
Influence of the lanthanide(III) ion in {[Cu3Ln2(oda)6(H2O)6]·nH2O}n (LnIII: La, Gd, Yb) catalysts on the heterogeneous oxidation of olefins†
P.
Cancino
ab,
V.
Paredes-García
bc,
C.
Aliaga
bd,
P.
Aguirre
*a,
D.
Aravena
bd and
E.
Spodine
*ab
aFacultad de Ciencias Químicas y Farmacéuticas, Universidad de Chile, Casilla 233, Santiago, Chile. E-mail: paguirre@ciq.uchile; espodine@uchile.cl; Fax: +56 2 29782868
bCentro para el Desarrollo de Nanociencia y Nanotecnología (CEDENNA), Santiago, Chile
cUniversidad Andrés Bello, Departamento de Ciencias Químicas, Santiago, Chile
dUniversidad de Santiago de Chile, Facultad de Química y Biología, Santiago, Chile
First published on 25th November 2016
Abstract
{[Cu3Ln2(oda)6(H2O)6]·nH2O}n (LnIII: La, Gd, Yb; odaH2: oxydiacetic acid) are reported as reusable heterogeneous catalysts in the oxidation of olefins. An influence of the LnIII ion on the catalytic performance of the series is observed, where the YbIII based framework presents a larger activity. The mentioned heteronuclear species are catalytically more active than the corresponding homonuclear catalyst {[Cu(oda)2]·0.5H2O}n. The use of t-butyl hydroperoxide (TBHP) as an oxidant gave conversions between 73–63% for styrene oxidation and between 57–48% for cyclohexene in dichloroethane/water (DCE/H2O). In four cycles, the loss of catalytic activity was less than 10%. Experimental data permit the consideration of the redox active CuII centres as the initiators of radical species generated from TBHP, which are responsible for the oxidation process of the studied olefins. Electron paramagnetic resonance (EPR) spectra of the reaction solutions, obtained in the presence of the spin trap PBN (N-tert-butyl-α-phenylnitrone), corroborate radical species formation during the oxidation process. DFT calculations support an electronic influence of the LnIII ions on the reactivity of the CuII centre, associated with changes in the stabilization of the empty CuII 3dx2−y2 orbital, affecting the reduction of CuII with TBHP to CuI. Model calculations employing several density functionals support a higher electron affinity in the CuGdMOF system in comparison to CuLaMOF. In this way, electronic structure calculations agree with the interesting observed trend of the influence of the secondary metal centre (LnIII ions) on the properties of the active centre (CuII).
Introduction
The oxidative functionalization of olefins is an effective reaction both for organic synthesis and in the production of fine chemicals. Olefins can undergo several different modes of attack by the oxidant, with the products being derived from the epoxidation and oxidative cleavage of the double bond, as well as the oxidation of the allylic C–H bond.1 Many soluble metal complexes have been found to be active catalysts in the oxidation reaction in the last few decades.2–5 However, some problems such as corrosion, deposition on reactor walls, difficulty in recovery and separation of the catalyst from the reaction mixtures are associated with these homogeneous catalysts. These disadvantages can be minimised when the homogeneous catalysts are immobilised into insoluble supports or with the use of heterogeneous catalysts. In relation to this last issue, metal–organic frameworks (MOFs) have all the advantages of soluble metal complexes (selectivity, activities) and due to their low solubility in organic solvents allow their use in several catalytic cycles under heterogeneous conditions.Metal–organic frameworks (MOFs) are a subfamily of coordination polymers,6 which have been studied in the last twenty years in many different fields of chemistry, such as magnetism, non-linear optics, gas storage, luminescent devices and heterogeneous catalysis, among others.7–13 The chemical characteristics of the organic linkers can generate porous structures, thus increasing the active surface of the catalyst. Besides, one of the advantages of MOFs, as compared with the supported catalysts, is that they are in many cases quite stable to leaching and therefore can be recycled. For these reasons, MOFs are being widely studied as heterogeneous catalysts.14 Several authors report in different reviews MOFs that have been used as catalysts in different reactions: the synthesis of MeOH, polymerization reactions, C–C coupling reactions (Heck or Suzuki), hydrogenation, asymmetric reactions and oxidation reactions.14–16
One of the reactions where MOFs have been used as heterogeneous catalysts is the oxidation of olefins, since the obtained products have great importance in the pharmaceutical and agrochemical industries.17–19 Homonuclear MOFs containing 3d or 4f metal ions have been reported as good catalysts in the oxidation of olefins, and several 3d based MOFs can be found in the literature as examples of this type of heterogeneous catalyst. Metal–organic frameworks (MOFs) based on copper(II) with different linkers, such as tris(4′-carboxybiphenyl)amine, 2,2′ or 4,4′-bipyridine, btec and H2btec (btec: 1,2,4,6-benzenetetracarboxylate anion), have been studied as catalysts in the oxidation of olefins.20–23 Recently, our group reported a recyclable [Cu2(bipy)2(btec)]n catalyst for the oxidation reaction of cyclohexene and styrene, showing high catalyst recovery after five cycles of reaction.23 Aerobic olefin epoxidation using cyclooctene as a substrate and {(NH4)2[Co3(Ina)(BDC)3(HCOO)]}n as a catalyst (Ina: isonicotinate and BDC: 1,4-benzenedicarboxylate) was reported by Sha et al. to produce cyclooctene oxide as the main product.24 Moreover, Sen et al. reported high conversions for the epoxidation of styrene, using NiII and MnII based MOFs with malonate ligands.25 Thus, similar high conversions can be obtained with catalytic systems that have different metal centres and ligands. Moreover, it becomes evident that the metal centre and the coordination sphere of the metal ion in the catalyst, together with the nature of the substrate and that of the oxidant, determine the final products which are obtained in the catalytic oxidation reaction of olefins.26
Moreover, several mixed MOFs (two different metals or a metal with different oxidation states) have been used as heterogeneous catalysts in reactions such as aerobic oxidation of 3,5-di-tert-butylcathecol, diastereoselective synthesis of E-α,β-unsaturated ketones, cyanosilylation of aldehydes, cyclohexene oxidation, and ring opening of epoxides. The catalytic performance of these species has been reviewed by Dhakshinamoorthy et al.27
Although several efforts have been made to obtain new rare earth based catalysts for the oxidation of olefins, only a few examples using rare earth MOFs have been reported as catalysts for this type of reaction. These catalysts have shown different catalytic activities; conversion and yields are strongly dependent on the metal centre of the catalyst and the substrate. As an example, a homonuclear yttrium(III) based MOF with nicotinate ligands as organic linkers can be used as a catalyst in the oxidation of cyclooctene (86% conversion; 100% selectivity for epoxide). When the used substrate was styrene, the conversion increased to 92%, while the epoxide selectivity decreased to 41%.28 A 3D gadolinium(III) based framework, {[Gd26(μ6-CO3)9-(NIC)32(μ3-OH)26](NO3)2·2H2O}n (NIC = nicotinate anion), was also used by Sen et al. as a heterogeneous catalyst in the epoxidation of olefinic substrates, including α,β-unsaturated ketones. The conversion for the oxidation of styrene was 57%, the principal product being epoxide with a yield of 100%.29 A cerium(IV) MOF, based on 2,5-dimethyl-1,4-benzenedicarboxylate, Ce-UiO-66-(CH3)2, was shown by Dalapati et al. to be a reusable catalyst for the oxidation of styrene and cyclohexene. The principal oxidation product for the first cycle was benzaldehyde, with epoxide in 29% yield.30
Additionally, heterometallic 3d–4f MOFs have been less studied as heterogeneous catalysts in olefin oxidation, and within the scarce examples of 3d–4f MOFs, the activity of the heterogeneous catalyst {[Cu0.5La2(HPDC)(PDC)2(SO4)(H2O)2]H2O}n (PDCH2: 3,5-pyridinedicarboxylic acid) in the oxidation of olefins and benzylic substrates has been recently reported by our group.31
Another interesting family of 3d–4f MOFs is {[Cu3Ln2(oda)6(H2O)6]·nH2O}n (Ln: lanthanide(III) ions and odaH2: oxydiacetic acid), which has been widely studied to be mostly characterised both structurally and magnetically.32–39 However, the corresponding catalytic behaviour of these compounds has not been reported to date. For this reason, we considered {[Cu3Ln2(oda)6(H2O)6]·nH2O}n with LnIII: La, Gd, Yb as good candidates to be investigated as heterogeneous catalysts, in order to have a preliminary understanding of the possible role of the lanthanide(III) ions in the redox properties of the copper(II), which can be considered as the initiator of the catalytic cycle in the oxidation of olefins.
The herein reported work presents the catalytic properties of the above-mentioned isostructural heterometallic 3d–4f frameworks in the oxidation of cyclohexene and styrene, using tert-butylhydroperoxide as an oxidant, and DFT calculations which assess the importance of the lanthanide ions in the redox properties of the copper(II) centre which acts as the initiator of the catalytic reactions.
Experimental
Synthesis of {[Cu3Ln2(oda)6(H2O)6]·nH2O}n (LnIII: La (1), Gd (2), Yb (3))
The synthesis and structural characterization of the tested heterometallic 3d–4f catalysts have been described elsewhere.39 CuO (3.1 × 10−3 mol), Ln2O3 (9.7 × 10−4 mol) and oxydiacetic acid (odaH2) (7.5 × 10−2 mol) were mixed in H2O (350 mL) and refluxed for 8 h. The formed crystalline solids, {[Cu3La2(oda)6(H2O)6]·nH2O}n, CuLaMOF (1), {[Cu3Gd2(oda)6(H2O)6]·nH2O}n, CuGdMOF (2), or {[Cu3Yb2(oda)6(H2O)6]·nH2O}n, CuYbMOF (3), were filtered off and dried under vacuum.Synthesis of {[Cu(oda)2]·0.5H2O}n (4)
The synthesis was performed following the technique described by Whitlow et al.40 Using an excess of oxydiacetic acid (9 × 10−2 mol) and adding this to basic copper(II) carbonate (3 × 10−3 mol), the reaction mixture was stirred for 30 minutes. The obtained blue crystalline solid corresponds to {[Cu(oda)2]·0.5H2O}n, CuMOF (4).Elemental analysis
The C, N, and H contents were determined using a Carlo-Erba EA-1108 micro-analyser, and Cu analysis was performed using an atomic absorption spectrophotometer, Shimadzu model AA-6200.{[Cu3La2(oda)6(H2O)6]·12H2O}n (CuLaMOF); Calc: C: 17.8, H: 3.7, Cu: 11.8; Exp: C: 17.6, H: 3.4, Cu: 11.6%; {[Cu3Gd2(oda)6(H2O)6]·12H2O}n (CuGdMOF); Calc: C: 18.2, H: 3.9, Cu: 11.8%; Exp: C: 18.4, H: 3.7, Cu: 11.6%; {[Cu3Yb2(oda)6(H2O)6]}n (CuYbMOF); Calc: C: 20.0, H: 4.2, Cu: 13.0%; Exp: C: 20.9, H: 3.9, Cu: 13.0%; {[Cu(oda)2]·0.5H2O}n (CuMOF); Calc: C: 23.5, H: 2.4, Cu: 31.1; Exp: C: 23.9, H: 2.7, Cu: 31.3%.
Thermogravimetric analysis
Thermogravimetric analyses were performed using the NETZCH Iris equipment. The TGA curves were obtained in the 20–700 °C temperature range, under a nitrogen atmosphere (20 mL min−1), using a 10 °C min−1 heating rate. The catalysts were stable in the temperature range used for the catalytic reactions. The thermograms are reported in the ESI† (Fig. S1a–d). In addition, the thermograms confirmed the different amounts of water present in the studied catalysts. The number of solvate water molecules associated with CuLaMOF and CuGdMOF is twelve, while CuYbMOF does not present crystallization water molecules (Table S1†).Powder X-ray diffraction
A Siemens D5000 diffractometer was used to record all powder diffractograms. The obtained diffractograms for CuLaMOF (1), CuGdMOF (2), CuYbMOF (3), and CuMOF (4) were compared with the calculated patterns generated from the single crystal X-ray data reported in ref. 39 and 40 (ESI,† Fig. S2). It was not possible to compare the experimental powder diffraction data of CuLaMOF with that generated from single crystal data, since the cif file for this compound is not available from the CCDC. The attempts to obtain single crystals for CuLaMOF were unsuccessful. The similarity of both the experimental and the calculated diffractograms permit to assess the identity of the bulk material with the single crystals of the other studied catalysts.Catalytic studies
The catalysts were not activated by thermal treatment and, after grinding, were added directly to the reaction mixture, taking into account that some of the catalytic runs were performed using an aqueous solution of the oxidant (TBHP, 70% in water). Thus, the preformed channels obtained by thermal treatment would have been filled with water molecules once the catalysts were exposed to the solvent. The amount of used catalyst, both for the heterometallic ones and for the homometallic copper(II) species, was normalised to contain an equal amount of copper(II) ions, so that the obtained catalytic activity could be compared and analysed, in relation to the role of the respective lanthanide cation present in the studied catalysts.For the oxidation of styrene and cyclohexene, reactions were performed in a magnetically stirred two-necked round-bottom 25 mL flask fitted with a condenser and placed in a temperature controlled oil bath. All the reactions were carried out under nitrogen atmosphere. Most of the experiments were performed at 75 °C, using the following reaction conditions: the catalyst (variable mass) was added to the reactor together with 10 mL of solvent ((i) 1,2-dichloroethane or (ii) n-decane); when the reaction temperature was reached, the substrate, styrene (4.6 mL, 40 mmol) or cyclohexene (4.1 mL, 40 mmol), and the oxidant were added. The oxidant t-butyl hydroperoxide (TBHP) was incorporated into the reaction mixture, using TBHP 70% in water (3.9 mL, 40 mmol) or TBHP 5 M in decane (7.3 mL, 40 mmol). The oxidant used in the 1,2-dichloroethane catalytic system was (i) TBHP (70% in water) and for the n-decane system (ii) TBHP (5.5 M in n-decane). The studied molar ratios of the substrate/TBHP/catalyst were 400/400/1, 800/800/1, 1200/1200/1 and 2400/2400/1 for the CuGdMOF catalyst in (i), while for the rest of the experiments, including those using the CuLa and CuYb MOFs, the molar ratio is 2400/2400/1 (ESI,† Fig. S3 and Table S2). Aliquots of the solution (10 μL) were removed at different reaction times and analysed by gas chromatography (GC). Gas chromatographic analyses were carried out using a Hewlett Packard 5890 GC, equipped with a flame ionization detector (FID) and a Carbowax 20 M capillary column (25 m × 0.2 mm × 0.2 μm), using nitrogen as the carrier gas. The oxidation products were identified by spiking, using standard compounds, and by MS-GC.
The EPR spectrum of the solid catalyst was recorded on a Bruker EMX-1572, operating at the X-band (9.0–9.9 GHz), at 298 K. 5 mg of the catalyst were transferred to a quartz capillary tube (1 mm diameter) before being inserted into a regular tube (3 mm diameter) and then into the spectrometer cavity (microwave power 0.638 mW; modulation amplitude 10 G). The spectra showed a typical wide and intense band associated with a CuII species (data not shown). Reaction solutions using the catalytic conditions in the presence of the spin trap N-tert-butyl-α-phenylnitrone (PBN), added to form radical adducts (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between the spin trap and the oxidant), were monitored. These solutions were filtered before recording the spectra, in order to avoid the interference of CuII. These spectra were also obtained at room temperature (298 K), using capillary tubes with the same dimensions as those used for the recording of the spectrum of the catalysts.
:1 ratio between the spin trap and the oxidant), were monitored. These solutions were filtered before recording the spectra, in order to avoid the interference of CuII. These spectra were also obtained at room temperature (298 K), using capillary tubes with the same dimensions as those used for the recording of the spectrum of the catalysts.
ICP spectroscopy
The solutions obtained after catalytic reactions were analysed by optical ICP spectroscopy using a Perkin Elmer Optima 2000 DV model spectrometer. The free copper concentrations were determined using standards of different concentrations.Continuous shape measurements
Structural trends in CuLnMOF systems were analysed in terms of continuous shape measurements, employing the SHAPE 2.1 program.41,42 In short, SHAPE allows the determination of the deviation of an arbitrary geometry with respect to ideal reference shapes, such as octahedra, tetrahedra, etc. If the problem geometry exactly matches the reference shape, it will present an S parameter of 0. The value of S will continuously rise upon larger deviations of the problem geometry. It is worth noting that S does not depend on the scale or the orientation of the measured geometry.Electronic structure calculations
Depending on the system size and DFT functional, calculations were performed using either the all-electron FHI-AIMS (version 071914_7)43 or ORCA 3.0.3 codes.44 DFT calculations performed with FHI-AIMS considered the PBE functional45 in conjunction with the ‘tight’ basis set. ORCA calculations tested density functionals of different types (i.e. PBE, B3LYP,46 and TPSSh47) in conjunction with Def2-TZVP and Def2-QZVPP basis sets.48 Lanthanide atoms were described using the SARC2-QZVP basis set.49 Scalar relativistic corrections were included in FHI_AIMS (atomic_zora keyword) and ORCA (DKH2).To analyse the influence of the lanthanide ions, we studied the cases of CuLaMOF and CuGdMOF. Structural models were constructed directly from the crystallographic structures of CuPrMOF39 and CuGdMOF.39 We considered the CuPrMOF structure for the geometry of CuLaMOF (replacing the Pr position by La) as the crystal structure of CuLaMOF is not available and CuPrMOF is the compound of this family with the earliest lanthanide presenting a resolved crystal structure. Calculations for CuYbMOF were also attempted but presented severe convergence issues. To converge, they required large broadening parameters that resulted in several orbitals presenting partial electron occupations.
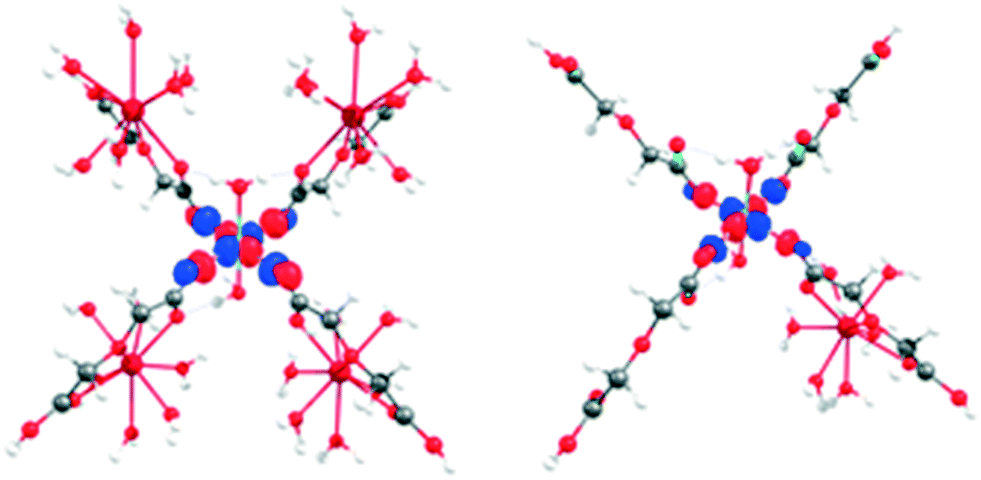
This is expected since the weak ligand field splitting of f orbitals leads to a manifold of low lying excited states associated with different f configurations, whose description is troublesome in a single reference treatment such as DFT. LaIII and GdIII-high spin do not present this problem as they have only one way to represent their f occupation (f0 and f7, respectively). To understand the role of LnIII and CuII ions and their interaction effect on the observed reactivity trends, molecular models of different sizes were constructed: (i) the large model, which considers a CuII centre surrounded by four LnIII ions (see Fig. 1). The two water molecules and four oda groups belonging to the coordination environment of CuII were kept intact from their crystallographic positions.
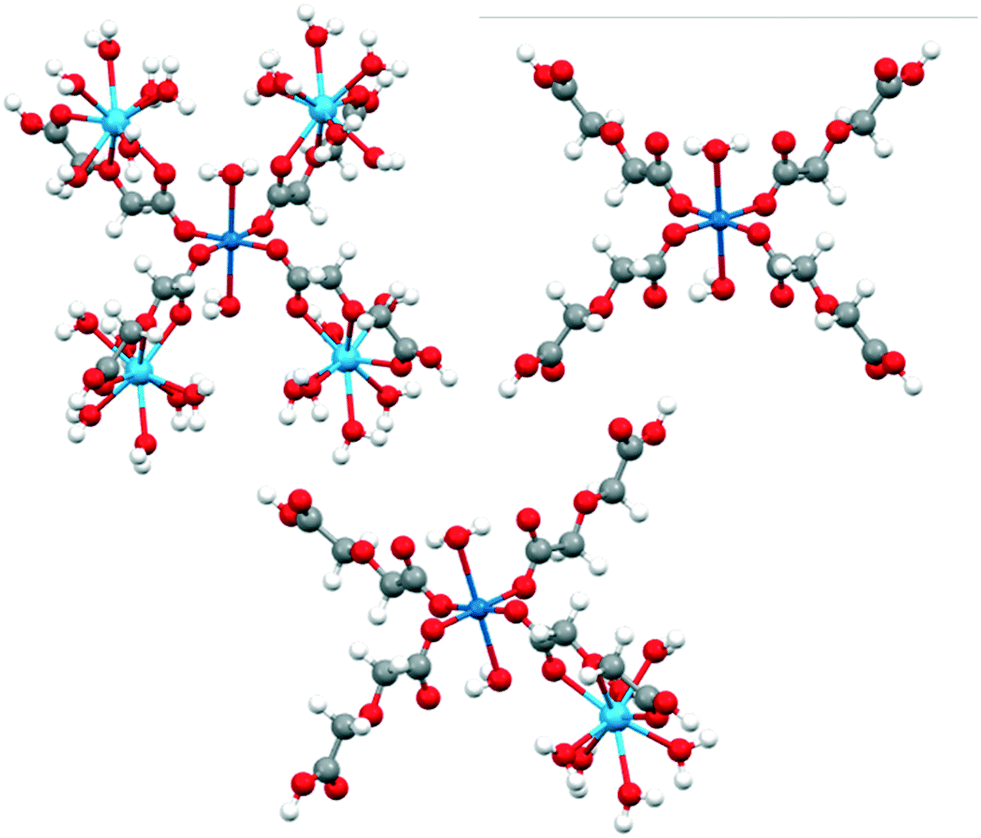 | ||
| Fig. 1 Large (top, left), small CuII (top, right) and small CuIILnIII (bottom) models used in DFT calculations. Color code: LnIII (light blue), CuII (blue), O (red), C (grey) and H (white). | ||
LnIII positions were not modified, although the two oda groups not connected to the central CuII were replaced by water ligands, with respect to the position of the donor oxygen atoms. After the construction of the model, hydrogen atoms were relaxed using FHI-AIMS (PBE/tight, as described above).
Truncation of the large model led to three smaller models: (ii) small CuII, including only the central CuII and its immediate coordination environment, and (iii) small CuIILnIII, designed to study the ligand mediated orbital interaction between metal ions. After the construction of the large model, all hydrogens were optimised.
Results and discussion
The results obtained by elemental analysis, powder X-ray diffraction and TGA confirm that the prepared microcrystalline compounds to be used as catalysts are the compounds reported in the literature.39,40 As shown in Fig. 2, the three heterogeneous catalysts have micrometric size and crystallize with different morphologies; (1) is mainly composed of irregular crystals with dimensions ranging from a length of 8 to 20 μm and a width of 5 to 10 μm, (2) appears as elongated crystals with a length of 10 to 15 μm and a width of 3 to 5 μm. Finally, (3) crystallizes as “microroses” with diameters of 20 to 25 μm (Fig. 2). | ||
| Fig. 2 SEM micrographs of CuLaMOF (1), CuGdMOF (2) and CuMOF (3). | ||
The studied isostructural MOFs present different coordination spheres for the CuII and LnIII centres; the hexacoordinated copper(II) is surrounded by four oxygen atoms belonging to carboxylate groups of different oda molecules and two oxygen atoms from two water molecules, while the LnIII centre is surrounded by nine oxygen atoms associated with three chelating oda ligands, thus generating the coordination polymers which extend in the three directions reaching a three-dimensional structure.32,39
Catalytic activity
The oxidation of styrene and cyclohexene was tested by varying the nature of the lanthanide ions present in the catalysts, solvent, and temperature.Different molar ratios of substrate/TBHP/catalyst were used in order to define the optimal reaction conditions. The studied molar ratios of substrate/TBHP/catalyst were 400/400/1, 800/800/1, 1200/1200/1 and 2400/2400/1 for the CuGdMOF catalyst in dichloroethane. Table 1 shows the TON and TOF values, which were calculated per copper(II) centre. From the data in Table 1, it is possible to infer that the most active substrate/oxidant/catalyst molar ratio is 2400/2400/1, associated with the highest values as referred to catalytic performance (TON) and reaction rate (TOF). Accordingly, the rest of the experiments were performed using this molar ratio.
| S/C ratio | |||||
|---|---|---|---|---|---|
| Substrate | 400:1 |
800:1 |
1200:1 |
2400:1 |
|
|
Reaction conditions: solvent: 1,2-dichloroethane; oxidant: tert-butylhydroperoxide in aqueous medium; temperature: 75 °C; substrate:oxidant ratio = 1:1. TON is calculated as moles of products per mole of active site (CuII ions); TOF is calculated as moles of products per mole of active site (CuII ions) per hour. Values in parentheses correspond to 24 h.
|
|||||
|
TON | 957 (2568) | 1957 (5347) | 3132 (7748) | 7202 (14950) |
| TOF (h−1) | 957 (107) | 1957 (223) | 3132 (323) | 7202 (623) | |
|
TON | 938 (2186) | 1630 (4147) | 2532 (5914) | 4365 (11349) |
| TOF (h−1) | 938 (91) | 1630 (173) | 2532 (246) | 4365 (473) | |
It is important to remark that independent of the experimental conditions the products for the catalysed oxidations remained unchanged. While the detected oxidation products for styrene were benzaldehyde (A), 1-phenylacetaldehyde (B), styrene epoxide (C), and 1,2-phenylethanediol (D), for cyclohexene these were 2-cyclohexene-1-one (A), 2-cyclohexene-1-ol (B), cyclohexene epoxide (C) and 1,2-cyclohexanediol (D) (Scheme 1). Conversion and yields for different detected oxidation products at 24 h for both substrates reacting in the biphasic DCE/H2O system in the presence of different CuLnMOFs are given in Table 2.
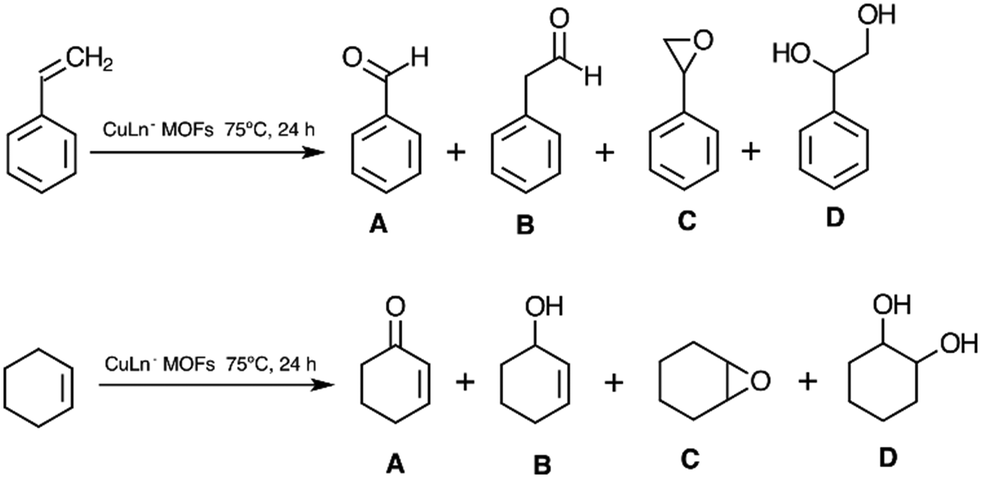 | ||
| Scheme 1 Detected oxidation products for styrene and cyclohexene. | ||
| Substrate | Catalyst | % Conversion (TOF, h−1) | (A) (%) | (B) (%) | (C) (%) | (D) (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: solvent: 1,2-dichloroethane; oxidant: tert-butylhydroperoxide in aqueous medium; temperature: 75 °C. For styrene: (A) benzaldehyde, (B) 1-phenylacetaldehyde, (C) styrene epoxide, and (D) 1,2-phenylethanediol; for cyclohexene: (A) 2-cyclohexene-1-one, (B) 2-cyclohexene-1-ol, (C) cyclohexene epoxide, and (D) 1,2-cyclohexanediol. | ||||||
|
CuLaMOF | 64 (552) | 20 | 47 | 33 | 0 |
| CuGdMOF | 69 (623) | 24 | 48 | 28 | 0 | |
| CuYbMOF | 73 (664) | 23 | 43 | 34 | 0 | |
|
CuLaMOF | 48 (436) | 56 | 31 | 5 | 8 |
| CuGdMOF | 52 (473) | 58 | 30 | 4 | 8 | |
| CuYbMOF | 57 (518) | 58 | 29 | 4 | 9 | |
The results were similar for the three catalysts under study, showing that while the conversion increased in late lanthanides (Yb) with respect to earlier ones (La and Gd), the yield of the different oxidation products remained similar.
Besides, the TOF values show the same trend, clearly indicating that the most active catalyst is CuYbMOF. The origin of this trend will be discussed in detail later. If the catalyst does not participate directly in the pathways to form the products, the product formation can be considered to proceed through a free radical pathway, with the initiator species being the same for the three catalysts, (CH3)3COO˙. This organic radical would be formed by a redox reaction between the oxidant TBHP and the copper(II) centres (Scheme 2).
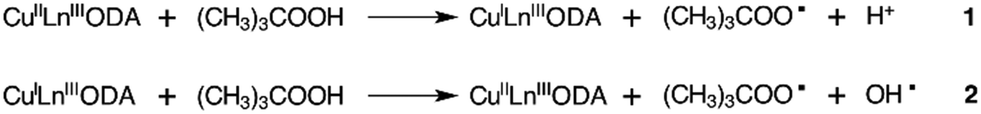 | ||
| Scheme 2 Activation of TBHP using CuLnMOF as a catalyst. | ||
The above-mentioned reactions take into account the lability of the water molecules of the first coordination sphere of copper(II), which enables the interaction of this metal centre with TBHP. On the other hand, the lanthanide ions were considered as species that were not directly involved in the catalytic reactions, since the corresponding coordination sphere is formed by three chelating oda ligands that isolate the lanthanide centres from the interaction with the oxidant or substrate.
However, as evidenced by the conversion data, the lanthanide(III) cation present in the framework affects the activity of the catalytic process (Table 2). This fact may be explained assuming that these ions are influencing the redox properties of the copper(II) centres, since the latter sites are considered to activate the oxidant (TBHP).
The catalytic activity of the studied 3d–4f catalysts, {[Cu3Ln2(oda)6(H2O)6]·12H2O}n (CuLnMOF), was also compared with that of the homonuclear catalyst, {[Cu(oda)2]·0.5H2O}n (CuMOF).38 The corresponding catalytic studies indicate that this homometallic catalyst is less efficient than the heterometallic catalysts; the conversion increased to ca. 35% for CuLnMOF (Fig. 3). These data make evident the influence exerted by the lanthanide(III) ions present in the studied 3d–4f catalysts
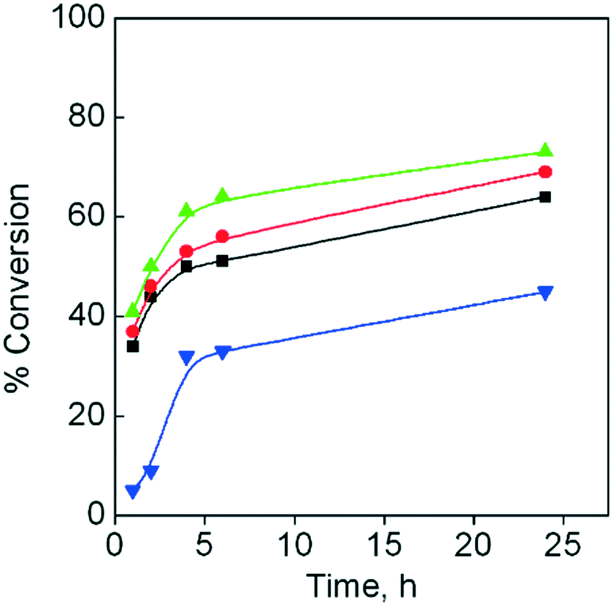 | ||
| Fig. 3 Temporal evolution of the conversion, using styrene as a substrate and TBHP as an oxidant; 1,2-DCE/water as reaction media; CuLaMOF (■); CuGdMOF (●); CuYbMOF (▲); CuMOF (▼). | ||
Reaction pathway
In order to test the viability of a radical mechanism, the CuGdMOF catalyst was used with TBHP (70% in water) as an oxidant at 75 °C and cyclohexene or styrene as a substrate with a 2400/2400/1 S/O/C ratio in 1,2-dichloroethane/water as solvent, in the presence of 10−2 moles of hydroquinone (HQ) as a free radical trapping agent.The results showed that with the use of hydroquinone, the conversion for the oxidation of styrene decreased from ca. 69 to 17%, while for cyclohexene the conversion decreased from ca. 52 to 10%. These results permit to infer that the mechanism has an important contribution from a radical pathway.50 In order to confirm this mechanism, EPR spectroscopy was used to investigate the nature of the radical species produced by the heterogeneous catalyst. In order to facilitate the detection of the reactive species, the measurements were run using the spin trap N-tert-butyl-α-phenylnitrone (PBN), which is known to form a long-lived adduct, useful to detect oxygen-centred radicals.51,52 Initially, control spectra were recorded, all in aerated solutions for different substrates and oxidants, in the presence of PBN.
The respective EPR spectra for the control samples did not evidence the formation of any adduct species with the spin trap. The PBN adduct was generated only in the presence of TBHP, using the catalytic conditions (Fig. 4).
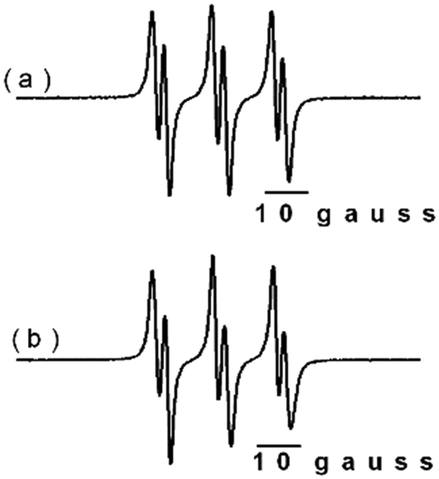 | ||
| Fig. 4 EPR spectra corresponding to the formed adduct species, using TBHP in the presence of PBN. Catalytic conditions: (a) cyclohexene and (b) styrene as substrates. Measurement conditions: centre field, 3495 gauss; time constant, 5.12 ms; conversion time, 20.48 ms; sweep field, 100 gauss; sweep time, 20.97 s; MW power, 25 mW; gain, 2.0 × 103; modulation amplitude, 1.0 gauss; modulation frequency, 100 kHz; time constant, 64 ms; MW frequency, 9877176 GHz. EPR parameters of the PBN radical, g = 2.012 gauss, line width = 1.49 gauss, AN = 15.06 gauss, AH = 3.03 gauss. | ||
The spectra clearly indicate that only one species has been trapped, specifically (CH3)3CO˙. This is due to the high reactivity of (CH3)3CO˙ as compared to that of (CH3)3COO˙.53,54 These spectra suggest that the catalytic processes begin with the formation of oxygen-centred radical species, as shown in Schemes 3 and 4. Thus, the prevailing mechanism can be assumed to be the radical route. Adopting this mechanism, it is possible to give an explanation to the different products detected during the catalytic processes.
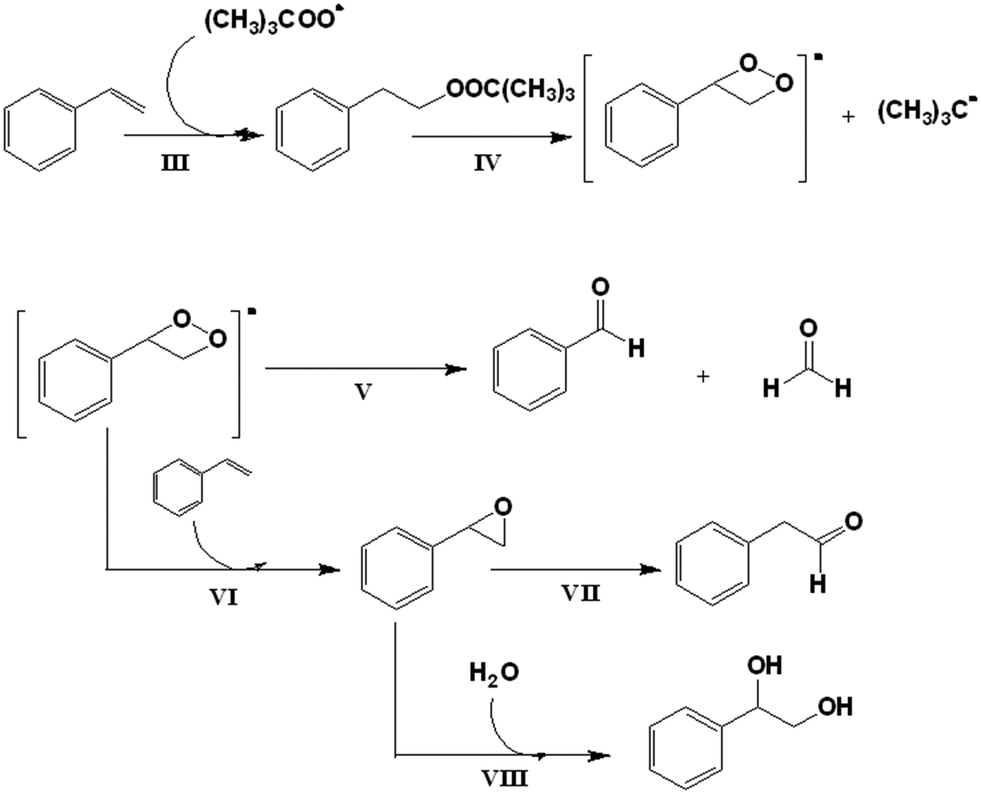 | ||
| Scheme 3 Mechanism for the catalysed oxidation of styrene with CuLnMOFs, in the presence of TBHP. | ||
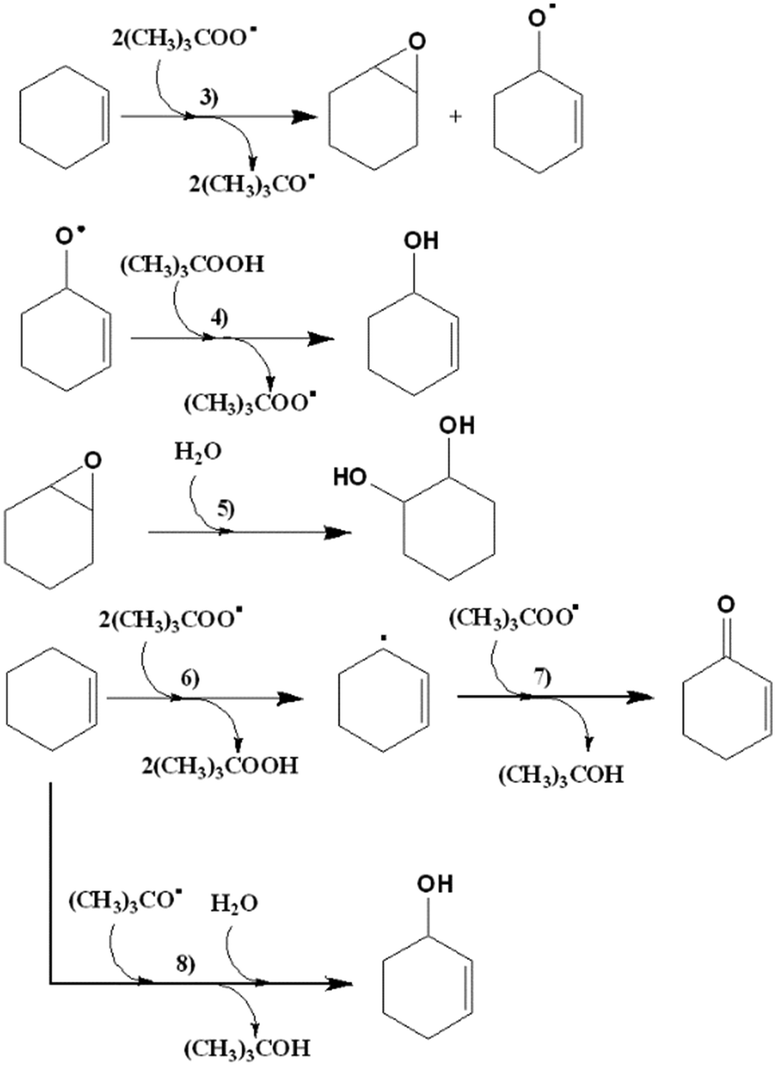 | ||
| Scheme 4 Mechanism for the catalysed oxidation of cyclohexene with CuLnMOFs, in the presence of TBHP. | ||
Ghosh et al.50 proposed a general scheme for the oxidation of olefins assuming a free radical pathway. For styrene oxidation in particular, Silva et al. and Sebastian et al.55,56 described the formation of a radical intermediate (III, IV) when styrene reacts with (CH3)3COO˙ and reported that this intermediate forms benzaldehyde and formaldehyde by internal decomposition (V). Another possibility to complement this mechanism is given by Sebastian et al.,56 who proposed that the intermediate radical species reacts with styrene to form styrene oxide (VI). The following step is the isomerization of styrene oxide to 1-phenylacetaldehyde (VII), or another possibility is that a ring opening reaction produces 1,2-phenylethanediol (VIII) (Scheme 3).
Assuming the radical pathway, the catalyst is supposed to act as an activator, which decomposes the oxidant (TBHP), through a redox reaction (1), giving (CH3)3COO˙.57 Then, the oxidation reaction starts by the attack of (CH3)3COO˙ on the substrate (III), followed by the sequence of steps, which leads to the final products. A new interaction between the reduced catalyst and the oxidant regenerates, by a redox reaction, the catalytic species and produces (CH3)3CO˙ (2). Also, the radical species formed in (1) and (2) (Scheme 2) can react with the substrates, dissolved oxygen, or between them, as is reported by different researchers.50,55,58 It is important to highlight that Liu et al. proposed a specific mechanism for the decomposition of alkyl hydroperoxides in the presence of copper(II) ions.59 A similar mechanism for the oxidation of cyclohexene can be formulated, that is, the reaction of the catalyst and the oxidizing agent (TBHP). Step (3) corresponds to the attack of the radical species on the olefinic position of cyclohexene, while steps (6), (7) and (8) correspond to the attack of the radical species on the allylic hydrogen. The attack on the allylic hydrogen is definitively responsible for the major products of the oxidation of cyclohexene (Scheme 4).
Reaction conditions
Two reaction media were tested for catalyst performance, dichloromethane/water and n-decane, considering the stabilization of the oxidant tert-butylhydroperoxide in n-decane as compared to the same oxidant in aqueous solution. A comparison of the effect of a completely anhydrous medium (n-decane) on the performance of the heterometallic catalysts, CuLaMOF (1), CuGdMOF (2) and CuYbMOF (3), with that of the biphasic system is given for CuLaMOF in Table 3 (additional information for CuGdMOF and CuYbMOF are given in the ESI,† Tables S3 and S4). For styrene and cyclohexene oxidation, the conversion increased ca. 25%, when the anhydrous non-polar solvent decane was used, as compared to the biphasic DCE/water system. However, the selectivity of cyclohexene oxidation appears not to be sensitive to solvent changes. On the other hand, when styrene was used as a substrate, the main product in DCE/water and n-decane was 1-phenylacetaldehyde (B), followed by benzaldehyde (A), and remained with similar yields. The variation in yields was only observed for the minor products.| Substrate | Temperature | % Conversion | (A) (%) | (B) (%) | (C) (%) | (D) (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: S/O/C ratios: 2400/2400/1; temperature: 75 °C; oxidant: tert-butylhydroperoxide in aqueous medium. For styrene: (A) benzaldehyde, (B) 1-phenylacetaldehyde, (C) styrene epoxide, and (D) 1,2-phenylethanediol; for cyclohexene: (A) 2-cyclohexene-1-one, (B) 2-cyclohexene-1-ol, (C) cyclohexene epoxide, and (D) 1,2-cyclohexanediol. | ||||||
|
DCE/Water | 64 | 27 | 44 | 29 | 0 |
| n-Decane | 81 | 35 | 40 | 8 | 17 | |
|
DCE/Water | 48 | 56 | 31 | 5 | 8 |
| n-Decane | 56 | 54 | 25 | 6 | 15 | |
With respect to the generation of epoxide, both systems showed a marked difference in the yield. While styrene epoxide was obtained in ca. 30% in DCE/water, an almost null yield of the same product was detected in n-decane. However, cyclohexene epoxide was not detected in either of the two reaction media. This last fact demonstrates the importance of the allylic oxidation of cyclohexene in the studied systems, which generates the corresponding ketone instead of the epoxide. Water has been reported by Alfayate et al. to act as an inhibitor of the radical route in the oxidation of cyclohexene.60,61 However, this issue was not observed in the studied systems.
In order to have a better understanding of the catalytic systems under study, several runs were carried out at room temperature (25 °C), 60 and 75 °C. The conversion and product yields for the oxidation of styrene are shown in Table 4, S5 and S6.† For styrene oxidation, the product yields are different for the reaction at 75 and 25 °C, with the main products being 1-phenylacetaldehyde and benzaldehyde, respectively. This effect can be explained by the proposed mechanism in Scheme 3, since benzaldehyde is formed by internal decomposition of the intermediary species (Bz-C˙CHOO˙) (Scheme 3, step IV).49 This process should require less energy than the formation of styrene oxide, since the formation of styrene oxide requires an interaction between the intermediate species with a molecule of styrene (Scheme 3, step V).62 This process is therefore favoured by a higher temperature, which provides energy to the system enhancing this reaction. When styrene oxide is formed, the next step is the isomerization of styrene oxide to generate 1-phenylacetaldehyde (Scheme 3, step VI);63 this process is an internal rearrangement and produces the main product whether the reaction is carried out at 60 or 75 °C.
| Substrate | Temperature | % Conversion | (A) (%) | (B) (%) | (C) (%) | (D) (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: S/O/C ratios: 2400/2400/1; solvent: 1,2-dichloroethane; oxidant: tert-butylhydroperoxide in aqueous medium. For styrene: (A) benzaldehyde, (B) 1-phenylacetaldehyde , (C) styrene epoxide, and (D) 1,2-phenylethanediol; for cyclohexene: (A) 2-cyclohexene-1-one, (B) 2-cyclohexene-1-ol, (C) cyclohexene epoxide, and (D) 1,2-cyclohexanediol. | ||||||

|
75 °C | 73 | 23 | 43 | 34 | 0 |
| 60 °C | 54 | 26 | 49 | 18 | 7 | |
| 25 °C | 26 | 68 | 12 | 12 | 8 | |
|
75 °C | 57 | 58 | 29 | 4 | 9 |
| 60 °C | 42 | 56 | 29 | 5 | 10 | |
| 25 °C | 18 | 69 | 29 | 0 | 2 | |
The thermal behaviour of the cyclohexene oxidation reaction is similar to that of styrene when the temperature is varied; as expected in both cases an increase in the conversion is observed as the temperature is increased. However, the yield of the different products varies when the temperature is decreased from 75 to 25 °C. For this system, the main product did not change and remained as 2-cyclohexene-1-one, but its yield increased from 58 to 69% as the temperature decreased from 75 to 25 °C. When the reaction took place at 25 °C, the yield of the products 2-cyclohexen-1-one and 2-cyclohexen-1-ol increased, together with some by-products. It is probable that a low temperature inhibits the route of formation of cyclohexene oxide and therefore its hydrolysis to 1,2-cyclohexendiol.
In order to study the reusability of the catalysts, after each catalytic cycle the catalyst was separated from the reaction mixture by filtration, washed with water and 1,2-dichloroethane and dried under vacuum. As the as-prepared catalysts were used without thermal treatment, the re-used catalysts were not activated by thermal treatment and were added directly to the following catalytic cycle.
Fig. 5a and b show the conversion obtained for both substrates after four catalytic cycles, using CuGdMOF as a catalyst. The catalytic results obtained for CuLaMOF and CuYbMOF, showing a similar trend, are given in the ESI† (Fig. S4a–d). For both styrene and cyclohexene, the catalyst remained active for four cycles (further catalytic cycles were not studied), showing a small decrease of ca. 10%, after the fourth catalytic run.
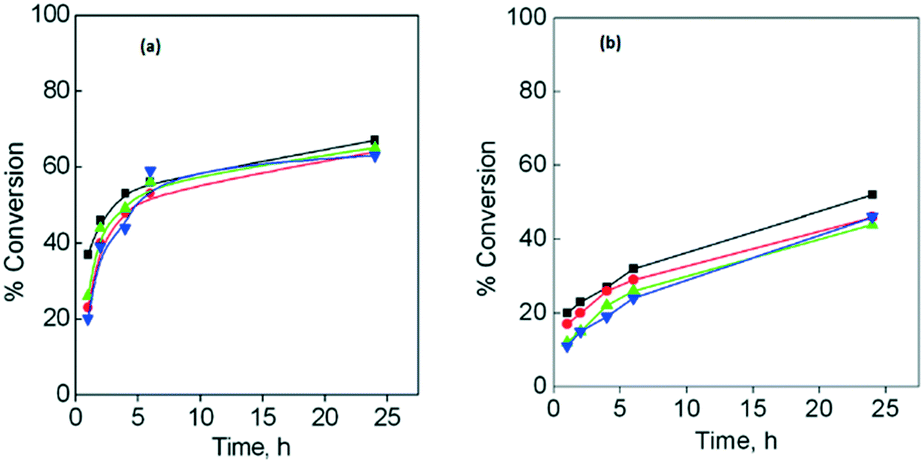 | ||
| Fig. 5 Reusability of CuGdMOF for styrene (a) or cyclohexene (b) oxidation. Run 1 (■); run 2 (●); run 3 (▲); run 4 (▼). | ||
The proposed mechanism considers the initial formation of radical species by the redox reaction of the catalyst with the oxidant, species that must be consumed in the reaction solution without the catalyst (Scheme 2).
During the first three catalytic runs, the re-used catalysts basically maintained their structure, as shown by the principal peaks of the corresponding recorded diffractograms (Fig. S5a–f†). After the fourth cycle, some new peaks became evident in the corresponding diffractograms of the reused catalysts, which obviated further catalytic studies.
Besides, using both substrates, the leaching test was carried out for the three catalysts. The amount of copper(II) in the respective solution after the first catalytic cycle was less than 0.2 wt% for CuLa-MOF, 0.1 wt% for CuGd-MOF and 0.1 wt% for CuYb-MOF of the initial amount of copper present in the catalyst. After the second, third and fourth catalytic cycles, the amount of copper(II) in solution was even lower than 0.02 wt% for all the studied systems. These results confirmed that the heterogeneous catalysts did not present significant leaching in the catalytic studies. Fig. 6 displays the hot filtration tests for (a) cyclohexene and (b) styrene, using CuYbMOF as a catalyst. The figures corresponding to the other catalysts are shown in the ESI† (Fig. S6a and b and S7a and b). These graphs show that when the reaction mixture is separated after one hour of reaction in two portions, the conversion of the filtered sample remains practically unchanged, while the unfiltered one that contains the heterogeneous catalyst increases its conversion. However, Fig. 6 also shows that once the reaction solution is filtered, the obtained solution shows a small increase in the conversion; the conversion reached a plateau after ca. 2 hours. It is possible to assume that the filtrate must have a remnant concentration of formed radical species for both studied systems.
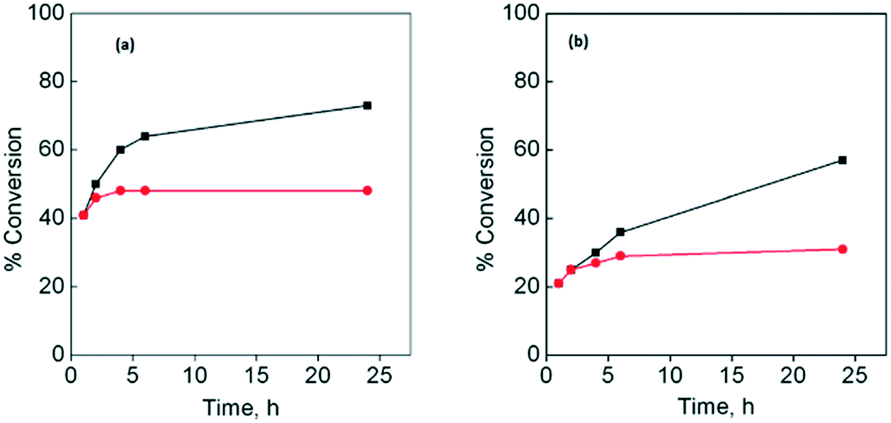 | ||
| Fig. 6 Hot test filtration using CuYbMOF as a catalyst for (a) cyclohexene oxidation and (b) styrene oxidation with a catalyst (■) and filtered after 1 hour (●). | ||
Influence of the lanthanide ions
To better understand the effect on the reactivity of the CuII centre, it is convenient to disentangle structural and electronic effects associated with each LnIII ion. In this way, we first analysed the structural trends in existing compounds of the CuLnMOF family to subsequently investigate them by DFT calculations. Crystal structures for Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er and Yb CuLnMOFs can be found in the literature.33,34,38,39,64 A smooth periodic trend is evident in the a and c cell parameters, as a diminishes from 15.199 Å to 14.344 Å for CuPrMOF to CuYbMOF, while the opposite trend is observed in c, rising from 14.584 Å to 15.470 Å (Fig. 7). These changes must affect the orientation of the four equatorial O donor atoms of the CuII coordination environment, as they belong to oda groups, directly connected to the LnIII ions. The most notable trend is the change in the Oeq–CuII–Oax angle, which monotonically increases from 82.69° (CuPrMOF) to 86.09° (CuYbMOF). Interestingly, CuII–Oeq bond distances are not particularly sensitive to the change in LnIII ions, ranging in an interval between 1.943 Å and 1.957 Å. On the other hand, CuII–Oax distances tend to be shorter in systems with later lanthanides (2.49 Å for CuYbMOF) in comparison with earlier systems (2.57 Å for CuPrMOF).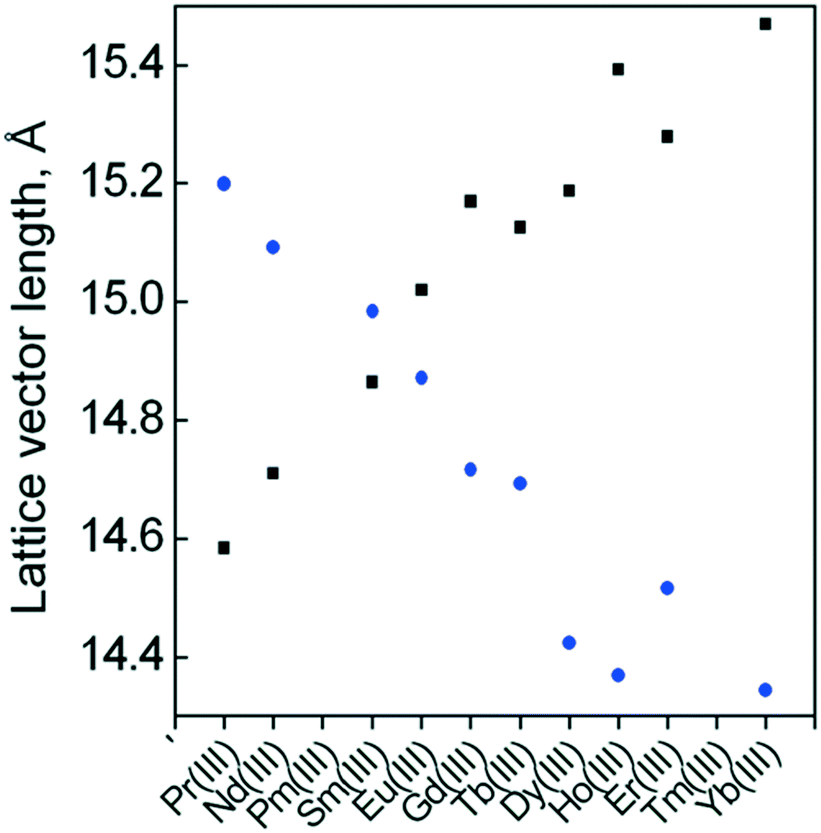 | ||
| Fig. 7 Periodic trend in the length of a (●) and c (■) lattice vectors for CuLnMOF crystalline structures through the lanthanide series. | ||
Continuous shape measurements describe the CuII coordination environment as nearly octahedral, with an S value of 2.29 for CuPrMOF that decreases towards late lanthanides (1.57 for CuYbMOF).
This is not unexpected since changes in Oeq–CuII–Oax angles (approaching 90°) and CuII–Oax distances (shortening of elongated axial distances) tend to a more regular octahedral shape.
To gain further information on the effect of these structural changes on the electronic structure of the CuII centre and the electronic role (if any) of the change in LnIII ion, we performed DFT calculations for structural models of CuLaMOF and CuGdMOF (see “Electronic structure calculations” section for further information on the construction of the models and the computational methodology).
As expected, large and small models predict that the unpaired electron of the CuII ion resides in the dx2−y2 orbital, in line with the elongated octahedral environment of this ion (Fig. 8).
 | ||
| Fig. 8 Molecular orbital representing the unpaired electron of large CuLaPOM (left) and small CuIILnIII (right). | ||
In the large models including the four surrounding LnIII ions, we observe a significant stabilization of the empty CuII dx2−y2 orbital (ca. 2 kcal mol−1) in CuGdMOF in comparison to CuLaMOF, consistent with the enhanced reactivity of the systems incorporating later lanthanides (Tables S7 and S8†), which will present a more favourable orbital to accept one electron from TBHP, promoting the reduction of CuII to CuI.
Furthermore, if we keep the CuGdMOF geometry and only change the Ln position for La, we observe some destabilization of the CuII dx2−y2 orbital (0.4 kcal mol−1).
The complementary calculations (i.e. using the CuLaMOF geometry and replacing the Ln for Gd) yield a marked stabilization of 6 kcal mol−1. In this way, the lanthanide ion is exerting a genuinely electronic effect on the regulation of the stability of the CuII dx2−y2 orbital.
To further check this conclusion, truncated (small CuII) models were constructed by removing the LnIII ions, just leaving the coordination environment of the CuII centre. It is interesting to note that the stabilization observed in the large models (larger than 2 kcal mol−1) is drastically reduced in the geometry excluding the lanthanides to 0.2 kcal mol−1. Then, the structural changes in the coordination environment of the CuII between CuLaMOF and CuGdMOF are not accounting for the CuII dx2−y2 stabilization, in line with the importance of the LnIII electronic effect.
Finally, a “dimeric” model (small CuIILnIII) was constructed to represent the interaction between CuII and LnIII. In this model, one of the lanthanide ions of the large model is kept alongside with the CuII coordination environment (Fig. 1). To check the robustness of our conclusions, electron affinity (EA) calculations were performed using several combinations of density functionals (PBE, B3LYP and TPSSh) and basis sets (Def2-TZVP and Def2-QZVPP), both in the gas phase and using water as a continuous solvent (COSMO model)65 (see Table S10† for more details).
In all cases, the additional electron associated with the EA is accommodated in the CuII dx2−y2 orbital, supporting the designation of CuII as the active redox centre for the catalytic process (Fig. 8). Furthermore, EA of CuGdMOF was larger than CuLaMOF, in line with the stabilization of the CuII dx2−y2 orbital in the case of CuGdMOF. It is interesting to observe how the characteristics of each density functional are reflected in the calculated EA values. PBE, being a pure GGA functional, presents the largest EA; TPSSh (hybrid meta-GGA, 10% HF exchange) shows intermediate values and B3LYP (hybrid, 15% HF exchange) the largest. This is clearly associated with the overestimation of low spin energies typical from GGA functionals, while B3LYP tends to be biased toward high spin states.66 We expect TPSSh to be more balanced in this respect.
The mechanism for the lanthanide effect can be clarified by comparing the molecular orbital corresponding to the empty CuII dx2−y2 orbital in the small LnIIICuII models of CuLaMOF and CuGdMOF. The GdIII ion is more strongly attracting the electronic density of the linking oda ligand than LaIII. In this way, the electron density of the oxygen donor atom bonded to CuII is slightly depleted (Fig. S8†) and the ligand field perceived by the 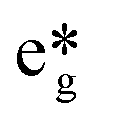 orbitals of CuII is weakened. This is in line with the higher Lewis acidity of GdIII in comparison to LaIII and could be rationalised in terms of a larger polarizing effect of GdIII, although the physical mechanism behind the effect is not purely electrostatic but orbital.
orbitals of CuII is weakened. This is in line with the higher Lewis acidity of GdIII in comparison to LaIII and could be rationalised in terms of a larger polarizing effect of GdIII, although the physical mechanism behind the effect is not purely electrostatic but orbital.
Conclusions
The heterometallic frameworks, {[Cu3Ln2(oda)6(H2O)6]·nH2O}n (LnIII: LaIII, GdIII, YbIII), have been shown to be active for several cycles as catalysts in the oxidation of the olefinic substrates, cyclohexene and styrene. All the obtained data for the catalytic systems can be rationalised by assuming a radical mechanism as the principal route for the oxidation processes.With respect to the generation of epoxide, both systems showed a marked difference in the yield. While styrene epoxide was obtained in ca. 30% in DCE/water, an almost null yield of the same product was detected in n-decane. Cyclohexene epoxide was not detected in an appreciable percentage in either of the two solvents. This last fact demonstrates the importance of the allylic oxidation of cyclohexene in the studied systems, which generates the corresponding ketone instead of the epoxide.
The lack of significant leaching, and the almost unaltered structures of the catalysts after several cycles, permits the conclusion that the CuLnMOFs can be considered as reusable heterogeneous catalysts.
The best catalytic results were obtained for CuYbMOF, as compared to the catalysts that contain LaIII or GdIII. Electronic structure calculations are consistent with this trend and predict the stabilization of the empty dx2−y2 orbital of CuII in the late lanthanides, thus favouring the reduction of the metal centre with TBHP. The mechanism of such effect can be understood by orbital stabilization of the dx2−y2 orbital of CuII, as influenced by the changes in Lewis acidity between GdIII and LaIII. Concretely, the stronger Lewis acidity of GdIII in comparison to LaIII results in a weaker ligand field around CuII, stabilising its 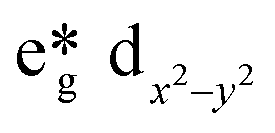 orbital.
orbital.
Acknowledgements
The authors thank Proyecto Anillo ACT1404 and Centres of Excellence with Basal/CONICYT Financing, FB0807 (CEDENNA) for financial support. PCR acknowledges financial support from CONICYT, Programa de Formación de Capital Humano Avanzado, Beca Doctorado Nacional 21110228. D. A. thanks CONICYT + PAI “Concurso nacional de apoyo al retorno de investigadores/as desde el extranjero, convocatoria 2014 – 82140014” for financial support. This research was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02) (Powered@NLHPC).References
- H. E. B. Lempers and R. A. Sheldon, Appl. Catal., A, 1996, 143, 137 CrossRef CAS.
- W. A. Herrmann, R. W. Fischer and D. W. Marz, Angew. Chem., Int. Ed. Engl., 1991, 30, 1638 CrossRef.
- S. Rayati, N. Sadeghzadeh and H. R. Khavasi, Inorg. Chem. Commun., 2007, 10, 1545 CrossRef CAS.
- D. Mohajer and S. Tangestaninejad, J. Chem. Soc., Chem. Commun., 1993, 240 RSC.
- R. M. Calvante, V. A. P. O'Shea, J. M. Campos-Martin, J. L. G. Fierro and E. Gutiérrez-Puebla, J. Mol. Catal. A: Chem., 2004, 214, 269 CrossRef.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3 CrossRef CAS.
- Y.-R. Peng, P.-H. Chien, H.-T. Chung, P.-Y. Pan, Y.-H. Liu and E.-C. Yang, J. Solid State Chem., 2014, 212, 159 CrossRef CAS.
- Y.-Q. Mu, M.-L. Han, B.-H. Wang, W. Xia, W.-W. Dong and D.-S. Li, Inorg. Chem. Commun., 2014, 46, 259 CrossRef CAS.
- X.-Q. Yan, M.-L. Han, G.-W. Xu, B. Liu, D.-S. Li and J. Zhang, Inorg. Chem. Commun., 2015, 58, 60 CrossRef.
- L. Ding and A. O. Yazaydin, Microporous Mesoporous Mater., 2013, 182, 185 CrossRef CAS.
- Y. Cui, B. Chen and G. Qian, Coord. Chem. Rev., 2014, 273–274, 76 CrossRef CAS.
- P. Valvekens, M. Vandichel, M. Waroquier, V. Van Speybroeck and D. De Vos, J. Catal., 2014, 317, 1 CrossRef CAS.
- I. Luz, F. X. Labrés i Xamena and A. Corma, J. Catal., 2012, 276, 134 CrossRef.
- A. Corma, H. García and F. X. Labrés i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Opanasenko, J. Cejka and H. García, Adv. Synth. Catal., 2013, 335, 247 Search PubMed.
- M. Yoon, R. Shirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed.
- R. A. Sheldon and J. K. Kochi, Metal Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- K. Jorgensen, Chem. Rev., 1989, 89, 431 CrossRef.
- J. R. Monnier, Appl. Catal., A, 2001, 221, 73 CrossRef CAS.
- K. Brown, S. Zolezzi, P. Aguirre, D. Venegas-Yazigi, V. Paredes-García, R. Baggio, M. A. Novak and E. Spodine, Dalton Trans, 2009, 1422 RSC.
- D. Shi, Y. Ren, H. Jiang, B. Cai and J. Lu, Inorg. Chem., 2012, 51, 6498 CrossRef CAS PubMed.
- D. Jiang, T. Mallat, D. M. Meier, A. Urakawa and A. Baiker, J. Catal., 2010, 270, 26 CrossRef CAS.
- P. Cancino, V. Paredes-García, P. Aguirre and E. Spodine, Catal. Sci. Technol., 2014, 4, 2599 CAS.
- S. Sha, H. Yang, J. Li, C. Zhuang, S. Gao and S. Liu, Catal. Commun., 2014, 43, 146 CrossRef CAS.
- R. Sen, S. Bhunia, D. Mal, S. Koner, Y. Miyashita and K. I. Okamoto, Langmuir, 2009, 25, 13667 CrossRef CAS PubMed.
- J. Hao, S. Li, L. Cheng, Q. Suo, Y. Xiao, X. Jiao, X. Feng, W. Bai and X. Song, Inorg. Chim. Acta, 2014, 421, 246 CrossRef CAS.
- A. Dhakshinamoorthy, A. M. Asiric and H. Garcia, Catal. Sci. Technol., 2016, 6, 5238 CAS.
- R. Sen, S. Koner, D. K. Hazra, M. Helliwell and M. Mukherjee, Eur. J. Inorg. Chem., 2011, 241 CrossRef CAS.
- R. Sen, D. K. Hazra, M. Mukherjee and S. Koner, Eur. J. Inorg. Chem., 2011, 2826 CrossRef CAS.
- R. Dalapati, B. Sakthivel, A. Dhakshinamoorthy, A. Buragohain, A. Bhunia, C. Janiak and S. Biswas, CrystEngComm, 2016, 18, 7855 RSC.
- P. Cancino, A. Vega, A. Santiago-Portillo, S. Navalon, M. Alvaro, P. Aguirre, E. Spodine and H. García, Catal. Sci. Technol., 2016, 6, 3727 CAS.
- J. G. Hao, L. Song and J. S. Huang, Chin. J. Struct. Chem., 1997, 16, 228 Search PubMed.
- J. G. Mao, L. Song, X. Y. Huang and J. S. Yuang, Polyhedron, 1997, 16, 963 CrossRef CAS.
- J. G. Mao and J. S. Huang, Transition Met. Chem., 1997, 22, 277 CrossRef CAS.
- J. Torres, F. Peluffo, S. Domínguez, A. Mederos, J. M. Arrieta, J. Castiglioni, F. Lloret and C. Kremer, J. Mol. Struct., 2006, 825, 60 CrossRef CAS.
- Q. D. Liu, J. R. Li, S. Gao, B. Q. Ma, F. H. Liao, Q. Z. Zhou and K. B. Yu, Inorg. Chem. Commun., 2001, 4, 301 CrossRef CAS.
- C. Kremer, J. Torres and S. Domínguez, J. Mol. Struct., 2008, 879, 130 CrossRef CAS.
- A. C. Rizzi, R. Calvo, R. Baggio, M. T. Garland, O. Peña and M. Perec, Inorg. Chem., 2002, 41, 5609 CrossRef CAS PubMed.
- R. Baggio, M. T. Garland, Y. Moreno, O. Peña, M. Perec and E. Spodine, J. Chem. Soc., Dalton Trans., 2000, 2061 RSC.
- S. H. Whitlow and G. Davey, J. Chem. Soc., Dalton Trans., 1975, 1228 RSC.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249, 1693 CrossRef CAS.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE v2.1, Universitat de Barcelona, 2013 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS.
- An ab initio, DFT, and semiempirical SCF-MO package-Version 3.0.3 Design and Scientific Directorship, F. Neese, Technical Directorship: F. Wennmohs, Max-Planck-Institute for Chemical Energy Conversion Stiftstr. 34, 45470 Mülheim a. d. Ruhr, Germany, Frank.Neese@cec.mpg.de. With contributions from: U. Becker, D. Bykov, D. Ganyushin, A. Hansen, R. Izsak, D. G. Liakos, C. Kollmar, S. Kossmann, D. A. Pantazis, T. Petrenko, C. Reimann, C. Riplinger, M. Roemelt, B. Sandhöfer, I. Schapiro, K. Sivalingam, B. Wezisla, contributions from our collaborators: M. Kallay, S. Grimme, E. Valeev and G. Chan Search PubMed.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- D. Aravena, F. Neese and D. A. Pantazis, J. Chem. Theory Comput., 2016, 12, 1148 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- R. Ghosh, Y.-C. Son, V. D. Makwana and S. L. Suib, J. Catal., 2004, 224, 288 CrossRef CAS.
- M. J. Davies, in Recent Developments in EPR Spin-Trapping. Electron Paramagnetic Resonance, ed. B. C. Gilbert, M. J. Davies and D. M. Murphy, The Royal Society of Chemistry, London, 2002, vol. 18 Search PubMed.
- Spin Trap Database – NIEHS – National Institute of Health, http://tools.niehs.nih.gov/stdb/ Search PubMed.
- W. A. Pryor, Annu. Rev. Physiol., 1986, 48, 657 CrossRef CAS PubMed.
- B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine, Oxford University Press, Oxford, 4th edn, 2008 Search PubMed.
- M. Silva, C. Freire, B. de Castro and J. L. Figueiredo, J. Mol. Catal. A: Chem., 2006, 258, 327 CrossRef CAS.
- J. Sebastian, K. M. Jinka and R. Vir Jasra, J. Catal., 2006, 244, 208 CrossRef CAS.
- U. Neuenschwander, E. Meler and I. Hermans, ChemSusChem, 2011, 4, 1613 CrossRef CAS PubMed.
- E. Spier, U. Neuenschwander and I. Hermans, Angew. Chem., Int. Ed., 2013, 52, 1581 CrossRef CAS PubMed.
- H. Liu, Y. Cheng, J. Lu, R. Li and K. Wang, J. Inorg. Biochem., 2006, 100, 1280 CrossRef CAS PubMed.
- A. Alfayate, C. Márquez-Alvarez, M. Grande-Casas, B. Bernardo-Maestro, M. Sanchez-Sanchez and J. Pérez-Pariente, Catal. Today, 2013, 213, 211 CrossRef CAS.
- A. Alfayate, C. Márquez-Alvarez, M. Grande-Casas, M. Sanchez-Sanchez and J. Pérez-Pariente, Catal. Today, 2014, 227, 57 CrossRef CAS.
- N. A. Porter, in Free Radicals in Biology, ed. W. A. Pryor, Academic Press Inc., 1980, ch. 8, vol. IV, pp. 281–289 Search PubMed.
- J. Haber, M. Klosowsky and J. Poltowiscz, J. Mol. Catal. A: Chem., 2003, 201, 167 CrossRef CAS.
- B. Barja, R. Baggio, M. T. Garland, P. F. Aramendia, O. Peña and M. Perec, Inorg. Chim. Acta, 2003, 346, 187 CrossRef CAS.
- S. Sinnecker, A. Rajendran, A. Klamt, M. Diedenhofen and F. Neese, Calculation of Solvent Shifts on Electronic G-Tensors with the Conductor-Like Screening Model (COSMO) and its Self-Consistent Generalization to Real Solvents (COSMO-RS), J. Phys. Chem. A, 2006, 110, 2235–2245 CrossRef CAS PubMed.
- M. Reiher, O. Salomon and B. Hess, Theor. Chem. Acc., 2001, 107, 48–55 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Structural and catalytic data; DFT geometries and energies. See DOI: 10.1039/c6cy02115h |
| This journal is © The Royal Society of Chemistry 2017 |