
A platform for more sustainable chitin films from an ionic liquid process†
Catherine
King
ac,
Julia L.
Shamshina
bc,
Gabriela
Gurau
bc,
Paula
Berton
 c,
Nur Farahnadiah Abdul Faruk
Khan
b and
Robin D.
Rogers
*ac
c,
Nur Farahnadiah Abdul Faruk
Khan
b and
Robin D.
Rogers
*ac
aDepartment of Chemistry, The University of Alabama, Tuscaloosa, AL 35487, USA
b525 Solutions, Inc., 720 2nd Street, Tuscaloosa, AL 35401, USA
cDepartment of Chemistry, McGill University, 801 Sherbrooke St. West, Montreal, QC H3A 0B8, Canada. E-mail: robin.rogers@mcgill.ca
First published on 26th September 2016
Abstract
A versatile platform for the preparation of chitin films with tunable strength, morphology, and efficacy of application has been designed from an ionic liquid process for the production of more sustainable high value materials. Films were prepared by a simple casting method from a solution of chitin in the ionic liquid 1-ethyl-3-methylimidazolium acetate ([C2mim][OAc]). The chitin source, the loading in ionic liquid, and the drying methods defined film properties such as strength, porosity, and water absorbency. Only chitin directly extracted from shrimp shells using the ionic liquid (rather than commercially available chitin) could be used to cast films strong enough to be handled and dried. The optimal loading of chitin in the ionic liquid was determined to be 2.5 wt% and different drying methods led to different film properties (e.g., hard and rigid vs. soft and porous). As an exemplary application, loading and release of a model drug (caffeine) was investigated. Interestingly, a burst release of the majority of the caffeine was observed in the first 20 minutes, followed by slow release of the remainder. Although more investigations are needed, the chitin film platform can be thought of as an attractive new tool in the development of packaging materials, biomedical devices, and absorbent materials (M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632) made from one of Nature's most abundant polymers.
1. Introduction
In our society, where environmental concern is increasing, there is heightened consumer demand for more sustainable alternatives to many petrochemical based synthetic polymers.2,3 Recycling of these materials is often impractical, but their disposal leads to accumulation in landfills and pollution of the environment.4 Due to their biodegradability, natural biopolymers can avoid many of these problems and are a promising alternative to petrochemical based materials. Biopolymer based materials, such as those made from cellulose (Fig. 1 left), are available on the market today, but are often produced using non sustainable processes. For example, the processing of cellulose into useful materials comprises the use of harsh or toxic solvent systems for chemical modification in order to obtain a more “soluble” form of the biopolymer.5 Cellophane, one such cellulose material, is a biodegradable packaging material made from cellulose, but the manufacturing process employs toxic chemicals such as carbon disulfide in order to modify the polymer into a form that is easy to manipulate.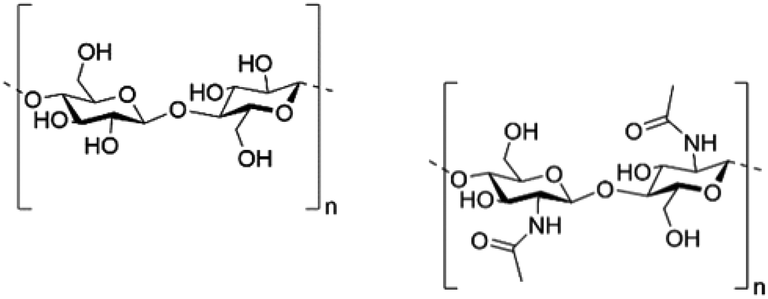 | ||
| Fig. 1 Structure of cellulose (left) and chitin (right). | ||
Chitin (Fig. 1, right) is the second most abundant biopolymer in the world after cellulose. It is biocompatible, biodegradable, biorenewable, and has long been known to possess regenerative properties.6 Despite its desirable properties, chitin has long been underutilized due to its insolubility in common solvent systems. In order to dissolve and manipulate chitin, harsh solvent systems are employed, such as N,N-dimethylacetamide (DMAc)/lithium chloride (LiCl),7 aqueous NaOH/urea,8 strong acids, and some fluorinated solvents.9,10
In recent years, it has been found that by using ionic liquids (ILs, defined as salts that melt below 100 °C),11 chitin can be dissolved and regenerated. Various imidazolium based ILs such as 1-butyl-3-methylimidazolium acetate and 1-butyl-3-methylimidzolium chloride,12 1-allyl-3-methylimidazolium bromide,13 and 1-ethyl-3-methylimidazolium propionate14 have been employed. Recently, IL systems other than those based on imidazolium cations have been investigated, such as tris(2-hydroxyethyl)methylammonium acetate with added ethylenediamine, which was shown to dissolve chitin without heating.15
In the last decade, our group has utilized 1-ethyl-3-methylimidazolium acetate as a tool for biopolymer dissolution.16–19 This IL has been used to manipulate high molecular weight chitin (regenerated from shrimp shells) into various architectures such as fibers,18,20 nanomats,21 and hydrogels.22 Another important architecture to be explored using the IL system is films. Biopolymer films are desirable for applications1,23 ranging from high value medical devices24 (such as wound dressings,25 drug delivery systems,26 and pharmaceutical coatings23) to packaging applications.27,28 We hypothesized that the IL process could be used to produce chitin films with different properties depending on the method of preparation, which would lead to the ability to also tune the film applications. Here we demonstrate a versatile platform for the preparation of chitin films, without the use of harsh solvents or chemical modification of the polymer, which allows the tailoring of film properties and thereby their utility.
2. Experimental
2.1 Chemicals and materials
All materials were used as supplied unless otherwise noted. The IL 1-ethyl-3-methylimidazolium acetate ([C2mim][OAc], purity >95%) was purchased from IoLiTec, Inc. (Tuscaloosa, AL, USA). Anhydrous ethanol, 200 Proof Pure Ethanol, was obtained from KOPTEC DLI, (King of Prussia, PA, USA). Acetone, phosphate buffered saline (PBS), and “Practical Grade” chitin (PG chitin) were purchased from Sigma-Aldrich Co. LLC (St. Louis, MO, USA). Anhydrous caffeine was obtained from Spectrum Chemical (Gardena, CA, USA). Deionized (DI) water was obtained from a commercial deionizer (Culligan, Northbrook, IL, USA) with specific resistivity of 16.82 MΩ cm at 25 °C.Dried shrimp shells, for laboratory use, were received from the Gulf Coast Agricultural and Seafood Cooperative in Bayou La Batre, AL, USA where they were dried by screw press to eliminate the majority of the water, heated to 160 °C in a fluidized bed dryer until the material had a final moisture content of less than 5 wt%, then pulverized with a hammer mill to particles 0.635 cm diameter and smaller. This pulverized material was shipped to The University of Alabama and used to extract chitin, according to the procedure described in ref. 18. The procedure for one batch was done according to the procedure given in ref. 20.
2.2 Viscosity testing
The viscosity of chitin/IL solutions at different chitin loadings was determined using a VISCOlab 3000 viscometer (Medford, MA, USA). Approximately 2 mL of each chitin solution was placed into the viscometer chamber and a piston corresponding to 500–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cP or 100–2000 cP (depending on the solution) was selected to obtain the measurements. Viscosities were recorded from 40 to 80 °C at 5 °C intervals.
000 cP or 100–2000 cP (depending on the solution) was selected to obtain the measurements. Viscosities were recorded from 40 to 80 °C at 5 °C intervals.
2.3 Film preparation details
For all the experiments described below, the regenerated chitin was re-dried overnight in the oven at 80 °C (Precision Econotherm Laboratory Oven, Winchester, VA, USA), ground to a fine powder using a mortar and pestle, and sieved to <90 μm particle size. A 2.5 wt% solution of regenerated chitin in IL was made by dissolving 0.258 g of chitin in 10 mL [C2mim][OAc]. The chitin and IL along with a magnetic stir bar were placed into a 20 mL vial, loosely capped, and placed into an oil bath. The mixture was heated at 90 °C and stirred at 760 rpm for 3 h until full dissolution of chitin was achieved, which was when the solution became homogeneous, honey-like in viscosity, and turned an amber color. The vial was then taken out of the bath, the stir bar removed, and the solution centrifuged (Dynac Becton Dickinson Centrifuge model 42010, Sparks, MD, USA) for 10 min at 3000 rpm to separate any undissolved residue. The vial was then placed into an oven (70 °C) until the solution was free flowing.The hot solution was then poured over a glass plate and cast using a gapped stainless steel rolling rod (R&D Specialties, Webster, NY, USA) of RDS 100, 135, or 150 which had wire diameters of 0.26 mm, 0.41 mm, and 0.50 mm, respectively. The steel rolling rods (made of a stainless steel cylindrical bar wrapped with a wire of a certain diameter) evenly distributed the solution, passing it through the grooves between the wires, and forming a layer with a thickness proportional to the wire diameter Once the solution had settled to uniform thickness (a few seconds), the glass plate was placed into a DI water bath (29.52 cm × 10.79 cm × 29.52 cm, length × width × height) to allow film coagulation. After about 5 min, the film detached from the glass plate and rose to the surface. The glass plate was carefully removed, and the DI water was replaced about 4 times to wash the IL from the film. The IL was considered washed out after four washes; after two to three washes the IL was no longer visible in the bath, then two additional washes were done to be sure that the IL was not detected visually. The film was removed from the solution by sliding a piece of wax paper underneath and gently lifting the paper from the water, with the film mounted onto the wax paper. At this point the film was transferred to non-stick parchment paper for drying. To do this, the wax paper with the film mounted on it was carefully placed onto a piece of parchment paper with the film side face down. The wax paper was then carefully peeled away, leaving the wet film mounted on parchment paper. The films were then dried and characterized.
2.4 Drying methods
Five different drying methods were attempted: air, oven, acetone, press, and sc-CO2 drying. For air drying, the wet film was placed onto a piece of parchment paper and then left out in the air for 24 h, or until dry to the touch. Oven drying was done by placing the wet film onto a piece of parchment paper and then placing into the oven (90 °C) for 24 h, or until dry to the touch. Acetone drying was done by placing the wet film into a small bath of acetone, then removing and placing it onto a piece of parchment paper, which was left on the bench top until dry to the touch. Only press drying and sc-CO2 drying yielded viable films, so these are described below in more detail.To press dry a film, the wet film was placed between two pieces of parchment paper. This was then placed under a flat weight of 5 lbs. The film was left under the weight for approximately 24 h, or until the film was dry to the touch, and then carefully removed.
For sc-CO2 drying, the wet film was placed in anhydrous ethanol bath, which was changed 5 times over a 24 h period, followed by supercritical drying for 3 h. Drying was done in a DCP-1 Critical Point Drying Apparatus (Denton Vacuum, Moorestown, NJ, USA) equipped with an 11 cm × 3.5 cm (height × diameter) drying chamber. The wet film was placed between two pieces of porous Teflon coated mesh (100 Mesh T304 Stainless 0.0045′′ Wire Dia. Green PTFE, Part # 100X100S0045W36_PTFE, TVP Inc., Berkeley, CA, USA), and the encased film was placed into the sealed chamber, which was immersed in room temperature water. Liquid CO2 was allowed to flow into the chamber with the chamber closed for 7 min, then the chamber was vented for another 7 min. These 7 min/7 min cycles were repeated up to 12 times. After the last cycle, the chamber was sealed and the CO2 flow stopped. The room temperature water bath was then replaced with a 60 °C water bath. As the chamber sat in the hot bath, the pressure in the chamber rose to about 1750 psi. Once this pressure was reached, the chamber was slowly vented at a rate of about 100 psi min−1. At pressure zero, the chamber was opened and the dry film removed.
2.5 Fourier transform infrared spectroscopy (FT-IR)
FT-IR spectra were recorded on ground films using a Bruker Alpha FT-IR instrument, Bruker Optics Inc. (Billerica, MA, USA) featuring an attenuated total reflection (ATR) sampler equipped with a diamond crystal. Spectra were obtained in the range of 400–4000 cm−1.2.6 Thermogravimetric analysis (TGA)
TGA analyses were performed on a Mettler-Toledo TGA/DSC (Columbus, OH, USA). The instrument's internal temperature was calibrated by observing the melting point of Au, Zn, and In standards (melting points of 1064 °C, 419.5 °C, and 155.6 °C, respectively). The samples were analyzed in 70 mg alumina pans using dried air as a purge gas. Samples between 2–5 mg were heated from room temperature to 1000 °C and measured in the dynamic heating regime, using a constant heating ramp of 5 °C min−1 with a 30 min isotherm at 75 °C. Decomposition temperatures are reported as 5 wt% mass loss (T5%Dec) and 50 wt% mass loss (T50%Dec), respectively.2.7 Shrinkage
The shrinkage was evaluated by measuring the initial and final lengths and widths of each film before and after drying. The areas before and after were calculated and the percent shrinkage was obtained using the following formula: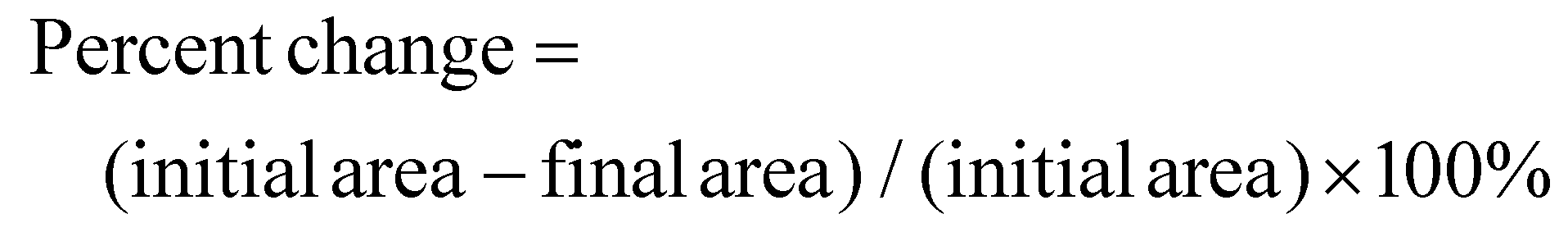
2.8 Optical microscopy
The thickness of the film was measured using a Motic BA 200 Microscope (Carlsbad, CA, USA) equipped with an XLI 2.0 camera (XL Imaging, Houston, TX, USA) at magnifications of 40× and 100×. The images were analysed with XLI-Cap image analysis software that was used to measure the thickness. The thickness of each of the films rolled with the different rolling rods was measured by focusing on an edge of the film and then measuring the distance across using the XLI-Cap image analysis software.2.9 Scanning electron microscope (SEM)
SEM was run using a LYRA3 TESCAN (Brno, Czech Republic). Films were mounted onto carbon tape and run in a low vacuum environment to prevent charging. All films were scanned using 15 kV electron beam, with a working distance of 10 to 12 mm.2.10 Tensile strength
Tensile testing was conducted on three different films of three different thicknesses. To obtain films for tensile testing, about 10 mL of 2.5 wt% IL/chitin solution was prepared and used to obtain a large enough film. Films for tensile testing were rolled with selected rolling rod to be 5.08 to 15.24 cm wide and 5.08 to 15.24 cm long. Films with uniform thickness and no obvious flaws were selected and used for testing. The films were cut into strips 2.54 cm wide and 5.08 to 15.24 cm long. Tensile strength (MPa) was calculated by dividing the load at break (Newtons) by the original cross cut area.2.11 Water content
Sc-CO2 dried films and press dried films were placed into separate 20 mL glass vials and were covered with aluminium foil with pin holes. These were weighed, placed in an oven at 80 °C overnight, and then reweighed the next day. The percentage difference in weight was calculated to determine the water content of the films.2.12 Caffeine loading and release
To prepare a saturated solution of caffeine, 16 g of caffeine was added to 1 L of DI water. The freshly prepared wet chitin film was immersed in a Petri dish filled with caffeine solution, covered and allowed to soak for ca. 24 h; the solution was replaced once. The film was then removed and press dried by the usual method. The remaining stock solution was kept in a bottle for further use.In order to load sc-CO2 dried films with caffeine, they were cast, coagulated and washed as usual. Before the films were sc-CO2 dried, they were placed in super-saturated ethanolic caffeine solution which was prepared by dispersing 6.50 g of caffeine into 500 mL of anhydrous ethanol. The ethanolic solution was placed in a 1 L bottle and capped until used. Approximately 15 mL of caffeine solution was added to the Petri dish, and the film was placed in. The caffeine solutions were replaced once within 72 h. After the loading period, the films were dried by the regular supercritical drying procedure outlined in the drying section.
For the release, a small piece of sc-CO2 dried film was weighed (2.0(1) mg) and then placed into a 3.5 mL spectrophotometer quartz cuvette (VWR, Radnor, PA, USA) along with 3.7 mL PBS solution. The PBS solution was prepared by dissolving one tablet in 200 mL DI water. The film was hooked to a syringe needle to prevent it from dropping to the bottom of the cuvette and to ensure the UV light would pass through the PBS solution without being blocked by the film, once in the spectrophotometer. The cuvette along with the film and solution was then placed into a UV-vis spectrophotometer (UV-1800 Shimadzu Corporation, Canby, OR, USA). The scanned wavelength range was set from 200 to 400 nm as caffeine has a molar absorptivity of 273 nm. The data was collected for approximately 24 h, with a 5, 10, 25, 35, 125 min, and finally around 14–16 h interval. The experiment was run in duplicate.
3. Results and discussion
To develop a platform for the preparation of biocompatible chitin films with tunable properties, the parameters that influence the final properties of the films need to be well understood. The most critical parameters to consider when preparing chitin films are: chitin source, loading of chitin in IL, size of the rod used to cast the film, and the drying method (Table 1).| Parameter | Effect |
|---|---|
| Chitin source | The molecular weight of the chitin (and entanglement of the polymer chains) determines the stability of the films once rolled and this depends on the chitin source |
| Loading of chitin in IL | The loading of the chitin in the IL determines the viscosity of the solution and thereby the ability to cast |
| Rolling rod size | Rolling rods of different size determine the thickness of the dried film |
| Drying method | The drying method determines the flexibility, strength, porosity, and morphology of the film |
3.1 Film preparation
The first step of chitin film preparation was to dissolve the chitin source in [C2mim][OAc], to form a homogenous chitin solution. Film preparation was attempted using commercially available PG chitin and IL-regenerated chitin (IL-chitin). Solutions of each were prepared by the dissolution of the chitin source in the IL to obtain 2.5 wt% chitin solutions (the mass loading used was based on our previous work preparing chitin fibers and hydrogels).18,20–22 The film cast from the PG-chitin solution yielded a very thin, web-like film which immediately broke apart after casting, likely due to its low molecular weight. This is a phenomenon that we have observed previously when preparing films from PG chitin/IL solution. One reason could be that the PG chitin contains more proteins, which increase the viscosity of the solution, but wash out once placed into the coagulation bath. During coagulation, the low molecular weight chains come apart due to lower entanglement density, and the film breaks apart. The regenerated IL-chitin can be cast into a film and stays together as the IL leaches out due to the longer polymer chains, which yield more chain entanglements.31 The film prepared from IL-chitin was stable during all steps of preparation, and yielded flexible films which could be handled with ease. With the choice of chitin source made, the ideal loading of chitin in the IL had to be determined. The loading of the solution determined the ease with which a film could be rolled and the ability of the cast film to be handled. Five chitin/IL solutions ranging from 2 wt% to 3 wt% chitin, in increments of 0.25 wt%, were prepared by thermal dissolution of chitin in IL. Each solution was used to cast a film using the procedure schematically shown in Fig. 2. For these initial steps, films were characterized only visually (appearance and thinness) and by resistance to manipulation (ability to handle the film). It was found that a chitin/IL solution of 2.5 wt% yielded films that appeared uniform, flexible, and could be easily handled. Chitin solutions with loading less than 2.5 wt% chitin gave free flowing solutions and yielded films that were excessively thin, weak, and that could not be removed from the water bath. Solutions with chitin loading of more than 2.75 wt% were too viscous and caused difficulty in rolling, resulting in films of non-uniform thickness (Table 2). The 2.5 wt% chitin/IL solution was thus selected for all subsequent film casting. | ||
| Fig. 2 Preparation chart for chitin films. | ||
| wt% Chitin | Results |
|---|---|
| 2.00 | Film was very thin, and fell to pieces |
| 2.25 | Tougher than 2.0 wt% chitin film, but was too difficult to handle |
| 2.50 | Film was easy to manipulate and was easy to roll |
| 2.75 | Film was tougher than 2.5 wt% chitin film, but very difficult to roll |
| 3.00 | Yields film, but pouring and rolling was very difficult |
After the solutions were tested for casting viability, small aliquots of the prepared solutions were used for viscosity measurements, which were conducted from 40–80 °C (solutions were heated in an oven at 80 °C immediately prior to casting). Viscosity was important for the casting of solution, though it was ultimately the ease of rolling of the solutions and the ability to handle the cast films that dictated the choice of chitin load. Viscosity curves for each of the solutions are shown in Fig. 3. As seen in the figure, solutions over 2.5 wt% had viscosities over 1000 cP at 40 °C, making manipulation difficult. Solutions under 2.5 wt% had viscosities less than 500 cP at 40 °C, which also led to difficulty of manipulation. The solutions cooled as they were cast, and, because of this, viscosity was not constant during the casting process. However, as mentioned above, the viscosity value itself during the process is not crucial.
 | ||
| Fig. 3 Viscosity of IL-chitin solutions from 40 to 80 °C. | ||
Seven rolling rods were tested, and three were selected to be used in film preparation. Rolling rods RDS 100 (wire diameter 0.26 mm), RDS 135 (wire diameter 0.41 mm), and RDS 150 (wire diameter 0.50 mm), yielded films that had visually uniform thickness and were strong enough to be submerged into a coagulation bath. Rods with wire diameters less than 0.26 mm yielded films that were too thin and difficult to manipulate, while rods with wire diameters larger than 0.50 mm resulted in films of non-uniform thickness.
Once rolled onto the glass plate, the plate with mounted chitin solution was placed into a water bath in order to allow coagulation of the film as the IL leached into the water. As the IL leached out, the film slowly released from the glass casting support, becoming free floating and transparent. Films were removed from the water by sliding wax paper underneath the film to mount, and then carefully removing both. The film was then transferred to another surface (parchment paper) for drying because films dried on the wax paper would adhere and could not be removed. Air drying attempts were made on glass, wax paper, aluminium foil, and parchment paper. This was a critical step, since the film must be mounted on a material for drying, but adhesion must be avoided. This was an issue with chitin films on surfaces likely due to the hydrogen bonds that the film forms with the surface as it dries. Parchment paper was chosen as it allowed for easy removal of films, likely because parchment paper is coated with a non-stick, non-polar silicone coating. With the other drying surfaces (glass, wax paper, and aluminium foil) the chitin film stuck once dry and could not be removed.
3.2 Drying of films
Once a drying mount was selected, five drying methods were attempted on films cast from the RDS 135 rod (Fig. 4). This rod was chosen because it was in the middle of the range of rods that produced uniform films. Drying of the film was crucial because the drying method determined the film's final properties, such as flexibility, uniformity, and porosity. Three of the tested drying methods, i.e. air drying, oven drying, and acetone drying, led to shrivelled films that were deemed unusable. We speculate that conventional ambient drying techniques, such as air drying or oven drying, comprise the removal of the solvent (water or acetone, in the case of acetone drying) by evaporation. This quick evaporation of solvent resulted in strengthening of the intermolecular hydrogen bonding between the biopolymer chains, which led to a strong 3D network, collapse of the pores, shrivelling, and much lower surface areas. The percent shrinkage from wet to dry for oven, air, and acetone was 46%, 48%, and 50%, respectively. | ||
| Fig. 4 Chitin films dried by different methods. | ||
Press drying and sc-CO2 drying yielded flexible films that could be easily handled and showed no visible signs of shrivelling. For press drying, severe shrivelling was prevented by placing ca. 5 lbs of weight on the film as it dried. This prevented hydrogen bonding other than that in-plane for the most part, though some shrinkage could be seen under SEM microscopy. For sc-CO2 dried films, structure collapse was prevented by means of highly pressurized supercritical fluid. It is a known phenomenon of supercritical fluid drying, where pore collapse is prevented by creating a mixture of the supercritical CO2 with the solvent which has no surface tension inside the structure, and the fluid is removed.32 Area of the dry films decreased compared to the wet film, although there was no shrivelling observed. The change in area was found by comparing the area of the wet film to the area of the dry film. Films obtained after press drying decreased in area less so (20%) than corresponding sc-CO2 dried film (28%), likely due to the heavy weight which prevented shrinkage. Once it was determined that press drying and sc-CO2 drying yielded flat and flexible films, these drying methods were used exclusively and subsequent films of different sizes were made.
3.3 Purity of dry films
FTIR on dry, ground press dried films revealed no peaks related to the IL, (see ESI, Fig. S1†) indicating that the IL concentration was below the sensitivity of the technique (this has been observed in our previously published chitin materials).20 Based on previously published FTIR characterization of the IL,30 five diagnostic peaks were identified, two of which overlap with chitin. Three of the five diagnostic peaks (the sp2 C–H stretching peaks at 3100 cm−1, the ring in-plane asymmetric stretching at 1190 cm−1, and the weak asymmetric bending of the HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH at 770 cm−1) were not observed in the chitin spectrum. Moreover, overlapping peaks (such as the IL peaks at 1560 cm−1 and 1390 cm−1) have significantly different intensities. Chitin has been extensively characterized by FTIR spectroscopy,29 and the spectra obtained here are consistent with chitin.
CH at 770 cm−1) were not observed in the chitin spectrum. Moreover, overlapping peaks (such as the IL peaks at 1560 cm−1 and 1390 cm−1) have significantly different intensities. Chitin has been extensively characterized by FTIR spectroscopy,29 and the spectra obtained here are consistent with chitin.
The thermal stability was also recorded. TGA demonstrated the loss of water and the normal chitin decomposition step, with T5%Dec = 253.2 °C, T50%Dec = 343.2 °C, and 6.2% ash. No proteins or CaCO3 were observed in the final film (see ESI, Fig. S2†).
3.4 Film Thickness
Once dry, the thickness of the film was measured using microscopic imaging software. Thickness depended not only on the rolling rod used, but also on the selected drying method (press or sc-CO2). As expected, larger wire sizes led to thicker films, and sc-CO2 dried films were more than twice as thick as their press-dried counterparts due to the lack of pore collapse. The thickness for each film is presented in Table 3.| Rolling rod | Press film thickness (μm) | sc-CO2 film thickness (μm) |
|---|---|---|
| RDS 100 (0.26 mm wire) | 2.9(8) | 6.7(8) |
| RDS 135 (0.41 mm wire) | 5.4(3) | 11.0(4) |
| RDS 150 (0.50 mm wire) | 5.7(4) | 14.2(8) |
3.5 Film morphology
To study and compare the surface morphology of press and sc-CO2 dried films, visual, optical microscopy, and SEM were used. Appearance varied drastically depending on the drying technique used. Films dried by pressing were translucent with a brown hue (Fig. 5, left). The brown hue was likely due to the denser packing of the polymer chains. Press drying flattens the layers together and forces the water out, leading to the removal of pores and a more condensed structure. Films dried by sc-CO2 drying appeared to have smooth surfaces, were completely white and opaque, and were sticky to the touch (Fig. 5, right). The stickiness was possibly due to the greater presence of free hydroxyl groups available for hydrogen bonding on the surface of the film. Because the sc-CO2 drying prevents structural collapse, many hydroxyl groups may stay free rather than interacting with one another, yielding a sticky surface. Though initially flat, when a sc-CO2 dried film was exposed to air for extended time, it gained a slight brown hue and began to curl up, possibly due to the interaction of the film with water molecules present in the atmosphere. | ||
| Fig. 5 Press dried (left) and sc-CO2 dried (right) films. | ||
Optical microscopy was used to assess the differences in the porosity of the two types of films (Fig. 6). Press dried films were lighter (thinner, so more light passed through). The press dried film exhibits a nonporous form, as expected due to the pressing of the film, while the sc-CO2 films exhibited a porous structure and 3D shape (a phenomenon similar to chitin and cellulose materials).33–35 The defects seen in the press dried films are surface defects, bumps, and shrinkage, which were confirmed by SEM. It should be noted that the color of the films under the microscope is not an indication of the actual color of the film, but of the thickness of the material; the sc-CO2 dried film appeared browner in color because it was thicker and less light passed through it.
 | ||
| Fig. 6 Press dried (left) and sc-CO2 dried (right) films at 100× magnification. | ||
SEM was used to better analyse and compare the surface morphology of the press dried and sc-CO2 dried films (Fig. 7). The surface of the press dried films had more defects, bumps, and wrinkling. These defects were likely due to the shrinkage that occurs during the drying of the film.
 | ||
| Fig. 7 Top row, from left to right: press dried film (5.4(3) μm thick) and sc-CO2 dried film (6.7(8) μm thick). Bottom film from left to right: sc-CO2 film (14.2(8) μm thick) and its backside. Image magnifications: 150×–250×. (The magnifications are not the same, as magnification was gradually increased while taking the SEM and the best pictures were selected.) | ||
The sc-CO2 dried films had a much smoother surface, but had more foreign objects on the surface. This was likely due to the nature of the drying method, whereby the structure of the film when wet was not lost upon drying, leading to a smoother surface. The foreign objects on the surface are likely broken pieces of chitin that became stuck to the film after drying due to the stickiness of the film. The pores of the sc-CO2 film could be seen on the backside of one film (Fig. 7).
3.6 Tensile strength
Tensile strength is also important since will dictate the material application field (e.g., packaging, biomedical materials, or membranes). Tensile strength was tested for both press dried and sc-CO2 dried films (Table 4). The strengths of the press dried films made from RDS 100, RDS 135, and RDS 150 were 19(2) MPa, 17(7) MPa, and 18(1) MPa, respectively (see ESI, Fig. S3†). There was no statistical variation in the tensile strength of the different films, the difference in MPa was due to the difference in thickness of the films, but the force in Newtons was the same (Pa = N m−2).| Rolling rod | Press film strength (MPa) | sc-CO2 film strength (MPa) |
|---|---|---|
| a The films tore very easily resulting in small holes thus the films were unsuitable for the testing. b The films curled up/difficult to unroll to be loaded for testing. | ||
| RDS 100 (0.26 mm wire) | 19(2) | N/Aa |
| RDS 135 (0.41 mm wire) | 17(7) | 2.6(0.2) |
| RDS 150 (0.50 mm wire) | 18(1) | N/Ab |
The average tensile strength of the RDS 135 sc-CO2 dried film was much less than that of press dried films, with a tensile strength of 2.6(0.2) MPa (see ESI, Fig. S4†). The lower tensile strength can be related to the presence of pores and the lower density of chitin chain packing that results from sc-CO2 drying. Tensile testing was attempted on sc-CO2 films made with both the RDS 100 and RDS 150 rod, but it was too difficult to obtain films that could be tested. The RDS 100 films were weak and tore very easily which resulted in films with small holes and were thus unsuitable for testing; the RDS 150 films curled up and became too difficult to unroll enough to load into the tensile testing machine. Though weaker, elongation (how much the film stretched) for sc-CO2 dried films was greater than that of press dried films (6% vs. 1%).
3.7 Water content
Water content was measured gravimetrically for press dried and sc-CO2 dried films. The percentage of weight loss for all films was less than 0.1 wt%. No significant differences on water content were observed between the two drying methods or film sizes.3.8 Film swelling
The swelling ability of the film was tested by conducting rehydration experiments on both press and sc-CO2 dried films rolled from RDS 100, RDS 135, and RDS 150. Water uptake was measured by weighing the film dry and after rehydrating in a water bath for 24 h. When placed into water, press dried films would quickly “spread,” due to the immediate absorbance of water, and maintained their color and opacity over 24 h. Sc-CO2 dried films looked drastically different when placed into water, immediately losing their white color and gaining the brown hue of the press dried films, likely due to water filling the pores that had been left open from drying.An inversely proportional relationship between water uptake and film thickness was observed (Table 5), suggesting limited access to the internal structure of the films once dried. No significant difference was observed on the rehydration percentage of the films made using the same wire rod but different drying method for thinner films. The thicker RDS 150 films had a significant difference in rehydration depending on drying method. For these, films dried by sc-CO2 absorbed more water than those press dried, suggesting that water can penetrate more easily into the internal structure of the sc-CO2 dried film due to the increased porosity.
3.9 Drug loading and release
Because the films showed the ability to swell in water, the ability of chitin to act as a drug eluting membrane was tested. Caffeine was used as a model drug compound to test the capacity of the films for uptake and release of an active substance. Caffeine loading and release was first attempted on press dried films. To load, the coagulated wet film was placed into a saturated aqueous caffeine solution and then dried normally. Once dry, the film was characterized by optical microscopy which revealed crystals of caffeine on the surface of the film, regardless of the concentration of the caffeine bath and the loading time (Fig. 8, left). In an attempt to avoid this, the films were quickly rinsed with cold water before drying, but this resulted in no caffeine embedded into the film (determined by quantitative Nuclear Magnetic Resonance, qNMR spectroscopy, see ESI, Fig. S5†). Because the caffeine was only present on the surface rather than inside, it washed off immediately upon coming into contact with water and their use for release studies was discarded. Another method, the addition of caffeine to the IL-chitin solution before casting, was attempted. This was done by adding both 10 wt% and 20 wt% caffeine to the prepared IL-chitin solutions, but these also resulted in no caffeine in the film, as it was all washed out in the coagulation step. | ||
| Fig. 8 Images obtained with optical microscopy, 40× of (left) caffeine crystals on the surface of a press dried film; (right) sc-CO2 film embedded with caffeine (no visible crystals). | ||
For caffeine embedded sc-CO2 dried films, drying and loading was done in a single step. Films were prepared, coagulated and washed as previously described. Loading of the film was done prior to drying due to the collapse of structure that occurs when the dry film is wetted. The loading of the caffeine was done before drying, where the films were placed in saturated ethanolic caffeine solution and covered for 72 h, during which time the solution was replaced twice. Films were then sc-CO2 dried as described in the Experimental section. Optical microscopy on the dry film revealed clean surfaces with no crystals, indicating that the remaining caffeine was in the film rather than on the surface (Fig. 8, right).
It is also important to note that, although this study demonstrated the ability to load caffeine into our film, this method is limited by the solubility of caffeine in ethanol, which is relatively poor (1.5 g per 100 mL),36 and due to the solubility of caffeine in sc-CO2, it is likely that a portion of the loaded caffeine was removed during the drying process.37
Since sc-CO2 allows the use of other solvents (e.g., acetone, methanol), the selection of solvent will permit loading of different active pharmaceutical ingredients (APIs) with different solubilities, and allow for control of the concentration of the API in the material.
In order to measure the release of caffeine, the film was placed into PBS solution and caffeine release was recorded over a period of 2100 min (ca. 36 h) using UV-vis. Over the 36 h period, 100% of the loaded caffeine was released into the solution. About 80% of the caffeine in the film was released almost immediately (so-called burst release) within the first 20 min, with the remaining 20% of the caffeine released much more slowly over the remaining 20 h (Fig. 9). The experiment was conducted in duplicate; the curves of release perfectly matched. This ability of the sc-CO2 dried films for load and release of active substances is promising for the use of these films as drug-releasing membranes.
 | ||
| Fig. 9 (Top) Percentage of caffeine released; and (bottom) mg of caffeine released. | ||
4. Conclusions
We have demonstrated a versatile platform for the preparation of biodegradable chitin films by using an ionic liquid process. By controlling the thickness, chitin loading, and drying methods, the resulting morphology and efficacy of the films could also be controlled. For both press dried and sc-CO2 films, the morphology, tensile strength, rehydration, and guest loading and release capabilities were studied, and the properties of each of the films compared. Press dried films, when compared to similarly prepared sc-CO2 dried films, are thinner, less porous, and stronger, yielding films with properties promising for use in packaging applications with further optimization. Sc-CO2 dried films, on the other hand, are porous, sticky, and capable of loading and release of guests, yielding a material that may have potential as a drug releasing membrane. Further optimization of each type of film could lead to high value, more sustainable chitin-based materials.Author information
R. D. Rogers has a partial ownership of 525 Solutions and is a named inventor on related patent applications. J. L. Shamshina and G. Gurau are consultants of 525 Solutions. The University of Alabama and McGill University maintain approved Conflict of Interest Management Plans. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.Acknowledgements
The authors would like to thank the U.S. Department of Energy (DOE) SBIR Office of Science (DE-SC0010152) for support. This research was undertaken, in part, thanks to funding from the Canada Excellence Research Chairs Program.Notes and references
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- R. A. Gross and B. Kalra, Science, 2002, 279, 803–807 CrossRef PubMed.
- M. Dicker, P. Duckworth, A. Baker, G. Francois, M. Hazzard and P. Weaver, Composites, Part A, 2014, 56, 280–289 CrossRef CAS.
- R. C. Thompson, C. J. Moore, F. S. vom Saal and S. H. Swan, Philos. Trans. R. Soc., B., 2009, 364, 2153–2166 CrossRef CAS PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- J. F. Prudden, P. Migel, P. Hanson, L. Freidrich and L. Balassa, Am. J. Surg., 1970, 119, 560–564 CrossRef CAS PubMed.
- M. Poirier and G. Charlet, Carbohydr. Polym., 2002, 50, 363–370 CrossRef CAS.
- X. Hu, Y. Du, Y. Tang, Q. Wang, T. Feng, J. Yang and J. F. Kennedy, Carbohydr. Polym., 2007, 70, 451–458 CrossRef CAS.
- B. M. Min, S. W. Lee, J. N. Lim, Y. You, T. S. Lee, P. H. Kang and W. H. Park, Polymer, 2004, 45, 7137–7142 CrossRef CAS.
- C. K. S. Pillai, W. Paul and C. P. Sharma, Prog. Polym. Sci., 2009, 34, 641–678 CrossRef CAS.
- Ionic Liquids. Industrial Applications for Green Chemistry, ed. R. D. Rogers and K. R. Seddon, ACS Symposium Series 818, American Chemical Society, Washington, DC, 2002 Search PubMed.
- Y. Wu, T. Sasaki, S. Irie and K. Sakurai, Polymer, 2008, 49, 2321–2337 CrossRef CAS.
- K. Prasad, M. Murakami, Y. Kaneko, A. Takada, Y. Nakamura and J. Kadokawa, Int. J. Biol. Macromol., 2009, 45, 221–225 CrossRef CAS PubMed.
- K. Mundsinger, A. Muller, R. Beyer, F. Hermanutz and M. R. Buchmeiser, Carbohydr. Polym., 2015, 131, 34–40 CrossRef CAS PubMed.
- M. Shimo, M. Abe and H. Ohno, ACS Sustainable Chem. Eng., 2016, 4, 3722–3727 CrossRef CAS.
- R. P. Swatlowski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4497 CrossRef.
- M. B. Turner, S. K. Spear, J. D. Holbrey and R. D. Rogers, Biomacromolecules, 2004, 5, 1379–1384 CrossRef CAS PubMed.
- Y. Qin, X. Lu, N. Sun and R. D. Rogers, Green Chem., 2010, 12, 968–971 RSC.
- M. L. Maxim, J. F. White, L. E. Block, G. Gurau and R. D. Rogers, in Ionic Liquids: Science and Applications, ed. A. E. Visser, N. J. Bridges and R. D. Rogers, ACS Symposium Series, American Chemical Society, Washington, DC, 2012, pp. 167–187 Search PubMed.
- J. L. Shamshina, G. Gurau, L. E. Block, L. K. Hansen, C. Dingee, A. Walters and R. D. Rogers, J. Mater. Chem. B, 2014, 2, 3924–3936 RSC.
- P. S. Barber, C. S. Griggs, J. R. Bonner and R. D. Rogers, Green Chem., 2013, 15, 601–607 RSC.
- X. Shen, J. L. Shamshina, P. Berton, J. Bandomir, H. Wang, G. Gurau and R. D. Rogers, ACS Sustainable Chem. Eng., 2016, 4, 471–480 CrossRef CAS.
- M. Vieira, M. da Silva, L. Santos and M. Beppu, Eur. Polym. J., 2011, 47, 254–263 CrossRef CAS.
- R. Jayakumar, D. Menon, K. Manzoor, S. V. Nair and H. Tamura, Carbohydr. Polym., 2010, 82, 227–232 CrossRef CAS.
- P. Agrawal, S. Soni, G. Mittal and A. Bhatnagar, Int. J. Lower Extremity Wounds, 2014, 13, 180–190 CrossRef PubMed.
- B. Ramos, in Biopolymers: Biomedical and Environmental Applications, ed. S. Kalia, Wiley, Hoboken, NJ, 2011, pp. 559–573 Search PubMed.
- K. V. H. Prashanth and R. N. Tharanathan, Trends Food Sci. Technol., 2007, 18, 117–131 CrossRef CAS.
- V. Siracusa, P. Rocculi, S. Romani and M. Dalla Rosa, Trends Food Sci. Technol., 2008, 19, 634–643 CrossRef CAS.
- M. L. Duarte, M. C. Ferreira, M. R. Marvao and J. Rocha, Int. J. Biol. Macromol., 2001, 28, 359–363 CrossRef CAS PubMed.
- J. Kiefer, K. Obert, A. Bosmann, T. Seeger, P. Wasserscheid and A. Leipertz, ChemPhysChem, 2008, 9, 1317–1322 CrossRef CAS PubMed.
- J. L. Fetters, D. J. Lohse, D. Richter, T. A. Witten and A. Zirkel, Macromolecules, 1994, 27, 4639–4647 CrossRef.
- H. D. Gesser and P. C. Goswami, Chem. Rev., 1989, 89, 765–768 CrossRef CAS.
- P. Ratanajiajaroen and M. Ohshima, J. Supercrit. Fluids, 2012, 68, 31–38 CrossRef CAS.
- C. Tsioptsias, A. Stefopoulos, I. Kokkinomalis, L. Papadopoulou and C. Panayiotou, Green Chem., 2008, 10, 965–971 RSC.
- M. Chili, C. Rodriguez, J. J. Letourneau, F. Espitalier and R. Calvet, Supercritical CO2 Drying of Polysaccharidic Membranes Produced in Ionic Liquid: Application to Cellulose and Chitin, 9th International Symposium on SuperCritical Fluids, 2009.
- S. Budavari, The Merck Index, Merck & Co., Inc., Whitehouse Station, NJ, 12th edn, 1996, p. 1674 Search PubMed.
- U. Kopcak and R. S. Mohamed, J. Supercrit. Fluids, 2005, 34, 209–214 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02201d |
| This journal is © The Royal Society of Chemistry 2017 |