
One-step synthesis of ultrathin nanobelts-assembled urchin-like anatase TiO2 nanostructures for highly efficient photocatalysis†
Xin
Yu‡
a,
Zhenhuan
Zhao‡
b,
Jian
Zhang
c,
Weibo
Guo
d,
Linlin
Li
*a,
Hong
Liu
*ae and
Zhong Lin
Wang
*a
aBeijing Institute of Nanoenergy and Nanosystems, Chinese Academy of Sciences, National Center for Nanoscience and Technology (NCNST), Beijing, 100083, P. R. China. E-mail: lilinlin@binn.cas.cn; zlwang@gatech.edu
bInstitute of Fundamental and Frontier Sciences, University of Electronics Science and Technology of China, Chengdu, 610054, P. R. China
cUniversite de lyon, ECL, INSA-Lyon, UCBL, CPE, CNRS, INL, UMR 5270, 36 Avenue Guy de Collongue, 69134 Ecully Cedex, France
dShandong Provincial Key Laboratory of Detection Technology for Tumor Markers, College of Chemistry and Chemical Engineering, Linyi University, Linyi 276005, P. R. China
eState Key Laboratory of Crystal Materials, Shandong University, Jinan, 250100, P. R. China. E-mail: hongliu@sdu.edu.cn
First published on 16th November 2016
Abstract
Nanostructured TiO2 materials with a controlled morphology and structure have drawn considerable attention to both fundamental research and practical applications owing to their unique characteristics. Herein, a novel, facile, and one-step hydrothermal approach was developed to synthesize urchin-like anatase TiO2 hierarchical nanostructures assembled from ultrathin nanobelts using urea as the morphology-directing agent. The effects of the urea concentration in the preparation process were discussed intensively. Photocatalytic experiments showed that the urchin-like anatase TiO2 nanostructures possessed a much higher degradation rate of methyl orange and phenol than the most successful commercial semiconductor photocatalyst P25. The reasons for the highly efficient photocatalytic activity was ascribed to the high specific surface area (171 m2 g−1) and ultrathin 1D nanobelts of anatase TiO2 self-assembled into the urchin-like hollow spheres. The urchin-like anatase TiO2 nanostructures as photocatalysts have potential applications in environmental and energy fields for photocatalytic degradation, hydrogen production, Li-ion batteries, and dye-sensitized solar cells. In addition, new hydrothermal method can be developed for synthesis of other hierarchical nanostructures.
Introduction
Semiconductor-based photocatalysis for pollutant degradation and hydrogen generation by water splitting has been considered as one of the most crucial approaches to solve the world's energy and environmental crisis.1–3 Titanium dioxide (TiO2) has been investigated extensively in several applications owing to its unique characteristics, including environmental benignancy, safety and stability.4,5 TiO2 possesses a wide bandgap of about 3.2 eV and thus possesses excellent photocatalytic activity under UV light illumination.6–8 It has been commercialized in many industrial fields and daily life, e.g., it is used as a white pigment in paints and as a UV absorber in sunscreens.9–11It has been generally accepted that the geometric morphology has tremendous effects on the physicochemical properties of TiO2.12,13 Controlling the morphology and size of TiO2 nanostructures has proven to be crucial for obtaining superior photocatalytic, photovoltaic, and electrochemical performance.14–18 Substantial research on the preparation, characterization, and fundamental understanding has boosted the utilization of TiO2 nanomaterials with outstanding performance in the past few decades.19–22 Synthesis methods for nanostructured TiO2 materials from zero to three-dimensional (3D) structures have been reported.23–27 In particular, one-dimensional (1D) nanostructures are of great importance owing to the particular exposed facets and anisotropic structures.28,29 A multitude of 1D TiO2 nanostructures, such as nanowires,30,31 nanorods,32,33 nanotubes34–36 and nanobelts,37 have been widely reported with enhanced performance in photocatalysis, dye-sensitized solar cells, and lithium-ion batteries.38–40
Several studies have reported the synthesis and properties of nanostructured anatase TiO2. For example, Lou's group developed a facile one-pot solvothermal method to synthesize anatase TiO2 nanowires with high productivity and yield.41 Wu's group developed W, N co-doped and Ni doped TiO2 nanobelts with high photocatalytic activity.42,43 A recent study by Zhao's group synthesized uniform 3D open macro/mesoporous TiO2 hollow microspheres with highly crystalline anatase thin shells by a simple solvent evaporation-driven confined self-assembly method.44 Chen and co-workers synthesized ordered TiO2 mesophyll cell-like microspheres with a prolonged electron lifetime for high performance photocatalytic water reduction and oxidation.45 These studies showed that high specific surface areas and 1D nanostructures with anatase crystals can improve the photocatalytic activity. Recently, remarkable efforts have been devoted to the synthesis of hollow structured TiO2,2,4,24 particularly yolk–shell structures,5,46 through non-templating hydrothermal approaches. However, it is still difficult to fabricate TiO2 nanomaterials with all these properties via a facile method.
To date, a few synthetic methods on the formation of TiO2 nanostructures have been developed via the hydrolysis of K2TiO(C2O4)2 under various conditions. For example, hierarchical TiO2 nanowires with anatase nanorod branches were obtained via hydrothermal treatment of K2TiO(C2O4)2 in diethylene glycol and H2O solution at 180 °C.47 Rutile TiO2 mesocrystals were achieved by a controlled dissolution and precipitation procedure in a HNO3, C3H6N6 and H2O2 aqueous solution at a temperature of 80 °C.21 Hierarchical nanosheet-assembled yolk–shell anatase TiO2 microspheres were synthesized by the slow hydrolysis of K2TiO(C2O4)2 in a mixed solution of H2O2 and HNO3, at temperatures as low as 40 °C.46
In this study, we propose a one-step strategic hydrothermal method for the synthesis of 3D biomimic urchin-like anatase TiO2 nanostructure, which is assembled from 1D TiO2 ultrathin nanobelts. Enhanced photodegradation performance was obtained due to the large surface area and ultrathin 1D nanobelt-assembled 3D urchin-like structures. Moreover, the urchin-like TiO2 structure is advantageous for facilitating electrolyte infiltration and promoting electron transportation.
Results and discussion
The morphology and microstructure of the 3D urchin-like TiO2 nanostructures were characterized by scanning electron microscopy (SEM) and high resolution transmission electron microscopy (HRTEM). With 1 g urea in the reaction system, the as-synthesized TiO2 nanostructures had a typical bionic urchin-like morphology assembled from several ultrathin nanobelts (Fig. 1a). From a broken sphere in a Fig. 1a arrow, it was found that the TiO2 nanostructures had hierarchical hollow structures. The diameter of the hollow sphere was about 1 μm. From the high magnification image (Fig. 1b), the hollow sphere was composed of radially assembled ultrathin nanobelts with a width of about 4–8 nm and length of about 200–300 nm. The inner wall of the hollow sphere was very compact, forming an urchin-like hollow structure. The TEM image in Fig. 1c shows that the hollow interior of the urchin-like TiO2 structure was about 300–400 nm. Moreover, the selected area electronic diffraction (SAED) pattern of the urchin-like TiO2 shows the appearance of a series of rings corresponding to the diffraction from the (101), (004), (200), (105), (204), (220), (215) and (224) planes, indicating the formation of anatase TiO2.48 The enlarged TEM images in Fig. 1e and S1† also confirmed that the shell of the urchin-like TiO2 nanostructures was composed of ultrathin nanobelts. In Fig. 1f, the high-resolution TEM (HRTEM) images showed the perfect continuous two-dimensional atomic lattices with a spacing of 0.35 nm, corresponding to the (101) planes of anatase TiO2. The urchin-like TiO2 structures assembled with numerous ultrathin nanobelts exposed more surface area, and the particular exposed facets and reactive sites may be advantageous for facilitating electrolyte infiltration and promoting electron transportation. | ||
| Fig. 1 (a and b) SEM images of urchin-like anatase TiO2 nanostructures, (c) TEM image of the urchin-like TiO2 nanostructure, (d) corresponding SAED pattern, (e and f) HRTEM image of TiO2 nanostructures. | ||
The crystallographic structure of the urchin-like TiO2 nanostructures was confirmed by powder X-ray diffraction (XRD). As shown in Fig. 2a, all diffraction peaks were assigned unambiguously to anatase TiO2 (JCPDS card no. 73-1764).49 This result was consistent with the SAED patterns in Fig. 1d. The chemical composition and valence state of urchin-like TiO2 nanostructures were characterized by X-ray photoelectron spectroscopy (XPS). The full scanned spectrum of the urchin-like anatase TiO2 in the range of 0–900 eV is shown in Fig. S2.† This overview spectrum demonstrates that C, Ti, O, and N exist in the nanostructures. C was from the system, and can be used to normalize the peaks. No other impurity peaks other than carbon's peak were found in the spectra. The X-ray photoelectron spectrum of Ti 2p can be deconvoluted into two peaks centered at 463.75 eV (Ti 2p 1/2) and 458.05 eV (Ti 2p 3/2), which were both assigned to the Ti4+ oxidation state.29,50 The O 1s XPS spectrum (Fig. 2c) showed two chemical states of oxygen. The sharp peak at 529.25 eV was assigned to the Ti–O bond, indicating the main lattice oxygen atoms. The other peak at 531.15 eV was assigned to O–H bonds from the interaction of the urchin-like TiO2 nanostructures shells with an ambient environment.51 Furthermore, from the XPS spectra, weak N 1s peaks at 399.65 eV and 395.05 eV related to N–H were found (Fig. 2d).52 This may be caused by urea during the surface adsorption in the reactants. However, in the XRD pattern, no peaks belonging to nitrogen containing compounds or other titanic polymorphs can be found. Furthermore, there was no evident shift of the XRD peaks indexed to the anatase phase. Thus, the crystallinity of the anatase TiO2 is not affected by the presence of nitrogen.
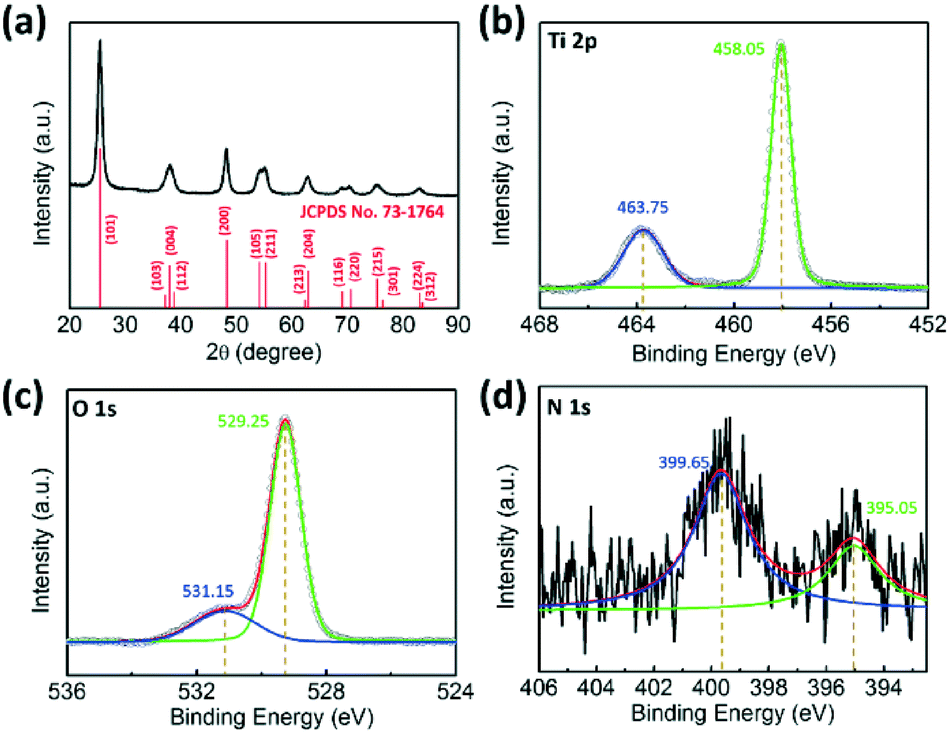 | ||
| Fig. 2 (a) XRD pattern and XPS spectra of (b) Ti 2p (c) O 1s (d) N 1s for the urchin-like anatase TiO2 nanostructures. | ||
To understand the formation mechanism of such a hollow sphere with a hierarchical structure, experiments under different urea amounts were conducted, which indicated that the amount of urea was crucial in controlling the morphology of the products. Fig. 3 presents SEM images of the TiO2 nanostructures obtained without and with different amounts of urea. As shown in Fig. 3a, TiO2 nanobelt crystals were obtained without urea in the synthetic system. The length of the nanobelts is up to several micrometers. The width and thickness of the nanobelts are in the range 50–100 nm and 20–30 nm, respectively. When the urea amount was 0.1 g, a spinous rod-like structure (several micrometers in length and about 200 nm in diameter) was formed (Fig. 3b). Enlarged images in Fig. S3† reveal that the ultrathin nanobelts were attached to each other at the end. When the synthesis was performed with 0.5 g of urea, the ultrathin nanobelts assembled together to form a ball with the nanobelts reaching out to the outer surface (Fig. 3c). The TiO2 sphere possessed a solid core (Fig. S4†). When the amount of urea further increases to 1.0 g, as shown in Fig. 3d, well-developed urchin-like nanostructures with a hollow interior were formed by self-assembly from the ultrathin nanobelts.
 | ||
| Fig. 3 SEM images of TiO2 sample with different synthesis conditions. (a) Without urea, (b) with 0.1 g urea, (c) with 0.5 g urea and (d) with 1 g urea. The insets in the SEM images show the corresponding morphology sketches. | ||
To investigate the growth kinetics of urchin-like TiO2 nanostructures, the time-dependent morphology evolution was monitored. In a short time of 30 min, nanoparticles with an irregular morphology and broad size distribution from 50 nm to 200 nm were observed (Fig. 4a). A hollow structure was found from a broken sphere, which consisted of several small nanoparticles (Fig. 4a insert). At 40 min (Fig. 4b), the nanospheres grew up into larger particles with a diameter of about 200 nm, still with a hollow structure. As the reaction time was prolonged further to 1 h, two types of spheres were co-existent in the reaction; the smaller one, similar to that obtained in 40 min, had a relatively smooth surface with a particle size of 100 nm. The larger urchin-like sphere began to appear with a diameter of about 500 nm. A few ultrathin nanobelts, 200–300 nm in length, formed starting from the surface of the urchin-like nanospheres, forming a hierarchical structure (Fig. 4c). When the hydrothermal reaction time was prolonged to 2 h, well-developed urchin-like TiO2 nanostructures were uniformly formed (Fig. 4d).
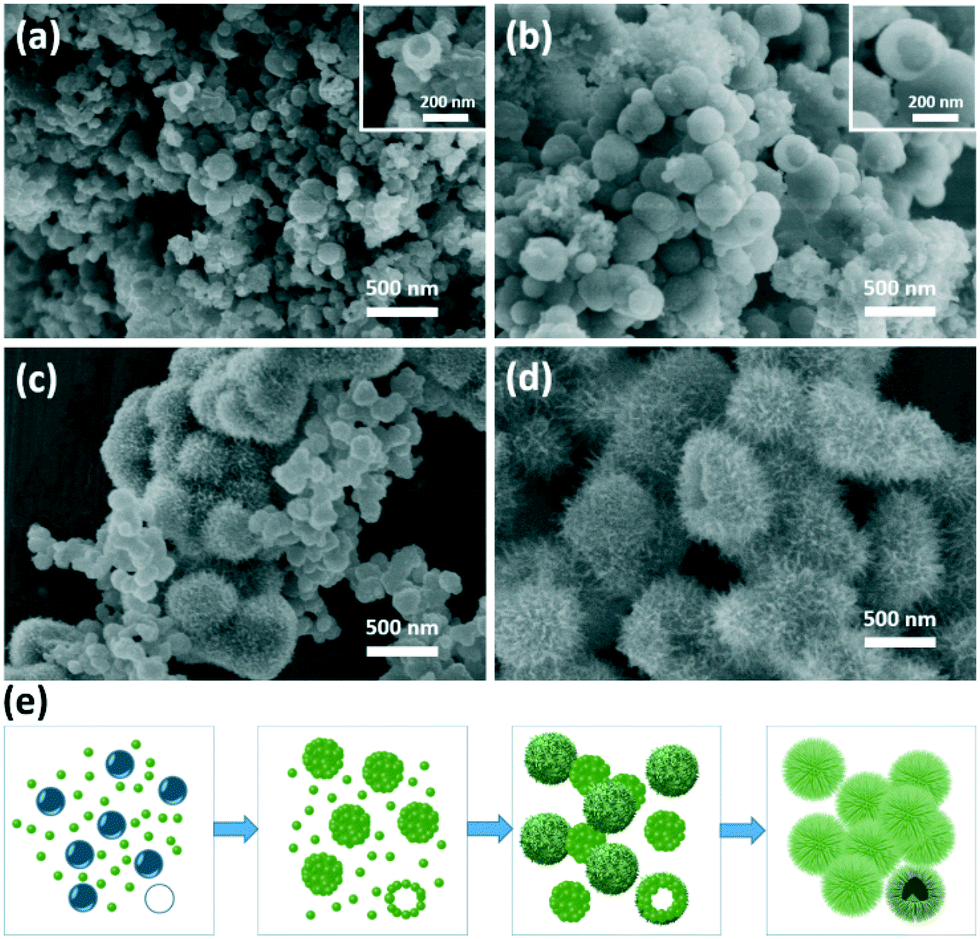 | ||
| Fig. 4 SEM images of urchin-like TiO2 nanostructures prepared at different reaction time intervals (a) 30 min, (b) 40 min, (c) 1 h, (d) 2 h at 180 °C with 1.0 g urea. (e) Schematic of the formation mechanism of urchin-like microspheres. | ||
According to the abovementioned results, urea played a key role in the formation of the urchin-like microsphere. The underlying mechanism for the formation of urchin-like TiO2 nanostructures was proposed, as shown in Fig. 4e. Urea was unstable and could decompose to NH3 and CO2 at temperatures higher than 90 °C.53 At a high hydrothermal reaction of 180 °C, urea that was introduced into the reaction rapidly decomposed. The decomposition product of NH3 acted as a basic catalyst to accelerate the formation of TiO2 seeds with burst-nucleation. In addition, Fig. S6† shows that when NH4OH was used instead of urea only TiO2 nanobelts formed, but the nanobelts agglomerated into blocks. Simultaneously, the generated gas bubbles in the aqueous solution acted as soft templates for providing aggregation centers of TiO2 seeds to form hollow structures (Fig. 4a). As time progressed, TiO2 grew further along the TiO2 seed and gradually assembled into TiO2 nanobelts. Finally, urchin-like hierarchical nanostructures were generated (Fig. 4d). In the process, diethylene glycol also assisted in nanobelt growth as “cosurfactant” and “cosolvent”.47,54 Without or with less diethylene glycol, the TiO2 nanoparticle agglomerated into blocks (Fig. S5†). At low urea concentrations (0.1 g), the gas bubble was not enough to support the formation of the hollow spherical structures. The co-influence of basic condition and diethylene glycol controlled the morphology of TiO2 nanorods with spinous rod-like structure (Fig. 3b). In conventional hydrothermal reaction without urea, it tended to form a nanobelt structure according to the crystal habit of TiO2 (Fig. 3a). In this study, TiO2 possessed an anatase structure. According to the symmetry of the crystal structure, the [010] crystal orientation grows fast and [101] crystal orientation grows slow. Therefore, it is easy to form the nanobelt structure. In the urea-assisted new hydrothermal method, the morphology of TiO2 could be controlled easily by varying the urea concentration and hydrothermal reaction time. An increase in the concentration of urea can improve the product yield and reduce the reaction time. From Table S1,† the TiO2 yield with 1.0 g urea and a 4 h hydrothermal reaction is 5 times of that without urea.
1D semiconductor ultrathin nanobelts have superior electron transport and light scattering ability compared to nanoparticles.55 In addition, the photocatalytic performance of anatase TiO2 is better than that of the rutile ones. Herein, we demonstrated the potential application of the hierarchical urchin-like anatase TiO2 nanostructures composed of nanobelts as an excellent photocatalyst for the degradation of methyl orange (MO). The result in Fig. S7† indicates that the degradation effect can be ruled out for the system containing photocatalyst without light irradiation, as well as the system with light irradiation without a photocatalyst. Fig. 5a shows the photocatalytic activity of TiO2 nanobelts synthesized with 0.1 g urea, urchin-like anatase TiO2 nanostructures synthesized with 1.0 g urea, commercial anatase TiO2 (A-TiO2) and Degussa P25 nanoparticles for comparison (Fig. S10b and c†). Under the 365 nm UV irradiation in a short period of 30 min, the urchin-like TiO2 could degrade over 95% of the original organic dye, whereas the degradation rate for the TiO2 nanobelt, A-TiO2 and P25 over the same time period was only about 35%, 50%, and 75%, respectively. When they were applied to degrade phenol, 10% phenol was retained in water after 120 min of UV illumination for the urchin-like TiO2, whereas the phenol remaining for the TiO2 nanobelt, A-TiO2 and P25 was about 70%, 50%, and 20%, respectively (Fig. 5b). P25 is thought as the most successful commercial semiconductor photocatalyst because of its high photocatalytic performance derived from its large specific surface area and anatase–rutile bi-phase structure. It was encouraging that the photocatalytic activity of urchin-like TiO2 nanostructures was much higher than that of P25. Furthermore, compared to other TiO2 samples, with different morphologies synthesized with different amounts of urea, the urchin-like TiO2 was still the best, as depicted in the Fig. S8.† To examine the photocatalytic stability and recyclability of the catalysts, the efficiency of the MO photodegradation was assessed with a repetitive mode by collecting the photocatalyst in the reaction solution and repeating the photocatalysis assessment. As shown in Fig. 5c and d, the MO dye and phenol was quickly decomposed after each injection of the MO or phenol solution and the photodecomposition rate was nearly constant after six repeated experiments. In addition, the urchin-like TiO2 can be easily separated from the aqueous solution by sedimentation for reuse, probably due to the large length-to-diameter ratio of the one-dimensional ultrathin nanobelt on the shell of the urchin-like TiO2 (Fig. S9†). In contrast, the Degussa P25 was extremely difficult to be collected and could only be separated by high-speed centrifugation, a high energy-assumption collection process. Thus, the urchin-like TiO2 was an effective and stable photocatalyst.
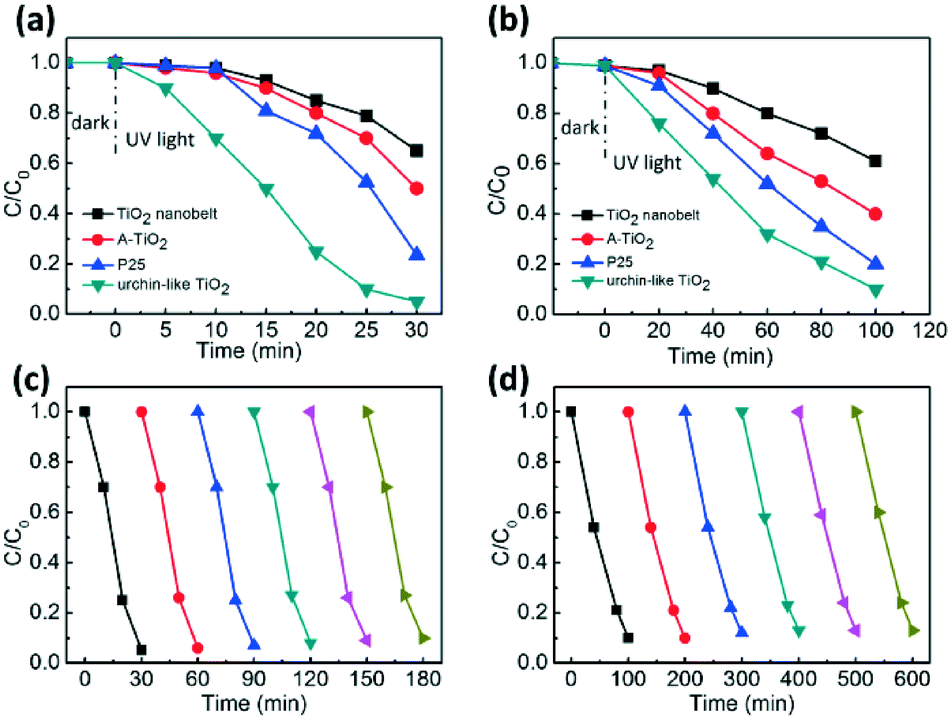 | ||
| Fig. 5 Photocatalytic degradation of (a) MO and (b) phenol with TiO2 nanobelt, commercial A-TiO2, P25 and urchin-like TiO2. Photocatalytic degradation of (c) MO and (d) phenol by the TiO2 hierarchical hybrid nanostructure in repeated experiments. | ||
The high photocatalytic performance of the urchin-like anatase TiO2 might arise for the following reasons. First, anatase itself has higher photocatalytic activity than other crystalline phases. Second, urchin-like anatase TiO2 has unique nanostructure composed of several single crystal ultrathin nanobelts. The single crystalline structure indicates fewer defects with fast charge transportation. In addition, the unique 1D nanostructure promotes charge separation in the nanocrystals. Third, the urchin-like anatase TiO2 has a small grain size because of the ultrathin nanobelts. The average crystallite size of the urchin-like anatase TiO2 was calculated to be 17 nm from 2θ = 25.3° in the XRD pattern, whereas TiO2 nanobelt, A-TiO2 and P25 samples showed a relatively larger half peak width with a diameter of 92 nm, 35 nm, and 39 nm, respectively. In addition, the hierarchical and hollow structure endows a large surface area to the material. Generally, when the catalyst size is smaller, there is a more per unit mass of catalyst particles. In addition, higher specific surface area induces more efficient surface contact to the pollutant, promoting the efficiency of photocatalytic degradation of the organic matter. According to the SEM images of the different samples (Fig. S10†) and the Debye–Scherrer formula (Fig. S11†), it can be concluded that the urchin-like TiO2 has the smallest catalyst size.
Optical properties of the TiO2 samples were investigated by diffuse reflectance ultraviolet-visible spectroscopy (UV-vis spectra) and the results are shown in Fig. 6a. Urchin-like anatase TiO2 showed a band-edge absorption around 395 nm, typical for anatase TiO2 with a band-gap of 3.15 eV. In contrast, P25 showed evident light absorption up to 410 nm, which should have originated from the heterogeneous junction between anatase and rutile, significantly reducing the absorption in visible light region.56
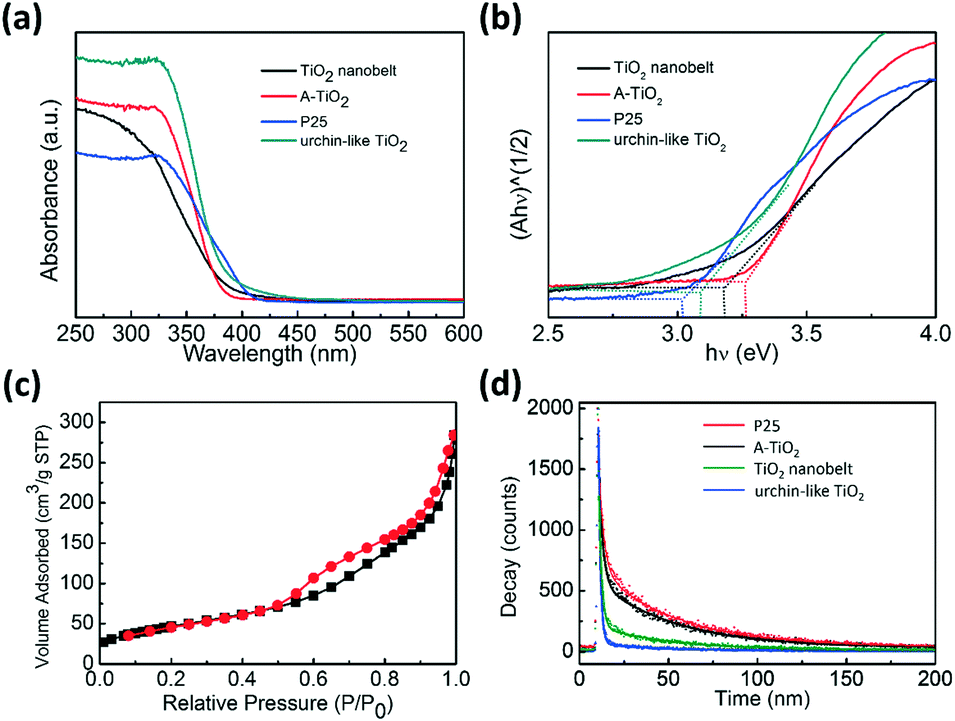 | ||
| Fig. 6 (a) UV-vis diffuse reflectance spectra of the TiO2 nanobelt, commercial A-TiO2, P25, and urchin-like anatase TiO2. (b) Kubelka–Munk plot derived from diffuse reflectance UV-vis spectroscopy experiments. (c) Nitrogen adsorption–desorption isotherms of the urchin-like anatase TiO2. (d) Time-resolved fluorescence decay of the samples at 450 nm of the samples exited by a 375 nm laser. | ||
The Kubelka–Munk function based on the diffuse reflectance spectra is employed to determine the band gap.57,58 For the direct band gap semiconductor, the relationship between the absorption coefficient (A) and photon energy (hν) can be written as
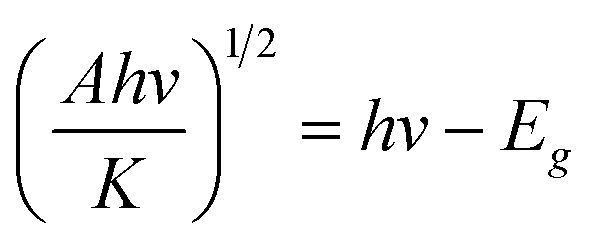
The excellent photocatalytic degradation of MO with urchin-like anatase TiO2 can also be attributed partially to their large specific area. Nitrogen adsorption–desorption measurements were performed to obtain the Brunauer–Emmett–Teller (BET) specific surface area. The result is shown in Fig. 6c, and all the samples' nitrogen adsorption–desorption curve and BET specific surface area are shown in Fig. S12 and Table S2.† The BET surface area of the urchin-like anatase TiO2 was the highest approximately 171 m2 g−1, which is 3.4 times of the P25.
Photoluminescence spectra are usually used to explore the efficiency of the charge carrier trapping, immigration, and transfer. In addition, it is quite helpful to understand the fate of the electron–hole pairs in semiconductor particles because photoluminescence emission is thought to have arisen from the recombination of photo-induced carriers.59 When electron and hole recombination occurs, it emits fluorescence. Therefore, the low fluorescent emission intensity indicates a low electron–hole recombination rate, which causes high photocatalytic activity of the semiconductor.60,61Fig. 6d shows the time-resolved fluorescence decay. The derived two components of the lifetime and their relative amplitudes are given in Table 1. An evident decrease in the photoluminescence lifetime of the urchin-like TiO2 (33.75 ns) was observed compared to that of TiO2 nanobelt (40.74 ns), P25 (43.76 ns) and the commercial A-TiO2 (43.72 ns), indicating that the urchin-like anatase TiO2 effectively diminished the recombination of photoinduced electron–hole pairs, which was beneficial to the photocatalytic activity.
| Sample | Decay time [ns] | Relative amplitude [%] | Average lifetime [ns] | ||
|---|---|---|---|---|---|
| τ 1 | τ 2 | f 1 | f 1 | τ | |
| a The average lifetime was calculated using equation: <τ> = (f1τ12 + f2τ22)/(f1τ1 + f2τ2). | |||||
| P25 | 2.32 | 44.02 | 10.80 | 89.20 | 43.76 |
| A-TiO2 | 2.76 | 44.51 | 22.33 | 77.67 | 43.72 |
| TiO2 nanobelt | 1.63 | 40.92 | 10.90 | 89.1 | 40.74 |
| Urchin-like TiO2 | 1.17 | 28.28 | 55.21 | 44.79 | 33.75 |
Experimental
Materials
All the reagents were of analytic grade and commercially available. Diethylene glycol (DEG), titanium oxalate K2TiO(C2O4)2, urea (H2NCONH2), and methyl orange were purchased from China National Medicines Corporation Ltd. The control titania P25 (TiO2; ca. 80% anatase, and 20% rutile) and anatase TiO2 (A-TiO2) were purchased from Sigma-Aldrich. All the chemicals were used as received without further purification.Synthesis
In a typical experiment for the synthesis of urchin-like TiO2, 0.35 g K2TiO(C2O4)2 and 1 g urea were added to a mixture of 10 mL of deionized water and 30 mL of diethylene glycol. After stirring vigorously for 1 h, the solution was transferred to a 50 mL Teflon-lined stainless steel autoclave, which was heated to 180 °C and maintained for 12 h. The autoclave was then cooled to room temperature. The obtained products were washed with deionized water and ethanol to remove ionic residue and then dried in an oven at 80 °C for 4 h. By varying the amount of urea, TiO2 with other morphologies were fabricated.Characterization
The XRD patterns of the samples were obtained by X-ray diffraction spectrometer (D8-advance, BrukerAXS, Germany) using Cu Kα radiation. The accelerating voltage and applied current were 40 kV and 40 mA, respectively. X-ray photoelectron spectroscopy (XPS) was conducted on an ESCALAB 250 photoelectron spectrometer (Thermo Fisher Scientific) at 2.4 × 10−10 mbar using a monochromatic Al Kα X-ray beam (1486.60 eV). All binding energies were referenced to the C 1s peak (284.60 eV) arising from adventitious hydrocarbons. The morphology and microstructure of the samples were examined by SEM (HITACHI S-8020). The TEM images were acquired on a JEOL JEM 2100 microscope with an operating voltage of 200 kV. The UV-vis diffuse reflectance spectra of the samples were recorded on a UV-vis spectrophotometer (UV-3600, Shimadzu) with an integrating sphere attachment within the range from 250 to 600 nm and with BaSO4 as the reflectance standard. The specific surface area was measured on Micromeritics, ASAP2020 and calculated using the Brunauer–Emmett–Teller (BET) method. The time-resolved fluorescence decay curve was taken out with combined steady state and time resolved fluorescence spectrometer (FLS980, Edinburgh).Photocatalysis
The photocatalytic activity was assessed by the photodegradation of methyl orange (MO) and phenol on a photochemical reaction apparatus. In a typical experiment, 20 mL aqueous suspensions of MO (20 mg L−1) or phenol (20 mg L−1) and 20 mg of the TiO2 sample were placed in a 50 mL beaker. Prior to irradiation, the suspensions were magnetically stirred in the dark for 30 min at room temperature to establish adsorption/desorption equilibrium between the dye and the surface of the catalyst. A 300 W mercury lamp with a maximum emission of 365 nm was used as the UV light source. For comparison, the photodegradation abilities of the other TiO2 control samples were evaluated under the same experimental conditions. At pre-determined irradiation time intervals, the residual MO or phenol concentration in the supernatant was analyzed by UV-vis spectroscopy (Shimadzu UV-3600). The normalized concentration of the solution equalled the normalized maximum absorbance, and hence we used C0/C in place of A0/A, where C0 and C are the initial and actual concentration of MO or phenol, respectively.Conclusions
We successfully synthesized the 3D urchin-like anatase TiO2 nanostructures by a one-step urea-assisted hydrothermal approach with urea as the morphology directing agent. The urea concentration plays a crucial role in determining the morphology and crystallite size. Compared to commercial anatase TiO2 and Degussa P25, the as-prepared urchin-like anatase TiO2 show the largest specific surface area, which enhances the photocatalytic activity for degradation of MO and phenol. The present study motivates us to use the new facile hydrothermal method for the preparation of new nanomaterials with a special morphology, which could be employed not only in photocatalysis, but also in solar water splitting systems and solar energy conversion systems, such as dye-sensitized solar cells with high efficiency.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 51372142, No. 81471784, No. 31270022, and No. 51402063), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (2015023), and the “Thousands Talents” program for pioneer researcher and his innovation team, China.Notes and references
- P. Kubiak, T. Fröschl, N. Hüsing, U. Hörmann, U. Kaiser, R. Schiller, C. K. Weiss, K. Landfester and M. Wohlfahrt-Mehrens, Small, 2011, 7, 1690–1696 Search PubMed.
- J. M. Wu, X. M. Song and M. Yan, J. Hazard. Mater., 2011, 194, 338–344 Search PubMed.
- D. Zhong, B. Cai, X. Wang, Z. Yang, Y. Xing, S. Miao, W.-H. Zhang and C. Li, Nano Energy, 2015, 11, 409–418 CrossRef CAS.
- J. M. Wu, X. M. Song, L. Y. Ma and X. D. Wei, J. Cryst. Growth, 2011, 319, 57–63 Search PubMed.
- L. L. Dai and J. M. Wu, Ceram. Int., 2015, 41, 12317–12322 Search PubMed.
- X. Yu, X. Han, Z. Zhao, J. Zhang, W. Guo, C. Pan, A. Li, H. Liu and Z. L. Wang, Nano Energy, 2015, 11, 19–27 Search PubMed.
- J. Chen, H. B. Yang, H. B. Tao, L. Zhang, J. Miao, H. Y. Wang, J. Chen, H. Zhang and B. Liu, Adv. Funct. Mater., 2016, 26, 456–465 Search PubMed.
- J. S. Chen, J. Liu, S. Z. Qiao, R. Xu and X. W. D. Lou, Chem. Commun., 2011, 47, 10443–10445 Search PubMed.
- H. Xu, S. Ouyang, L. Liu, P. Reunchan, N. Umezawa and J. Ye, J. Mater. Chem. A, 2014, 2, 12642–12661 CAS.
- S. A. Arabi, J. Dong, M. Mirza, P. Yu, L. Wang, J. He and C. Jiang, Cryst. Growth Des., 2016, 16, 2624–2630 Search PubMed.
- X. Wang, R. Long, D. Liu, D. Yang, C. Wang and Y. Xiong, Nano Energy, 2016, 24, 87–93 Search PubMed.
- Y. Liu, A. A. Elzatahry, W. Luo, K. Lan, P. Zhang, J. Fan, Y. Wei, C. Wang, Y. Deng and G. Zheng, Nano Energy, 2016, 25, 80–90 Search PubMed.
- Y. Yu, J. Li, D. Geng, J. Wang, L. Zhang, T. L. Andrew, M. S. Arnold and X. Wang, ACS Nano, 2015, 9, 564–572 CrossRef CAS PubMed.
- X. Yu, Z. Zhao, J. Zhang, W. Guo, J. Qiu, D. Li, Z. Li, X. Mou, L. Li and A. Li, Small, 2016, 12, 2759–2767 CrossRef CAS PubMed.
- M. Zhao, H. Xu, H. Chen, S. Ouyang, N. Umezawa, D. Wang and J. Ye, J. Mater. Chem. A, 2015, 3, 2331–2337 Search PubMed.
- Y. Xie, X. Zhang, P. Ma, Z. Wu and L. Piao, Nano Res., 2015, 8, 2092–2101 Search PubMed.
- Z. Sun, J. H. Kim, Y. Zhao, F. Bijarbooneh, V. Malgras, Y. Lee, Y.-M. Kang and S. X. Dou, J. Am. Chem. Soc., 2011, 133, 19314–19317 CrossRef CAS PubMed.
- J. H. Pan, X. Z. Wang, Q. Huang, C. Shen, Z. Y. Koh, Q. Wang, A. Engel and D. W. Bahnemann, Adv. Funct. Mater., 2014, 24, 95–104 Search PubMed.
- M. Ge, C. Cao, J. Huang, S. Li, Z. Chen, K.-Q. Zhang, S. Al-Deyab and Y. Lai, J. Mater. Chem. A, 2016, 4, 6772–6801 Search PubMed.
- W. Wen, J. M. Wu, Y. Z. Jiang, J. Q. Bai and L. L. Lai, J. Mater. Chem. A, 2016, 4, 10593–10600 Search PubMed.
- L. L. Lai, L. L. Huang and J. M. Wu, RSC Adv., 2014, 4, 49280–49286 Search PubMed.
- W. Q. Wu, H. S. Rao, Y. F. Xu, Y. F. Wang, C. Y. Su and D. B. Kuang, Sci. Rep., 2013, 3, 1892 Search PubMed.
- L. Li, S. Zhou, E. Chen, R. Qiao, Y. Zhong, Y. Zhang and Z. Li, J. Mater. Chem. A, 2015, 3, 2234–2241 Search PubMed.
- Z. Zheng, B. Huang, X. Qin, X. Zhang and Y. Dai, Chem. – Eur. J., 2010, 16, 11266–11270 Search PubMed.
- C. Cheng, H. Zhang, W. Ren, W. Dong and Y. Sun, Nano Energy, 2013, 2, 779–786 Search PubMed.
- B. Liu, L. M. Liu, X. F. Lang, H. Y. Wang, X. W. D. Lou and E. S. Aydil, Energy Environ. Sci., 2014, 7, 2592–2697 CAS.
- Y. Mao, M. Kanungo, T. H. Benny and S. S. Wong, J. Phys. Chem. B, 2006, 110, 702–710 Search PubMed.
- J. Tian, Z. Zhao, A. Kumar, R. I. Boughton and H. Liu, Chem. Soc. Rev., 2014, 43, 6920–6937 Search PubMed.
- L. Zheng, S. Han, H. Liu, P. Yu and X. Fang, Small, 2016, 12, 1527–1536 CrossRef CAS PubMed.
- Y. J. Hwang, C. Hahn, B. Liu and P. Yang, ACS Nano, 2012, 6, 5060–5069 Search PubMed.
- B. Li, J. M. Wu, T. T. Guo, M. Z. Tang and W. Wen, Nanoscale, 2014, 6, 3046–3050 Search PubMed.
- W. Wang, J. Dong, X. Ye, Y. Li, Y. Ma and L. Qi, Small, 2016, 12, 1469–1478 Search PubMed.
- J. Zhang, X. Yu, W. Guo, J. Qiu, X. Mou, A. Li and H. Liu, Nanoscale, 2016, 8, 9382–9389 Search PubMed.
- X. Yu, L. Wang, J. Zhang, W. Guo, Z. Zhao, Y. Qin, X. Mou, A. Li and H. Liu, J. Mater. Chem. A, 2015, 3, 19129–19136 Search PubMed.
- L. Yu, Z. Wang, L. Zhang, H. B. Wu and X. W. D. Lou, J. Mater. Chem. A, 2013, 1, 122–127 Search PubMed.
- V. Zhou, N. Haublein, N. Liu, T. Nguyen, E. M. Zolnhofer, H. Tsuchiya, M. S. Killian, K. Meyer, L. Frey and P. Schmuki, Angew. Chem., 2016, 55, 3763–3767 Search PubMed.
- Z. Zhao, J. Tian, Y. Sang, A. Cabot and H. Liu, Adv. Mater., 2015, 27, 2557–2582 Search PubMed.
- H. Liu, W. Li, D. Shen, D. Zhao and G. Wang, J. Am. Chem. Soc., 2015, 137, 13161–13166 Search PubMed.
- T. Zhao, W. Luo, Y. Deng, Y. Luo, P. Xu, Y. Liu, L. Wang, Y. Ren and W. Jiang, Nano Energy, 2016, 26, 16–25 Search PubMed.
- G. Lui, G. Li, X. Wang, G. Jiang, E. Lin, M. Fowler, A. Yu and Z. Chen, Nano Energy, 2016, 24, 72–77 Search PubMed.
- H. B. Wu, H. H. Hng and X. W. D. Lou, Adv. Mater., 2012, 24, 2567–2571 Search PubMed.
- J. Q. Bai, W. Wen and J. M. Wu, CrystEngComm, 2016, 18, 1847–1853 Search PubMed.
- L. L. Lai and J. M. Wu, J. Mater. Chem. A, 2015, 3, 15863–15868 Search PubMed.
- Y. Liu, K. Lan, A. A. Bagabas, P. Zhang, W. Gao, J. Wang, Z. Sun, J. Fan, A. A. Elzatahry and D. Zhao, Small, 2016, 12, 860–867 Search PubMed.
- Y. Zhang, B. Wu, Y. Tang, D. Qi, N. Wang, X. Wang, X. Ma, T. C. Sum and X. Chen, Small, 2016, 12, 2291–2299 Search PubMed.
- L. L. Lai, W. Wen and J. M. Wu, CrystEngComm, 2016, 18, 5195–5201 Search PubMed.
- W.-Q. Wu, B.-X. Lei, H.-S. Rao, Y.-F. Xu, Y.-F. Wang, C.-Y. Su and D.-B. Kuang, Sci. Rep., 2013, 3, 1352 Search PubMed.
- Y. Chimupala, P. Junploy, T. Hardcastle, A. Westwood, A. Scott, B. Johnson and R. Brydson, J. Mater. Chem. A, 2016, 4, 5685–5699 Search PubMed.
- X. Yu, J. Zhang, Z. Zhao, W. Guo, J. Qiu, X. Mou, A. Li, J. P. Claverie and H. Liu, Nano Energy, 2015, 16, 207–217 Search PubMed.
- J.-Y. Hwang, S.-T. Myung, J.-H. Lee, A. Abouimrane, I. Belharouak and Y.-K. Sun, Nano Energy, 2015, 16, 218–226 Search PubMed.
- J. Ni, S. Fu, C. Wu, J. Maier, Y. Yu and L. Li, Adv. Mater., 2016, 28, 2259–2265 Search PubMed.
- J. Wang, D. N. Tafen, J. P. Lewis, Z. Hong, A. Manivannan, M. Zhi, M. Li and N. Wu, J. Am. Chem. Soc., 2009, 131, 12290–12297 Search PubMed.
- P. Madhusudan, J. Ran, J. Zhang, J. Yu and G. Liu, Appl. Catal., B, 2011, 110, 286–295 Search PubMed.
- W.-Q. Wu, Y.-F. Xu, C.-Y. Su and D.-B. Kuang, Energy Environ. Sci., 2014, 7, 644–649 Search PubMed.
- S. Chang, X. Yang, Y. Sang and H. Liu, Chem. – Asian J., 2016, 11, 2352–2371 Search PubMed.
- P. Yan, X. Wang, X. Zheng, R. Li, J. Han, J. Shi, A. Li, Y. Gan and C. Li, Nano Energy, 2015, 15, 406–412 Search PubMed.
- S. Wang, L. Yi, J. E. Halpert, X. Lai, Y. Liu, H. Cao, R. Yu, D. Wang and Y. Li, Small, 2012, 8, 265–271 Search PubMed.
- J. Dong, P. Yu, S. A. Arabi, J. Wang, J. He and C. Jiang, Nanotechnology, 2016, 27, 275202–275210 Search PubMed.
- B. Gao, Y. Lin, S. Wei, J. Zeng, Y. Liao, L. Chen, D. Goldfeld, X. Wang, Y. Luo and Z. Dong, Nano Res., 2012, 5, 88–98 Search PubMed.
- H. Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 Search PubMed.
- Y. Kang, Y. Yang, L. C. Yin, X. Kang, L. Wang, G. Liu and H. M. Cheng, Adv. Mater., 2016, 28, 6471–6477 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ce02241c |
| ‡ Xin Yu and Zhenhuan Zhao are contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |