
Ligand-assisted thickness tailoring of highly luminescent colloidal CH3NH3PbX3 (X = Br and I) perovskite nanoplatelets†
Ievgen
Levchuk
*ab,
Patrick
Herre
c,
Marco
Brandl
d,
Andres
Osvet
a,
Rainer
Hock
d,
Wolfgang
Peukert
c,
Peter
Schweizer
e,
Erdmann
Spiecker
e,
Miroslaw
Batentschuk
a and
Christoph J.
Brabec
abf
aFriedrich-Alexander University Erlangen-Nürnberg, Materials for Electronics and Energy Technology (i-MEET), Martensstrase 7, 91058 Erlangen, Germany. E-mail: ievgen.levchuk@fau.de
bEnergy Campus Nürnberg (EnCN), Fürther Str. 250, 90429 Nürnberg, Germany
cFriedrich-Alexander University Erlangen-Nürnberg, Institute of Particle Technology, Cauerstraße 4, 91058 Erlangen, Germany
dFriedrich-Alexander University Erlangen-Nürnberg, Chair for Crystallography and Structural Physics, Staudtstrasse 3, 91058 Erlangen, Germany
eFriedrich-Alexander University Erlangen-Nürnberg, Center for Nanoanalysis and Electron Microscopy (CENEM), Department Werkstoffwissenschaen, Cauerstraße 6, 91058 Erlangen, Germany
fZAE Bayern, Renewable Energies, Haberstr. 2a, 91058 Erlangen, Germany
First published on 28th November 2016
Abstract
Quantum size-confined CH3NH3PbX3 (X = Br and I) perovskite nanoplatelets with remarkably high photoluminescence quantum yield (up to 90%) were synthesized by ligand-assisted re-precipitation. Thickness-tunability was realized by varying the oleylamine and oleic acid ligand ratio. This method allows tailoring the nanoplatelet thickness by adjusting the number of unit cell monolayers. Broadly tunable emission wavelengths (450–730 nm) are achieved via the pronounced quantum size effect without anion–halide mixing.
Hybrid organic–inorganic and all-inorganic metal halide perovskite nanocrystals with the general formula of ABX3 (A = Cs+, NH2CHNH2+ and CH3NH3+; B = Sn2+ and Pb2+; X = Cl−, Br− and I−) have been rapidly developed during the last few years.1–6 The possibility of simple halide compositional mixing enables a wide wavelength tunability (400–800 nm) of the narrow-band emission.1,3 Furthermore, the high photoluminescence quantum yield (PLQY) of these colloidal nanocrystals reaches up to over 90% without additional surface passivation, demonstrating that dangling bonds do not lead to non-radiative relaxation.1,7
Recently, the synthesis of quasi-2D cesium lead halide nanoplatelets (NPLs) with layered structures has been performed using colloidal methods varying the starting ligand ratio, the temperature of the synthesis, the HBr amount, as well as the length of alkylammonium cations and carboxylic acids.8–13 Concerning the hybrid nanoplatelets (NPLs), only a few papers have demonstrated their thickness tailoring and quantum size effect in suspensions with PLQY <45%.10,14,15 For example, Cho et al.15 utilized alkylamines with different lengths to achieve the thickness modulation. Sichert et al.10 tuned the ratio of octylammonium bromide and methylammonium bromide for the same purpose. Furthermore, due to the lower stability and sensitivity to moisture of the organolead iodide perovskite colloids,3,6 the synthesis of the CH3NH3PbI3 NPLs with different thicknesses is more challenging.
In this contribution, we present a fast synthesis of nearly monodisperse colloidal CH3NH3PbX3 (X = Br and I) organometallic halide perovskite NPLs via a ligand-assisted method, enabling monolayer thickness control and leading to a high PLQY of up to 90%.
The synthesis of colloidal CH3NH3PbX3 (X = Br and I) NPLs was performed via controllable precipitation at room temperature, following a modified approach of Zhang et al.3 The main difference to the method presented in the paper of Zhang is using chloroform as anti-solvent for the CH3NH3PbI3 NPLs, and tailoring the size and thickness by varying the oleic acid/oleylamine ligand ratio only for both bromide and iodide NPLs (see the details in the Methods section, ESI†).
The injection of a DMF solution of the CH3NH3PbBr3 precursor into toluene rapidly forms highly luminescent NPLs (see the supplementary video, ESI†). By changing the ratio of oleic acid (OA) and oleylamine (OAm) as ligands, we obtained different band gaps as well as luminescence wavelengths of the colloids (Fig. 1a). Upon fixation of the amount of OA in the precursor solution, a certain volume of OAm was added (Table S1, ESI†). This enables a precise tuning of the absorption edge and photoluminescence (PL) of the NPLs from green (514 nm) to blue (447 nm) color. However, beginning from the OA/OAm ratio of 200 μl/30 μl, no further PL shift is observed (peak wavelength at 447 nm, Fig. S1, ESI†). Only the lateral size of the NPLs was further reduced from 10 to 5 nm at OA/OAm = 200 μl/60 μl (shown in Fig. 1d and Fig. 2a). The most challenging task in the synthesis was the cleaning process. Since the organometallic halide perovskite material is highly sensitive to polar solvents and especially water, the conventional alcohol/non-polar solvent (e.g. ethanol/toluene) washing approach, which is common for quantum dots (QDs) such as CdSe,16 destroys the perovskite particles. We found that a certain ratio of acetonitrile/toluene (Table S1, ESI†) allows the precipitation of NPLs without decomposition, which was already successfully used for CsPbBr3 NCs.17 Other mixtures such as isopropanol/toluene, acetone/toluene or ethanol/toluene completely destroyed and/or dissolved the perovskite NPLs. Washed NPLs can be re-dissolved in non-polar solvents such as toluene or hexane and form a stable colloidal solution at least for one week under ambient conditions. Fig. 1c shows the optical absorption and the PL spectra of a toluene solution of CH3NH3PbBr3 NPLs of various sizes that clearly demonstrate the quantum size confinement of the optical band gap. The change from 2.36 to 2.74 eV is in good agreement with the theoretical calculations of Sapori et al.18 Since the reaction kinetics are so fast due to the ionic nature of the metathesis,9 some of the PL spectra reveal a double peak. This is explained by the formation of NPLs with two different thicknesses at the same time, confirmed by electron microscopy analysis discussed below. Fig. 1d shows a representative bright-field transmission electron microscopy (TEM) micrograph of the square particles having a mean side length of 10.7 nm ± 1.5 nm. The size distribution is shown in Fig. S1d (ESI†). Measurements of the photoluminescence quantum yield (details listed in the ESI†) revealed an exceptionally high efficiency (89%) for the thickest platelets with a band gap of 2.36 eV (green PL, 514 nm). This efficiency gradually decreases with the thickness of the plates, reaching 50% for NPLs with a band gap of 2.59 eV (cyan PL, 488 nm) and 32% for the platelets with the largest band gap of 2.74 eV (blue PL, 447 nm). To the best of our knowledge, these are the highest reported values for the CH3NH3PbBr3 NPLs.6,10,14 The decrease in the quantum yield is accompanied by the reduction of the luminescence decay times from 12 to 5 ns (Fig. 1b). While the radiative decay rate remains as a rule almost unchanged, the non-radiative rate, calculated from the quantum yield and the PL decay time, increases from 0.9 × 107 s−1 to 11 × 107 s−1 (Fig. S7a and Table S3, ESI†). Thus, the faster decay in thinner platelets is caused by additional nonradiative relaxation related to the influence of the surface of the platelets.
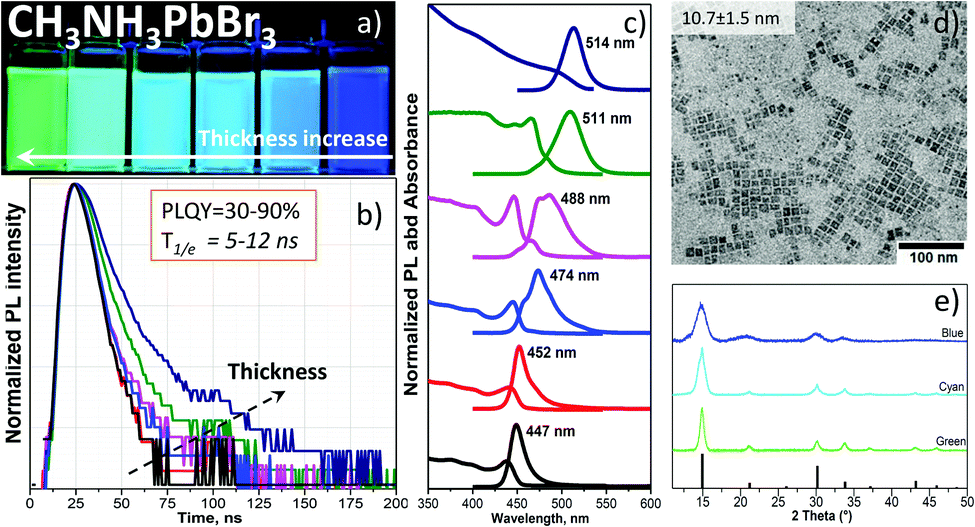 | ||
| Fig. 1 CH3NH3PbBr3 NPLs with different thicknesses in toluene solutions. (a) Digital photo of the NPLs under UV-365 nm illumination. (b) PL decay. (c) Absorbance and PL spectra. (d) Typical bright-field TEM image of the NPLs with a PL peak at 447 nm obtained with a ligand ratio of OA/OAm = 200 μl/30 μl. (e) XRD pattern of NPLs with a characteristic PL (green, cyan and blue). | ||
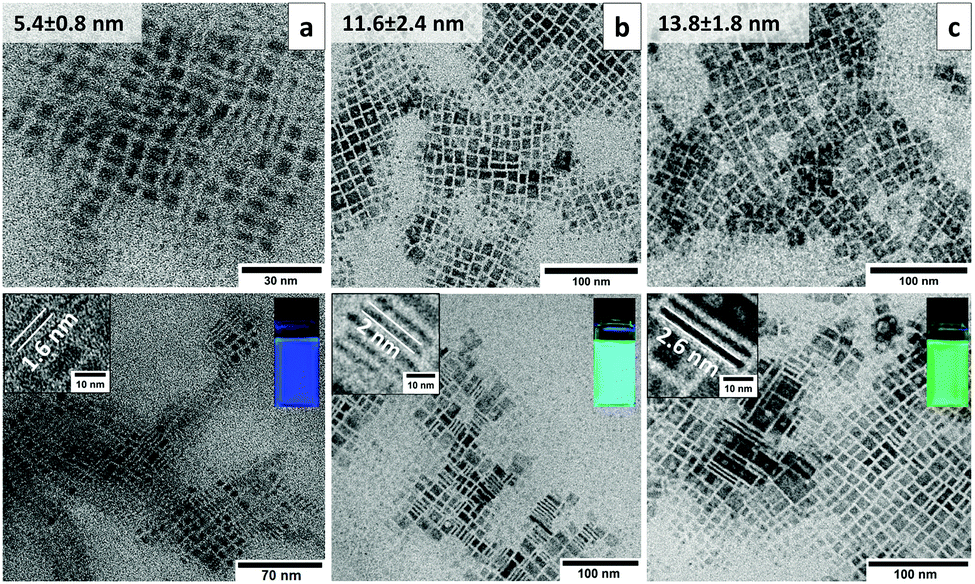 | ||
| Fig. 2 Bright-field TEM characterization of the ligand-assisted layer control of CH3NH3PbBr3 NPLs. Lateral particle size and thickness determination are derived from the upper and lower images, respectively, of (a) blue (447 nm), (b) cyan (488 nm) and (c) green (514 nm) emitting NPLs. Insets: selected standing nanoplatelets and their corresponding photoluminescence color. | ||
A typical electron diffraction pattern of the NPLs shown in Fig. 1d is depicted in Fig. S3a and b (ESI†), nicely confirming the perovskite crystal structure of CH3NH3PbBr3. The corresponding line profile obtained by rotational averaging of the ring pattern is shown in Fig. S3b (ESI†). The theoretical peak intensities for a random orientation of the NPLs are indicated as red bars. A comparison with the experimentally observed peak intensities indicates a preferred (001) orientation of the NPLs on the carbon support grid consistent with the situation where most NPLs lie flat on the carbon support film. This can be seen from the almost complete absence of the (111) peak and the reduced intensity of the (210) peak.
TEM studies of colloidal CH3NH3PbBr3 NPLs with blue, cyan and green PLs clearly link the observed optical emission to the platelet thickness. Consistent with the preferred crystal orientation deduced from electron diffraction patterns, the NPLs in all of the three samples exhibit a square shape (Fig. 2a–c, upper row). The average lateral sizes for blue, cyan and green emitting NPLs are 5.4 ± 0.8 nm, 11.6 ± 2.4 nm and 13.8 ± 1.8 nm, respectively. The size statistics are shown in Fig. S2a–d (ESI†). In order to evaluate the thickness of the NPLs, the platelets have to be imaged in an edge-on orientation. It turned out that by using diluted hexane (∼0.5 mg ml−1) instead of the concentrated toluene (∼10 mg ml−1) solution during the TEM sample preparation a considerable number of standing NPLs are observed on the carbon support film. In particular, the NPLs show upright stacked assemblies on the TEM grid allowing even for a statistical thickness determination directly from the micrographs (Fig. 2a–c, bottom row). The average thicknesses for the blue, cyan and green emitting NPLs are 1.6 ± 0.1 nm, 2 ± 0.1 nm and 2.6 ± 0.2 nm, respectively (Fig. S2e, ESI†). According to the CH3NH3PbBr3 unit cell size (5.93 Å) and the measured thickness by TEM, the number of unit cell layers was estimated to be n = 3, 4 and 5 for the blue, cyan and green emitting NPLs, respectively. The thickness of these platelets is comparable, or less than the 3D Bohr radius in the CH3NH3PbBr3 perovskites (∼2.2 nm19), below which quantum confinement occurs. Our data on the measured NPL thickness is in excellent agreement with the Bohr radii and with the data previously reported for suspension type CH3NH3PbBr310 NPLs as well as quantum dots.20 Furthermore, the 5 nm NPLs form a self-assembled superstructure on the grid (Fig. S4a, ESI†). The distances between the vertically stacked NPLs (2.82 nm) are related to the overlap of two OAm molecules with a total length of 2.85 nm (Fig. S4b and c, ESI†).
In addition to the electron diffraction analysis (Fig. 2a and b), X-ray diffraction (XRD) was used to determine the crystal structure of all NPLs (Fig. 1e). The blue, cyan and green emitting NPLs were crystalized in the cubic perovskite structure according to the data obtained from a single crystal.21 In contrast to the samples prepared for electron diffraction (on carbon support film), the samples prepared for XRD (on glass substrate) show a rather random orientation of the NPLs, as can be seen from the relative peak intensities in the θ−2θ profile, which nicely correspond to the theoretical values (Fig. 1e). Since the NPLs are highly anisotropic, the Scherrer equation cannot be employed for an independent determination of the platelet dimensions from the observed peak widths. Therefore, concerning the dimension (thickness and lateral size) of the NPLs, we rely on the direct measurement by TEM (Fig. 2a–c, bottom row).
In the case of CH3NH3PbI3 NPLs, only chloroform worked as a re-precipitation medium for thicker NPLs with their PL in the orange spectral range, and toluene for the thinner NPLs with yellow-green emission (Table S2, ESI†). Nanocrystals were formed within a second. A washing process was performed for the red and NIR emitting NPLs only by rapid centrifugation at −10 °C. NPLs with their PL in the green-orange range were destroyed during the centrifugation due to the higher sensitivity and instability of the material compared to CH3NH3PbBr3 NPLs, as well as due to the presence of DMF in the crude solution.
Fig. 3a shows the visual appearance of the pure chloroform and chloroform/toluene solutions of CH3NH3PbI3 NPLs under a UV-365 nm light; the thickness, band gap and emission wavelength were tailored by varying the OA/OAm ratio (Table S2, ESI†) in the same manner as for CH3NH3PbBr3 NPLs. The PL maximum and absorption edge gradually shift to shorter wavelengths, depending on the thickness of the NPLs (Fig. 3c). This phenomenon is demonstrated for the first time experimentally for CH3NH3PbI3 colloids. The PL peaks are in the range of 547–722 nm (optical band gap: 2.5–1.70 eV), being in good agreement with theoretical calculations performed by Sapori et al.18 with the estimated monolayer number n between 1 and 8. The colloidal stability of the NIR emitting NPLs from the crude solution is higher compared to the washed NPLs due to the partial ligand washing after purification. The non-washed NPLs start to agglomerate within one day after preparation. Tailoring the CH3NH3PbI3 NPLs thickness is slightly different compared to CH3NH3PbBr3 NPLs. The ligand-assisted approach in chloroform worked properly in the case of thicker platelets with PL between NIR and orange wavelengths.
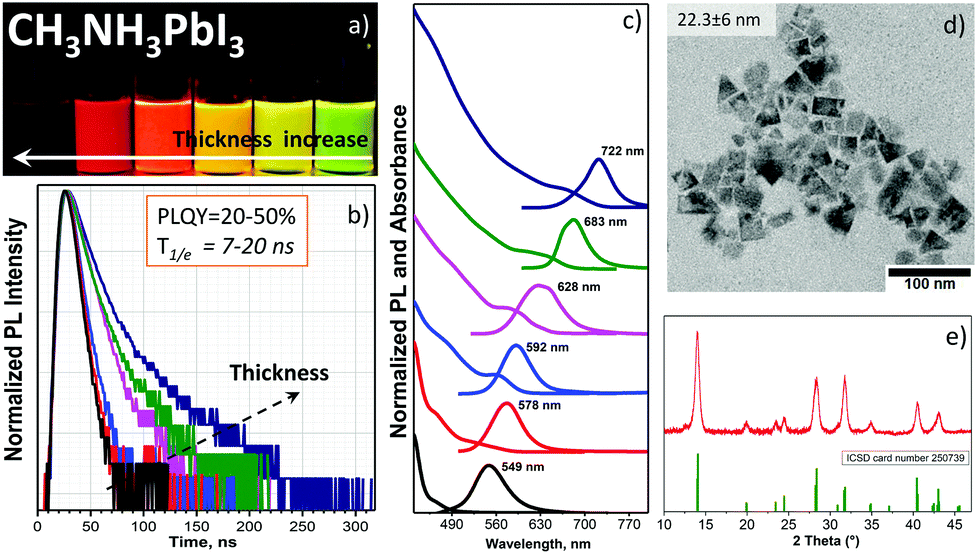 | ||
| Fig. 3 (a) CH3NH3PbI3 NPL solutions under UV-365 nm illumination. (b) PL decay. (c) Absorbance and PL spectra. (d) Typical bright-field TEM image of the NPLs with a PL peak at 722 nm obtained with a ligand ratio of OA/AOm = 200 μl/50 μl. (e) XRD pattern of purified NPLs with a PL of 722 nm (ICSD card No. 250739). | ||
For the thin yellow and green emitting NPLs we used chloroform/toluene or toluene only for precipitation to obtain stable colloids. This phenomenon can be explained by the difference between the dielectric constants of chloroform (ε = 4.81), toluene (ε = 2.38) and DMF (ε = 37.8). In this way, chloroform blends better and faster with DMF if compared to toluene, and forms red and NIR emitting stabilized NPLs only, due to the fast nucleation kinetics of the reagents. In the case of toluene, the reaction kinetics is comparably slow, and this is enough to stabilize the just-formed yellow or green emitting NPLs. The quantum yield of these NPLs lies between 20% and 50% with the highest value observed for the red emitting (683 nm) sample. This is the highest reported value observed for the colloidal CH3NH3PbI3 perovskite according to the best of our knowledge.3,6 Along with the decreasing PLQY in thinner platelets, the photoluminescence decay time τ1/e decreases (Fig. 3b) as well, explained by the enhanced non-radiative relaxation. The rate of the latter increased from 3.5 × 107 s−1 in the platelets with a 722 nm emission peak to 11.3 × 107 s−1 in the platelets with a 546 nm peak wavelength (Fig. S7b and Table S4, ESI†). The minimum thickness and the largest band gap achieved in our experiments are very similar to the quantum size effect for CdTe quantum dots,22,23 where the band gap of the bulk material is almost similar (1.5 vs. 1.48 eV) to the value of the bulk CH3NH3PbI324 (Fig. S5a and b, ESI†).
Structural analysis. NIR-emitting NPLs have a square-like shape in the TEM micrograph (Fig. 3d) with a lateral size of 22.3 ± 6 nm. The size statistics are shown in Fig. S2f (ESI†). Since it was possible to extract only the NIR and red emitting NPLs, the decreased contrast in the red emitting sample confirms its reduced NPL thickness (Fig. S6a and b, ESI†), in agreement with previous reports on perovskite NPLs.10,25 In Fig. S6a (ESI†), we can see square plates as well as circle dots, which are a result of NPL degradation of the last one due to the higher sensitivity of the thinner red emitting NPLs to high voltage electron irradiation26 compared with the NIR emitting NPLs. Importantly, the XRD analysis of the NIR emitting NPL sample demonstrates a pure tetragonal phase of the CH3NH3PbI3 perovskite (the pattern from the structure database (ICSD card No. 250739) is shown in Fig. 3d).
In conclusion, we report on a facile and fast method for the synthesis of colloidal perovskite CH3NH3PbX3 (X = Br and I) NPLs by ligand-assisted re-precipitation, which allows to tailor the nanoplatelet thickness between 1 and 8 unit cell monolayers. The quantum confinement effect was successfully demonstrated using this easy approach yielding high colloidal stability. The remarkable PLQY of CH3NH3PbBr3 (max. 90%) and CH3NH3PbI3 (max. 50%) NPLs and their simple band gap tuning make this method very attractive for the synthesis of perovskite NPLs for optoelectronic applications and printable electronics.
This research was performed as a part of the Energie Campus Nürnberg (EnCN) and supported by funding through the “Bavaria on the Move” initiative of the state of Bavaria and the project 1006-11 of the Bavaria Research Foundation (BFS). We gratefully acknowledge financial support from the Cluster of Excellence “Engineering of Advanced Materials” at the University Erlangen-Nürnberg and from the Federal Ministry for Economic Affairs and Energy (MNPQ program BMWI 11/12). The authors also gratefully acknowledge financial support of Joint Projects Helmholtz-Institute Erlangen Nürnberg (HI-ERN) under project number DBF01253. I. L. thanks César Omar Ramírez Quiroz for help with photography. P. H. acknowledges financial support by the DFG through the research training group 1896 ‘‘In situ microscopy with electrons, X-rays and scanning probes’’. Electron microscopy resources have been kindly provided by the Center for Nanoanalysis and Electron Microscopy (CENEM).
Notes and references
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- L. C. Schmidt, A. Pertegás, S. González-Carrero, O. Malinkiewicz, S. Agouram, G. Mínguez Espallargas, H. J. Bolink, R. E. Galian and J. Pérez-Prieto, J. Am. Chem. Soc., 2014, 136, 850–853 CrossRef CAS PubMed.
- F. Zhang, H. Zhong, C. Chen, X.-G. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- M. F. Aygüler, M. D. Weber, B. M. D. Puscher, D. D. Medina, P. Docampo and R. D. Costa, J. Phys. Chem. C, 2015, 119, 12047–12054 Search PubMed.
- T. C. Jellicoe, J. M. Richter, H. F. J. Glass, M. Tabachnyk, R. Brady, S. E. Dutton, A. Rao, R. H. Friend, D. Credgington, N. C. Greenham and M. L. Böhm, J. Am. Chem. Soc., 2016, 138, 2941–2944 CrossRef CAS PubMed.
- O. Vybornyi, S. Yakunin and M. V. Kovalenko, Nanoscale, 2016, 8, 6278–6283 RSC.
- H. Huang, A. S. Susha, S. V. Kershaw, T. F. Hung and A. L. Rogach, Adv. Sci., 2015, 2, 1500194 CrossRef.
- Q. A. Akkerman, S. G. Motti, A. R. Srimath Kandada, E. Mosconi, V. D'Innocenzo, G. Bertoni, S. Marras, B. A. Kamino, L. Miranda, F. De Angelis, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 1010–1016 CrossRef CAS PubMed.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS PubMed.
- J. A. Sichert, Y. Tong, N. Mutz, M. Vollmer, S. Fischer, K. Z. Milowska, R. García Cortadella, B. Nickel, C. Cardenas-Daw, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6521–6527 CrossRef CAS PubMed.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943–7954 CrossRef CAS PubMed.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS PubMed.
- P. Tyagi, S. M. Arveson and W. A. Tisdale, J. Phys. Chem. Lett., 2015, 6, 1911–1916 CrossRef CAS PubMed.
- Z. Yuan, Y. Shu, Y. Xin and B. Ma, Chem. Commun., 2016, 52, 3887–3890 RSC.
- J. Cho, Y.-H. Choi, T. E. O'Loughlin, L. De Jesus and S. Banerjee, Chem. Mater., 2016, 28, 6909–6916 CrossRef CAS.
- O. Chen, X. Chen, Y. Yang, J. Lynch, H. Wu, J. Zhuang and Y. C. Cao, Angew. Chem., Int. Ed., 2008, 47, 8638–8641 CrossRef CAS PubMed.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun, 2015, 6, 8056 CrossRef CAS PubMed.
- D. Sapori, M. Kepenekian, L. Pedesseau, C. Katan and J. Even, Nanoscale, 2016, 8, 6369–6378 RSC.
- K. Tanaka, T. Takahashi, T. Ban, T. Kondo, K. Uchida and N. Miura, Solid State Commun., 2003, 127, 619–623 CrossRef CAS.
- L. Peng, A. Tang, C. Yang and F. Teng, J. Alloys Compd., 2016, 687, 506–513 CrossRef CAS.
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- A. L. Rogach, T. Franzl, T. A. Klar, J. Feldmann, N. Gaponik, V. Lesnyak, A. Shavel, A. Eychmüller, Y. P. Rakovich and J. F. Donegan, J. Phys. Chem. C, 2007, 111, 14628–14637 CAS.
- G. B. Sukhorukov, A. L. Rogach, M. Garstka, S. Springer, W. J. Parak, A. Muñoz-Javier, O. Kreft, A. G. Skirtach, A. S. Susha, Y. Ramaye, R. Palankar and M. Winterhalter, Small, 2007, 3, 944–955 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- V. A. Hintermayr, A. F. Richter, F. Ehrat, M. Döblinger, W. Vanderlinden, J. A. Sichert, Y. Tong, L. Polavarapu, J. Feldmann and A. S. Urban, Adv. Mater., 2016, 28, 9478–9485 CrossRef CAS PubMed.
- H. Yuan, E. Debroye, K. Janssen, H. Naiki, C. Steuwe, G. Lu, M. Moris, E. Orgiu, H. Uji-i, F. De Schryver, P. Samorì, J. Hofkens and M. Roeffaers, J. Phys. Chem. Lett., 2016, 7, 561–566 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental section with supplementary Fig. S1–S7 and Tables S1–S4. See DOI: 10.1039/c6cc09266g |
| This journal is © The Royal Society of Chemistry 2017 |