
Exploring nanoparticle porosity using nano-impacts: platinum nanoparticle aggregates†
Xue
Jiao
a,
Stanislav V.
Sokolov
a,
Eden E. L.
Tanner
a,
Neil P.
Young
b and
Richard G.
Compton
 *a
*a
aDepartment of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QZ, UK. E-mail: richard.compton@chem.ox.ac.uk
bDepartment of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, UK
First published on 8th December 2016
Abstract
The porosity of platinum nanoparticles (PtNPs) is explored for the first time using tag-redox coulometry (TRC). This is achieved by monitoring the reduction of the 4-nitrobenzenethiol (NTP)-tagged PtNPs on carbon electrodes via both immobilisation and nanoimpacts. The average charge per impact is measured and attributed to the reduction of NTP adsorbed on individual PtNPs. The number of NTP molecules and thus the “active surface area” of the PtNPs is calculated and compared with two models: fully solid and porous nanoparticles, and the extent of the particle porosity is revealed. This allows a fuller understanding of the (electro-)catalytic behaviour of nanoparticles by providing insight into their porosity and “true/active surface areas”.
Nanoparticles find ever increasingly diverse applications embracing the catalysis of chemical and biochemical reactions, the electrocatalysis of electrode reactions, especially those important for energy transformation processes and environmental remediation.1–7 Key to many uses, both fundamental and applied, is the availability of very high surface areas, in relation to mass, in comparison with conventional materials. This can significantly promote the adsorption of molecular species which often lies at heart of the physical and chemical process of interest. Whilst this is encouraged by the intrinsic nanoscale of the particles it can be further enhanced if the nanoparticles themselves show significant nanoporosity. This can be realised synthetically by the aggregation during formation of ultra-small nanoparticles clumping together to form porous larger nanoparticles8 or by the initial formation of alloys followed by a dealloying protocol which selectively removes one of the alloy components.9 Nanoporosity has been suggested variously to lead to improved adsorption in environmental applications,10–14 catalysed oxygen reduction kinetics,15–18 changed optical properties19,20 and enhanced pseudocapacitor performance.21
Many of the applications of porous nanoparticles involve their being exposed to a solution phase where they might form a coating on an electrode, adsorb environmental targets or catalyse a desired reaction. It is therefore of interest to develop experiments to probe the extent to which the internal surfaces of a porous nanoparticle ‘see’ the surrounding solution phase, or to put the issue in a different form, to find the extent to which the internal surfaces of a porous nanoparticle or nanoparticle aggregate are accessible to solution phase species. Traditionally such nanoporosity has been accessed via ex situ gas phase adsorption isotherm approaches, such as the Brunauer–Emmett–Teller (BET) method. However the qualitative contrast between gas/solid and liquid/solid interfaces coupled with the very small size of the molecules (such as nitrogen) used in gas adsorption measurements makes the development of in situ approaches desirable.
In this paper we report on the use of the nanoimpacts method to measure the surface coverage of redox active molecules adsorbed on the surface of platinum nanoparticles (PtNPs) (ca. 50 nm diameter) which are themselves composed of aggregates of much smaller particles and for which transmission electron microscopy (TEM) reveals significant nanoporosity. In the nanoimpacts method22–25 single nanoparticles suspended in solution from time to time impact on the surface of a microelectrode by virtue of their Brownian motion. If the electrode is held at a suitable potential the nanoparticle itself may be oxidised or reduced26,27 or, in the case of the experiments reported below, surface layers of absorbate on the particles may be electrolysed. Measurement of the charge then, in the latter case, shows the extent of the surface coverage which, when compared with TEM data, allows an assessment of the particle porosity and the scale to which the internal surfaces of the porous particles are accessible to the solution phase.
The PtNPs used were found to be made up of an aggregation of smaller particles (around 2.5 nm radius from TEM), and typical images are shown in Fig. 1(a) and (b). A total of 165 PtNP aggregates were sized and their radii calculated, as shown in Fig. 1(c). A mean radius of 24.3 ± 1.6 nm was obtained, close to the value of 25 nm provided by the manufacturer. To clarify, in this paper, “PtNP” refers to the platinum nanoparticle aggregate (ca. 25 nm radius), and “small nanoparticle” is the component of a PtNP (ca. 2.5 nm radius).
 | ||
| Fig. 1 (a) TEM bright field image of PtNPs. (b) High-resolution TEM image of one PtNP. (c) Size distribution of the PtNPs with an average radius of 24.3 ± 1.6 nm.‡ | ||
The porosity of PtNPs was next established by tag-redox coulometry (TRC). This was achieved through the electrochemical analysis of 4-nitrobenzenethiol (NTP) reduction using both cyclic voltammetric and nanoimpact techniques. First, a glassy carbon macroelectrode was modified either by immersion into an NTP solution or drop casting a NTP-tagged PtNP suspension and recording cyclic voltammograms (CVs). Second, an overpotential was carefully selected from voltammetric measurements in order to sufficiently reduce the NTP-tagged nanoparticles. Applying the chosen potential to the TRC, reductive transients were observed. From this, the number of NTP molecules was calculated and compared with two theoretical models: a solid sphere, and a completely porous nanoparticle formed by aggregation of a large number of smaller particles. This therefore allows an estimation of the PtNP porosities, which can be further confirmed by the TEM images (Fig. 1(a) and (b)).
Initial experiments were conducted to explore the redox properties of molecular NTP adsorbed on both glassy carbon electrode and PtNP surfaces. First, a glassy carbon electrode was modified by NTP molecules (ESI†) before being transferred to an aqueous solution of 10.0 mM HClO4 and 30.0 mM NaClO4. CVs were recorded and a reductive peak was obtained at ca. −0.4 V (vs. SCE), as shown in Fig. 2(a). In the absence of NTP, no signal was observed (red line in Fig. 2(a)), indicating the peak was due to the NTP reduction. This response likely corresponds to a four-electron, four-proton reduction of the nitro group to the hydroxylamine28–30 (Scheme 1).
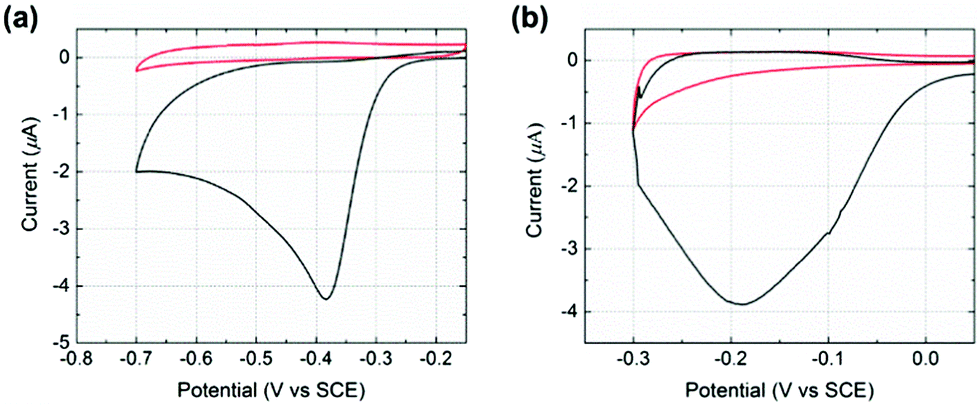 | ||
| Fig. 2 CVs for a glassy carbon macroelectrode with (black) or without (red) modification of (a) NTP and (b) NTP-tagged PtNPs. All scans were performed in a nitrogen saturated solution of 10.0 mM HClO4 and 30.0 mM NaClO4 at a scan rate of 25 mV s−1. | ||
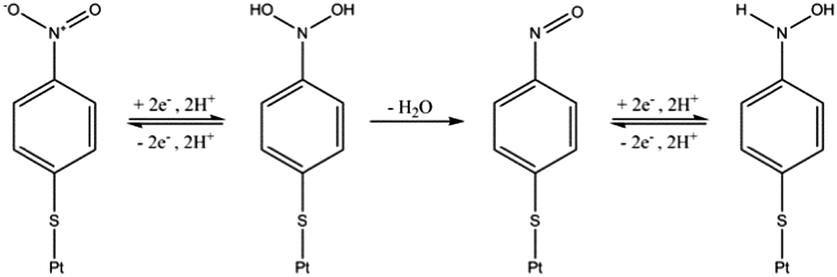 | ||
| Scheme 1 The four-electron reduction of adsorbed NTP. | ||
Next, a glassy carbon electrode was modified with NTP-tagged PtNPs (ESI†) and analogous experiments were performed. A reductive wave was observed at ca. −0.2 V (vs. SCE) and again attributed to the reduction of NTP, as shown in Fig. 2(b).§ No peak was seen when drop casting stock PtNPs (red line in Fig. 2(b)). The NTP reduction peak potentials varied between glassy carbon to PtNP surfaces, representing the different environments influencing possibly both the thermodynamics and kinetics of the process. Therefore, based on the voltammetric response of NTP adsorbed on PtNPs, an applied potential of −0.25 V (vs. SCE) was chosen for the nanoimpact experiments such that measurements were recorded at sufficiently negative potentials in order to maximise the obtained reductive current by ensuring full reduction of the surface layers.
Finally, nanoimpact measurements were conducted to investigate the reductive charge of NTP per nanoparticle. Chronoamperograms (CAs) were recorded in an aqueous electrolyte of 10.0 mM HClO4 and 30.0 mM NaClO4 at −0.25 V (vs. Ag/AgCl (1.0 M KCl)). This potential was carefully selected from the drop-casting experiment (Fig. 2(b)) and was sufficient for a complete reduction of the adsorbed NTP. In the experiments, 60 pM NTP-tagged PtNPs were added and spikes were clearly observed with an approximately millisecond duration, as shown in Fig. 3 (black line). These are attributed to the reduction of the nitro group taking place when the NTP-tagged PtNPs make contact with the carbon fibre substrate. No impact spikes were observed in the case of untagged PtNP or electrolyte only, confirming that the reduction of NTP on the surface of the nanoparticles is the source of the transients (red line in Fig. 3(a)).
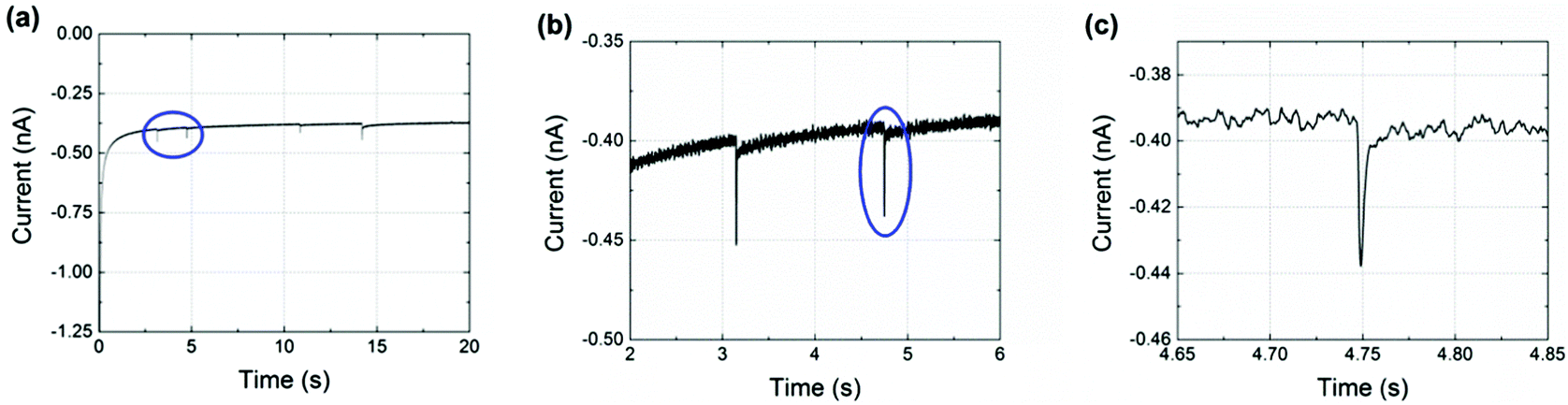 | ||
| Fig. 3 (a) Chronoamperometric profiles with NTP-tagged PtNPs. (b) A zoom of circled reductive transients in (a). (c) Further amplification of the circled spike in (b). All scans were recorded in a nitrogen saturated solution of 10.0 mM HClO4 and 30.0 mM NaClO4 and potentiostatted at −0.25 V vs. Ag/AgCl (1.0 M KCl). | ||
The average charge passed per transient (Q/C) was found to vary with the NTP modification time of the nanoparticles: it increased up to 5 h and then leveled off as shown in Fig. 4. The reason for this is probably because the tag molecule continually adsorbs onto the PtNP surface until a complete self-assembled monolayer (SAM) forms.31,32 The surface was saturated after 5 h and thus no additional NTP can be inserted at a longer modification times (t/h).
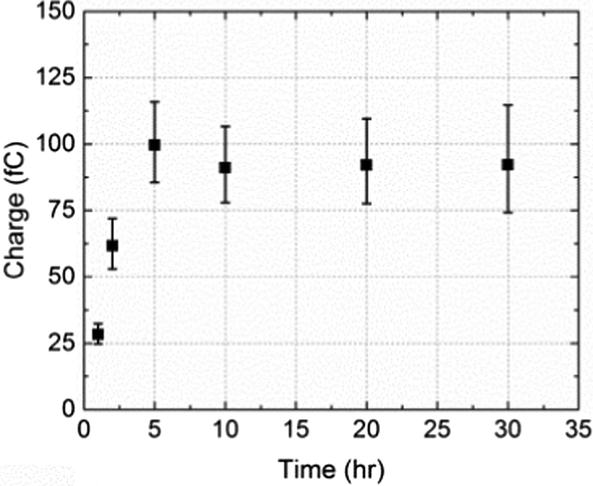 | ||
| Fig. 4 The variation of the average charge per impact transient, from overall 251 measurements, as a function of time allowed for NTP molecules to adsorb onto PtNPs. | ||
A total of 160 spikes obtained at 5, 10, 20 and 30 h modification times were analysed, corresponding to impacts of particles with a full NTP monolayer coverage. A mean charge of 0.094 ×/ 1.1 pC passed per impact transient (Q/C) was calculated,33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ¶ which is related to the number of tag molecules (NNTP/molecules) via the electronic charge (e/C) according to
¶ which is related to the number of tag molecules (NNTP/molecules) via the electronic charge (e/C) according to
| Q = 4eNNTP | (1) |
For Model 1, the assumption was made that the PtNP is a solid, smooth sphere of radius 25 nm and its surface area (SPtNP,solid/m2) is
| SPtNP,solid = 4πR2 | (2) |
Assuming optimal close-packing was adopted by the NTP molecules on a two-dimensional surface plane, the fractional filling efficiency (f1) is 0.91 for spheres or ellipses34 and the surface area of a PtNP (SPtNP/m2) can be written as
| f1 × SPtNP = ANTP × NNTP | (3) |
| NNTP = (4πR2 × f1)/ANTP | (4) |
In Model 2, each PtNP (ca. 25 nm radius) was an aggregate of identical small nanoparticles (ca. 2.5 nm radius, estimated from Fig. 2(b)). As it is difficult to know the inner configuration of the aggregates from the TEM images, these small solid spheres were assumed to arrange in a densest close-packing in space and the fractional filling efficiency (f2) is 0.74.34
As the volume of a spherical PtNP (VPtNP/m3) is
| VPtNP = (4π/3)R3 | (5) |
| f2 × VPtNP = (4π/3)r3 × n | (6) |
| r = 0.1R | (7) |
| SPtNP,porous = 1000f2 × 4πr2 | (8) |
| NNTP = (1000f1f2 × 4πr2)/ANTP | (9) |
Comparing the number of NTP obtained from experiments (1.5 × 105 molecules) with the ones from solid (6.5 × 104 molecules) and porous (4.8 × 105 molecules) PtNP models, the experimental result is between the two extremes and closer to the fully porous case, suggesting a significant degree of porosity in the PtNPs used. TEM images also reveal clusters of small nanoparticles (Fig. 1(a)), but the knowledge of their internal packing was limited. We conclude the NTP molecules can partially insert into the PtNP aggregates, and undergo adsorption at their inner surface.
This result is significant as first it extends the method of TRC to investigation of the porosity of nanoparticles. Electrons are transferred via either direct electron transfer or a “hopping” mechanism,36,37 enabling an in situ quantification of inner structures for porous nanoparticle. In addition to this, electroactive tag molecules can be suitably selected and reduced or oxidised without destroying the underlying core. Second and most importantly, it can be realised that the electrocatalytic ability and other properties of the nanomaterials cannot be correctly understood without realising their porosities.8,16,38–40 In particular, the extent to which “catalytic” properties can be attributed to a change of effective area or to altered intrinsic activity resulting from either quantum confinement or surface morphology effects, requires a realistic estimation of the “active surface area” of the particle in solution. The assumption that the internal surfaces do not contribute to any (electro-)catalysis may lead to incorrect inferences.
In summary, porous PtNPs formed of an aggregate of many (102–103) much smaller nanoparticles have been shown to present internal surfaces available for electrocatalytic reaction and hence can be inferred to be accessible to the solution phase in which the particles are suspended. The nano-impacts method provides a methodology for the in situ measurement of the “active surface area” of the nanoparticles which is essential in understanding the origin of any catalytic behaviour shown by the nanoparticles.
Acknowledgements
The research is sponsored by the funding from the European Research Council under the European Union Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement No. [320403]. Xue Jiao thanks the China Scholarship Council for supporting her DPhil research.Notes and references
- K. Taeho and H. Taeghwan, Nanotechnology, 2014, 25, 012001 CrossRef PubMed.
- P. R. Bandaru, H. Yamada, R. Narayanan and M. Hoefer, Mater. Sci. Eng., R, 2015, 96, 1–69 CrossRef.
- F. Raimondi, G. G. Scherer, R. Kötz and A. Wokaun, Angew. Chem., Int. Ed., 2005, 44, 2190–2209 CrossRef CAS PubMed.
- R. Tong, H. D. Hemmati, R. Langer and D. S. Kohane, J. Am. Chem. Soc., 2012, 134, 8848–8855 CrossRef CAS PubMed.
- A. Chen and P. Holt-Hindle, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS PubMed.
- A. Chen, D. J. La Russa and B. Miller, Langmuir, 2004, 20, 9695–9702 CrossRef CAS PubMed.
- R. A. Thearle, Z. Sofer, D. Bouša and M. Pumera, ChemPhysChem, 2016, 17, 2096–2099 CrossRef CAS PubMed.
- N. C. Bigall, T. Härtling, M. Klose, P. Simon, L. M. Eng and A. Eychmüller, Nano Lett., 2008, 8, 4588–4592 CrossRef CAS PubMed.
- P. Mani, R. Srivastava and P. Strasser, J. Power Sources, 2011, 196, 666–673 CrossRef CAS.
- H. B. Jung, H. Xu, H. Konishi and E. E. Roden, J. Geochem. Explor., 2016, 169, 80–88 CrossRef CAS.
- Y. Chen, Q. Ma, H. Jia and Y. Wang, J. Mater. Sci.: Mater. Electron., 2016, 1–7, DOI:10.1007/s10854-016-5102-4.
- J. Liu, Z. Wang, A. Sheng, F. Liu, F. Qin and Z. L. Wang, Environ. Sci. Technol., 2016, 50, 5606–5613 CrossRef CAS PubMed.
- S. V. Sokolov, K. Tschulik, C. Batchelor-McAuley, K. Jurkschat and R. G. Compton, Anal. Chem., 2015, 87, 10033–10039 CrossRef CAS PubMed.
- B. Ni and X. Wang, CrystEngComm, 2015, 17, 6796–6808 RSC.
- J. Snyder, I. McCue, K. Livi and J. Erlebacher, J. Am. Chem. Soc., 2012, 134, 8633–8645 CrossRef CAS PubMed.
- H. Guo, X. Liu, C. Bai, Y. Chen, L. Wang, M. Zheng, Q. Dong and D.-L. Peng, ChemSusChem, 2015, 8, 486–494 CrossRef CAS PubMed.
- B. Han, C. E. Carlton, A. Kongkanand, R. S. Kukreja, B. R. Theobald, L. Gan, R. O'Malley, P. Strasser, F. T. Wagner and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 258–266 CAS.
- J. Li, A. Wang and Y. Yang, China Pat., CN 104003404, 2014 Search PubMed.
- E. P. Chang and J. S. Evans, Biochemistry, 2015, 54, 5348–5355 CrossRef CAS PubMed.
- D. Wang and P. Schaaf, Germany Pat., DE 102014003993, 2015 Search PubMed.
- X. Lang, A. Hirata, T. Fujita and M. Chen, Adv. Energy Mater., 2014, 4, 1301809 CrossRef.
- W. Cheng and R. G. Compton, TrAC, Trends Anal. Chem., 2014, 58, 79–89 CrossRef CAS.
- M. Pumera, ACS Nano, 2014, 8, 7555–7558 CrossRef CAS PubMed.
- N. V. Rees, Electrochem. Commun., 2014, 43, 83–86 CrossRef CAS.
- P. H. Robbs and N. V. Rees, Phys. Chem. Chem. Phys., 2016, 18, 24812–24819 RSC.
- Y.-G. Zhou, B. Haddou, N. V. Rees and R. G. Compton, Phys. Chem. Chem. Phys., 2012, 14, 14354–14357 RSC.
- M. Giovanni, A. Ambrosi, Z. Sofer and M. Pumera, Electrochem. Commun., 2015, 56, 16–19 CrossRef CAS.
- I. Rubinstein, J. Electroanal. Chem. Interfacial Electrochem., 1985, 183, 379–386 CrossRef CAS.
- Y.-G. Zhou, N. V. Rees and R. G. Compton, Chem. Commun., 2012, 48, 2510–2512 RSC.
- B. J. Plowman, N. P. Young, C. Batchelor-McAuley and R. G. Compton, Angew. Chem., Int. Ed., 2016, 55, 7002–7005 CrossRef CAS PubMed.
- J. U. Nielsen, M. J. Esplandiu and D. M. Kolb, Langmuir, 2001, 17, 3454–3459 CrossRef CAS.
- F. Cecchet, D. Lis, J. Guthmuller, B. Champagne, Y. Caudano, C. Silien, A. Addin Mani, P. A. Thiry and A. Peremans, ChemPhysChem, 2010, 11, 607–615 CrossRef CAS PubMed.
- E. Limpert, W. A. Stahel and M. Abbt, BioScience, 2001, 51, 341–352 CrossRef.
- T. Matsumoto and W. Nowacki, Z. Kristallogr., 1966, 123, 401 CrossRef.
- N. V. Rees, Y.-G. Zhou and R. G. Compton, Chem. Phys. Lett., 2012, 525–526, 69–71 CrossRef.
- C. Amatore, Y. Bouret, E. Maisonhaute, J. I. Goldsmith and H. D. Abruña, ChemPhysChem, 2001, 2, 130–134 CrossRef CAS PubMed.
- C. Amatore, Y. Bouret, E. Maisonhaute, J. I. Goldsmith and H. D. Abruña, Chemistry, 2001, 7, 2206–2226 CrossRef CAS PubMed.
- Z. Guo, S. J. Percival and B. Zhang, J. Am. Chem. Soc., 2014, 136, 8879–8882 CrossRef CAS PubMed.
- H. S. Ahn and A. J. Bard, Angew. Chem., Int. Ed., 2015, 54, 13753–13757 CrossRef CAS PubMed.
- J. Kim, B.-K. Kim, S. K. Cho and A. J. Bard, J. Am. Chem. Soc., 2014, 136, 8173–8176 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp07910e |
| ‡ To measure the nanoparticle size we assumed each PtNP aggregate is a sphere and a circle was drawn to include all the fuzzy small nanoparticles at the aggregate surface. |
| § A background subtraction was performed to reduce the capacitative contribution to the signal. |
| ¶ The transient charges from each modification time were dispersed in a lognormal distribution, with the mean μ and the standard error of mean σ/√n, as measurements of the minimum current restricted by the potentiostat. To clarify, the “back-transformed” values in terms of charge (Q/C) can be written in a mathematical expression according to the lognormal law, with the median μ* = eμ and the multiplicative standard error of mean σ*/√n = eσ*/√n. Therefore the sign ×/ (times/divide) was employed to denote the error, analogous to the ± notation used in a Gaussian distribution. |
| This journal is © the Owner Societies 2017 |