M) upon applying a large amplitude oscillation (shear rate: 100 s−1) for 30 seconds resulted in a decrease of the complex viscosity value (η*) from 4.58 to 0.02 Pa s, indicating the disruption of the network structure. Upon removing the applied stress, η* instantaneously recovered to 1.0 Pa s, which then gradually increased up to the original value over 150 seconds (Fig. 4i).
In conclusion, we have demonstrated that tethering two π-conjugated units with an odd-numbered alkylene linker is a promising supramolecular monomer design to provide elongated fibers that show polymeric properties in aqueous media. Detailed spectroscopic and AFM studies of a dyad revealed that the elongation of supramolecular polymers does not progress linearly with increasing water content in THF, but occurs abruptly at a certain solvent composition with time evolution due to kinetically trapped aggregates. The transformation of nonhelical to helical morphologies also occurs at a specific water content, below which dynamic rotation of stacked dyad molecules is allowed due to a weaker solvophobic effect. The water-induced growth of supramolecular polymers also occurs from the precipitated state in THF, affording viscoelastic gel-like liquids with self-healing properties against mechanical agitation. Our simple molecular design to produce supramolecular polymers in aqueous media can be applied to various related functional dyes and π-conjugated systems.
This work was supported by KAKENHI (no. 26102010), a Grant-in-Aid for Scientific Research on Innovative Areas “π-Figuration” (no. 26102001) of The Ministry of Education, Culture, Sports, Science, and Technology, Japan. We thank Dr Daiki Kuzuhara and Prof. Dr Hiroko Yamada for fluorescence quantum yield measurements.
Notes and references
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- (a) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed; (b) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (c) S. Yagai, Bull. Chem. Soc. Jpn., 2015, 88, 28–58 CrossRef; (d) C. Rest, R. Kandanelli and G. Fernández, Chem. Soc. Rev., 2015, 44, 2543–2572 RSC.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- (a) L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2007, 36, 941–967 RSC; (b) V. K. Praveen, S. S. Babu, C. Vijayakumar, R. Varghese and A. Ajayaghosh, Bull. Chem. Soc. Jpn., 2008, 81, 1196–1211 CrossRef CAS.
- (a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS PubMed; (b) S. Bhattacharya and S. K. Samanta, Langmuir, 2009, 25, 8378–8381 CrossRef CAS PubMed; (c) J. W. Steed, Chem. Commun., 2011, 47, 1379–1383 RSC; (d) G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC; (e) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- (a) K. J. C. van Bommel, A. Friggeri and S. Shinkai, Angew. Chem., Int. Ed., 2003, 42, 980–999 CrossRef CAS PubMed; (b) C. Rest, M. J. Mayoral and G. Fernández, Int. J. Mol. Sci., 2013, 14, 1541–1565 CrossRef CAS PubMed; (c) M. R. Molla and S. Ghosh, Phys. Chem. Chem. Phys., 2014, 16, 26672–26683 RSC.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- (a) F. Würthner, Chem. Commun., 2004, 1564–1579 RSC; (b) T. Seki, X. Lin and S. Yagai, Asian J. Org. Chem., 2013, 2, 708–724 CrossRef CAS; (c) F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- X. Lin, M. Hirono, T. Seki, H. Kurata, T. Karatsu, A. Kitamura, D. Kuzuhara, H. Yamada, T. Ohba, A. Saeki, S. Seki and S. Yagai, Chem. – Eur. J., 2013, 19, 6561–6565 CrossRef CAS PubMed.
- A. Wicklein, A. Lang, M. Muth and M. Thelakkat, J. Am. Chem. Soc., 2009, 131, 14442–14453 CrossRef CAS PubMed.
- A. D. Shaller, W. Wang, A. Li, G. Moyna, J. J. Han, G. L. Helms and A. D. Q. Li, Chem. – Eur. J., 2011, 17, 8350–8362 CrossRef CAS PubMed.
- Assuming the absorption spectrum of PBI 1 in THF corresponds to monomeric species (α = 0).
- (a) R. B. Martin, Chem. Rev., 1996, 96, 3043–3064 CrossRef CAS PubMed; (b) Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- Because dyad 2 is not completely monomeric in pure THF upon dilution to 1 × 10−6 M, we also obtained concentration-dependent data in 20% THF–chloroform mixture. As a result, the isodesmic model process of dyad 2 was also confirmed in the range of 0.0 < α < 1.00. (Fig. S2, ESI†).
- D. Görl and F. Würthner, Angew. Chem., Int. Ed., 2016, 55, 12094–12098 CrossRef PubMed.
- (a) K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS PubMed; (b) M. Kumar and S. J. George, Chem. Sci., 2014, 5, 3025–3030 RSC.
- (a) X. Zhang, D. Görl, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 1270–1274 CrossRef CAS PubMed; (b) D. Görl, X. Zhang, V. Stepanenko and F. Würthner, Nat. Commun., 2015, 6, 7009 CrossRef PubMed.
- S. Ghosh, X. Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef CAS PubMed.
- Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. A. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem. – Eur. J., 2007, 13, 436–449 CrossRef CAS PubMed.
- D. Görl, X. Zhang and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6328–6348 CrossRef PubMed.
- (a) E. Krieg, E. Shirman, H. Weissman, E. Shimoni, S. G. Wolf, I. Pinkas and B. Rybtchinski, J. Am. Chem. Soc., 2009, 131, 14365–14373 CrossRef CAS PubMed; (b) E. Krieg, H. Weissman, E. Shimoni, A. B. On and B. Rybtchinski, J. Am. Chem. Soc., 2014, 136, 9443–9452 CrossRef CAS PubMed; (c) D. Görl, B. Soberats, S. Herbst, V. Stepanenko and F. Würthner, Chem. Sci., 2016, 7, 6786–6790 RSC.
- (a) I. K. Iverson and S.-W. Tam-Chang, J. Am. Chem. Soc., 1999, 121, 5801–5802 CrossRef CAS; (b) G. A. Bhavsar and S. K. Asha, Chem. – Eur. J., 2011, 17, 12646–12658 CrossRef CAS PubMed; (c) S. Yagai, M. Usui, T. Seki, H. Murayama, Y. Kikkawa, S. Uemura, T. Karatsu, A. Kitamura, A. Asano and S. Seki, J. Am. Chem. Soc., 2012, 134, 7983–7994 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis and characterization of PBI dyads; absorption spectra and AFM and TEM images of dyad 2. See DOI: 10.1039/c6cc08995j |
| This journal is © The Royal Society of Chemistry 2017 |