
Acetaldehyde as an ethanol derived bio-building block: an alternative to Guerbet chemistry†
Cameron M.
Moore
a,
Orion
Staples
a,
Rhodri W.
Jenkins
a,
Ty J.
Brooks
a,
Troy A.
Semelsberger
b and
Andrew D.
Sutton
*a
aChemistry Division, MS K558, Los Alamos, New Mexico, USA
bMaterial Physics Applications Division, MS K793, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: adsutton@lanl.gov
First published on 15th November 2016
Abstract
In this work, we describe a highly selective poly-aldol condensation of acetaldehyde, which can readily be obtained via dehydrogenation of ethanol. The process operates under mild temperatures (100 °C or less) using commercially available catalysts and exhibits excellent total carbon yield of C4+ products with good selectivity for C6 products. The products derived from the reactions described herein are shown to be candidate drop-in fuel replacements for compression ignition engines and precursors to valuable chemicals.
Global ethanol production from corn and cellulosic materials has grown massively over the past decade with worldwide production rising above 25 billion gallons (95 billion litres) per year in 2015.1 In the US, the Renewable Fuels Standard (RFS2) mandated by the Environmental Protection Agency (EPA) requires 36 billion gallons (136 billion litres) of renewable fuel to be blended with transportation fuel per year by 2022.2 Blending ethanol with gasoline, however, is unlikely to rise significantly above the current 10% (E10) “blend wall” in the US in the near future. This is partly due to the potential corrosiveness of ethanol toward common engine technologies and refueling infrastructure, as well as a low energy density when compared to gasoline (ca. 65%) and the high miscibility of ethanol with water.3 While some cars could utilise higher blends of ethanol fuel, such as E85 vehicles, they comprise less than 10% of the cars on the road in the US.4
One possibility for utilising the large surplus of bioethanol is through catalytic upgrading to provide higher molecular weight molecules that have physical and fuel performance properties that more closely match gasoline and diesel fuel. The Guerbet reaction is one such approach for upgrading ethanol and has been studied since 1899 when Marcel Guerbet first described the conversion of 1-butanol to 2-ethyl-1-hexanol using a strong base and metal oxide catalyst at high temperature.5 The Guerbet reaction is a complex sum of elementary reaction steps requiring a heterogeneous catalyst that contains acidic, basic, hydrogenation and dehydrogenation functionality all at once.6–8 Alternatively, homogeneous catalysts have also been employed for Guerbet upgrading of ethanol,9–11 however these systems are typically limited by low catalyst lifetimes and poor ethanol conversions. The fine tuning of catalyst properties and active sites required to achieve high selectivity and activity for both heterogeneous and homogeneous catalysts is therefore a significant challenge for employing Guerbet chemistry toward upgrading ethanol.
The Guerbet reaction sequence has been highly debated, but is generally accepted to proceed through three distinct reaction steps, (dehydrogenation, aldol condensation, and hydrogenation) each with specific thermodynamic considerations. For example, in the Guerbet reaction of 1-butanol to produce 2-ethyl-1-hexanol, the initial dehydrogenation of 1-butanol to produce butyraldehyde is calculated to be endergonic (“uphill”) at temperatures below 200 °C, while all other steps have been calculated to be exergonic (“downhill”) at these temperatures.6 Therefore, at lower temperatures, the Guerbet reaction is thermodynamically limited by the initial alcohol dehydrogenation reaction. Alternatively, at higher temperatures where alcohol dehydrogenation is exergonic, the aldol condensation step becomes less favoured and significant side reactions are observed leading to by-products such as aromatics, esters and acids.6
Other notable routes to produce fuels from ethanol include the proposed oligomerisation of ethylene12,13 (derived from the dehydration of ethanol) and high temperature ethanol processing over zeolites to yield complex mixtures of hydrocarbons.14 These methods are limited by their applicability toward ethanol or their propensity to form large quantities of C2 and C3 products, respectively. Acetone alkylation with ethanol and butanol has also been investigated as a method for producing higher molecular weight molecules that can be subsequently defunctionalized to produce fuels.15,16 We envisioned an alternative approach where the thermodynamic constraints of the Guerbet process could be alleviated by separating alcohol dehydrogenation and aldol condensation into separate steps to provide a selective process for producing higher molecular weight condensates from ethanol. In this way, the hydrogen produced from alcohol dehydrogenation could be employed for subsequent aldehyde upgrading reactions, which would positively impact the economics of this process since external hydrogen would not be required for upgrading. Given recent literature demonstrating the selective dehydrogenation of ethanol to acetaldehyde,17–19 as well the potential to directly produce acetaldehyde from biomass fermentation,20 we pursued the poly-aldol condensation of acetaldehyde as a means to selectively produce C4+ condensates that are candidate drop-in fuel replacements and valuable chemical precursors.
Previous studies on the self-condensation of acetaldehyde have largely focused on the selective dimerisation of acetaldehyde to yield crotonaldehyde, which is used in the production of numerous commodity chemicals.21 Investigations employing heterogeneous catalysts in flow reactors have targeted both acidic and basic catalysts to promote the aldol condensation reaction,22–24 and selectivities approaching 100% for crotonaldehyde have been reported.25 While there are numerous reports of dimerisation of acetaldehyde, there are comparatively fewer describing the formation of higher molecular weight products.26 In fact, the vapor phase aldol condensation of acetaldehyde catalysed by titanium dioxide was reported to be inhibited by the formation of C4+ products.25 In this case, catalyst deactivation was caused by the deposition of non-volatile, high molecular weight organic products onto the surface of the catalyst which inhibits further reaction of acetaldehyde. As a means to avoid catalyst inhibition by the formation of C4+ products, we therefore examined acetaldehyde condensation with heterogeneous catalysts in liquid phase batch reactors.
We first examined the condensation of acetaldehyde in ethanol solutions, which can be envisioned as a feedstock of partially dehydrogenated ethanol. Based on our prior investigations into poly-aldol condensations of carbonyl compounds,27 we employed Amberlyst 15 as a solid acid catalyst capable of promoting condensations, in combination with Pd/C as a hydrogenation catalyst to drive the aldol condensation reaction. Under a low partial H2 pressure (0.3 MPa), we observed formation of poly-aldol condensates with carbon chains ranging from C4 to C8 (Fig. 1A). Employing these conditions, we also observed the formation of diethyl-acetals, resulting from the reaction of ethanol with the aldehydes under acidic conditions. The total carbon yield of the observed products, including the diethyl-acetal derived from acetaldehyde, was 46%. The low observed total yield could be due to vaporisation of acetaldehyde or hydrogenation of acetaldehyde to yield ethanol, both of which would be indistinguishable pathways under the above reaction conditions. Moreover, the yield of C4+ aldol condensation products is likely further impeded by the acid-catalysed acetalisation of the aldehydes (Fig. 1B), which would limit the effective concentration of aldehydes in solution available for condensation.
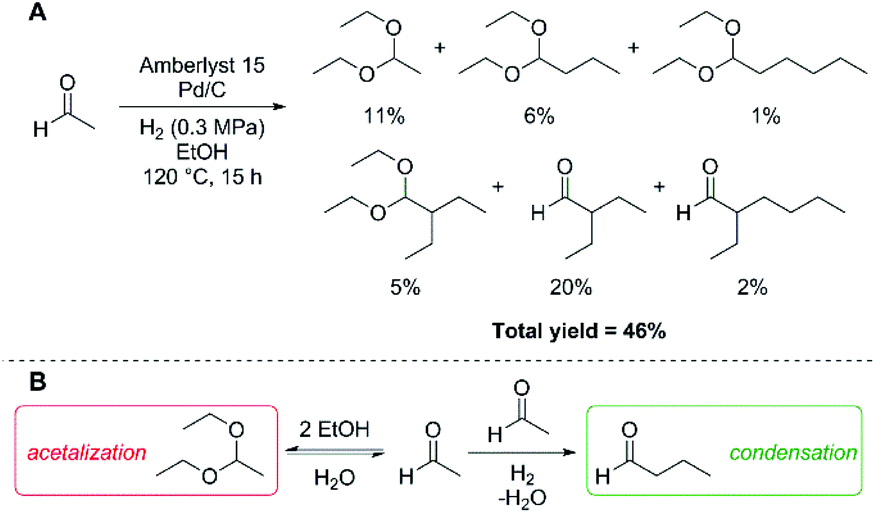 | ||
| Fig. 1 Poly-aldol condensation of acetaldehyde in ethanol solution (A; yields are based on carbon and determined by GC) and competition between acetalisation and condensation of acetaldehyde in ethanol solution (B). Conditions: acetaldehyde (8.90 mmol), Amberlyst 15 (0.30 g), Pd/C (10 wt%; 0.05 g), EtOH (4.0 mL). | ||
As an alternative to ethanol solutions, acetaldehyde condensation reactions were pursued in water solutions under low H2 pressures (Table 1, entries 1–5). Under these conditions, acetals were not formed from the condensation reaction, however the total yields of C4+ aldehydes were low (≤15%) after prolonged reaction times. These results are likely a consequence of an inhibitory effect of water (see ESI† for more details). To circumvent the issues encountered in ethanol and water solutions, we hypothesised that the acetaldehyde condensation reactions could be performed in an aprotic, organic solvent solution where acetalisation could not take place.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | P (MPa) | Time (h) | Yieldsa (%C) | |||
| C4 | C6 | C8 | Total | |||||
| a Yields are based on carbon and determined by GC, see ESI for more details. b 94% Ar, 6% H2. | ||||||||
| 1 | H2O | 60 | 0.2, H2 | 24 | <1 | 2 | <1 | 3 |
| 2 | H2O | 80 | 0.2, H2 | 24 | <1 | 13 | 2 | 15 |
| 3 | H2O | 80 | 0.3, H2 | 24 | <1 | 10 | 1 | 11 |
| 4 | H2O | 120 | 0.3, H2 | 3 | <1 | 7 | 1 | 8 |
| 5 | H2O | 120 | 0.7, H2 | 3 | <1 | 7 | <1 | 7 |
| 6 | C6H12 | 80 | 0.3, H2 | 1 | 9 | 38 | 5 | 51 |
| 7 | C6H12 | 100 | 0.3, H2 | 1 | 5 | 36 | 5 | 46 |
| 8 | C6H12 | 120 | 0.3, H2 | 1 | <1 | 21 | 4 | 25 |
| 9 | C6H12 | 40 | 1.7, Ar/H2b | 1 | 5 | 5 | 5 | 15 |
| 10 | C6H12 | 40 | 1.7, Ar/H2b | 3 | 6 | 9 | 2 | 17 |
| 12 | C6H12 | 60 | 1.7, Ar/H2b | 1 | 11 | 30 | 3 | 44 |
| 13 | C6H12 | 60 | 1.7, Ar/H2b | 3 | 7 | 45 | 4 | 56 |
| 14 | C6H12 | 80 | 1.7, Ar/H2b | 1 | 7 | 47 | 6 | 60 |
| 15 | C6H12 | 80 | 1.7, Ar/H2b | 3 | 2 | 53 | 8 | 62 |
| 16 | C6H12 | 100 | 1.7, Ar/H2b | 1 | 3 | 26 | 3 | 32 |
| 17 | C6H12 | 100 | 1.7, Ar/H2b | 3 | 1 | 34 | 5 | 40 |
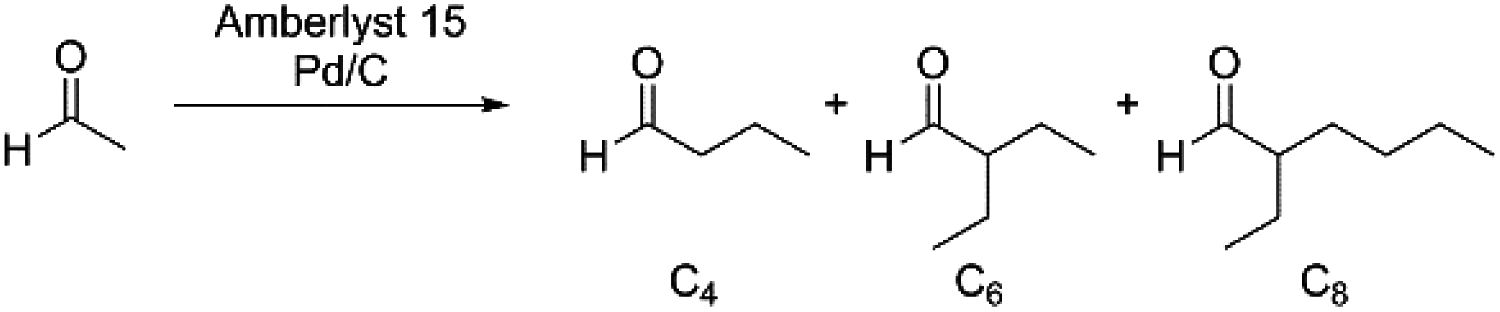
To this end, we examined the reaction of acetaldehyde in cyclohexane solution using Amberlyst 15 and Pd/C as catalysts at various temperatures under an H2 atmosphere (0.3 MPa). Under these conditions, we observed the formation of C4+ aldehydes as the major products, with a high selectivity for the C6 branched aldehyde. Increasing reaction temperatures were found to have a detrimental effect on the overall yield of the condensates (Table 1, entries 6–8). Alternatively, we examined the effect of using dilute H2 in Ar as the headspace gas, so as to increase the overall pressure of the reaction and avoid unwanted C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond reduction. Under these conditions, the overall yields of the products were higher in general than observed using low pressure H2. Again, increasing reaction temperatures had a detrimental effect on the overall yield of the condensate products (cf. entry 12 vs. entry 16). Heating at 80 °C for 3 h provided the best overall carbon yield of 62%, with a 53% yield of the C6 aldehyde product and an 8% yield of the C8 product (entry 15).28 The formation of the observed products can be explained by the initial self-condensation of acetaldehyde to produce butyraldehyde, which undergoes condensation with either acetaldehyde or butyraldehyde to yield the branched C6 or C8 aldehyde products, respectively (Scheme 1).
O bond reduction. Under these conditions, the overall yields of the products were higher in general than observed using low pressure H2. Again, increasing reaction temperatures had a detrimental effect on the overall yield of the condensate products (cf. entry 12 vs. entry 16). Heating at 80 °C for 3 h provided the best overall carbon yield of 62%, with a 53% yield of the C6 aldehyde product and an 8% yield of the C8 product (entry 15).28 The formation of the observed products can be explained by the initial self-condensation of acetaldehyde to produce butyraldehyde, which undergoes condensation with either acetaldehyde or butyraldehyde to yield the branched C6 or C8 aldehyde products, respectively (Scheme 1).
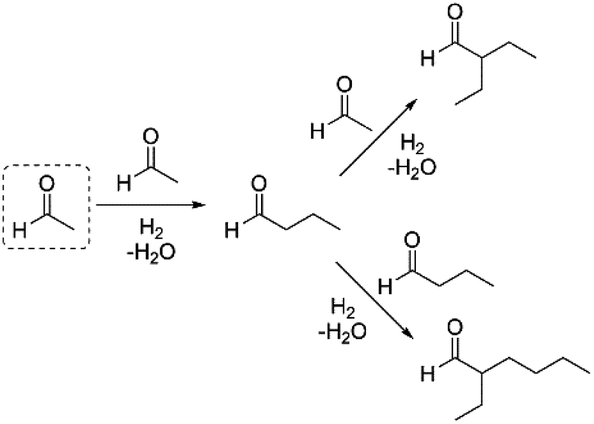 | ||
| Scheme 1 Pathways for formation of observed acetaldehyde condensation products. | ||
Increasing temperature was found to have a detrimental effect on overall yield under all reaction conditions screened. We therefore examined the acetaldehyde condensation reaction using a two-stage heating scheme, where an initial condensation was performed at a lower temperature, then immediately heated to an elevated temperature. We examined an initial stage of heating for one hour at 60 °C; which had provided the highest yield of butyraldehyde during our initial screening (Table 1, entry 6). Heating the acetaldehyde reaction mixture for one hour at 60 °C, followed by heating at 100 °C for three hours resulted in an overall carbon yield of 92% (Fig. 2A). The C6 branched aldehyde was again observed as the major product (79%), while the majority of the rest of the carbon was comprised of the C8 branched aldehyde (13%). Under these conditions, only 7% of the carbon remained unaccounted for by these three products. As a comparison, the best yield for the Guerbet upgrading of ethanol in a batch reactor we have encountered in the open literature is a 38% combined yield of C4+ condensates with 55% ethanol conversion at 230 °C using a lanthanum-modified nickel catalyst supported on alumina.29
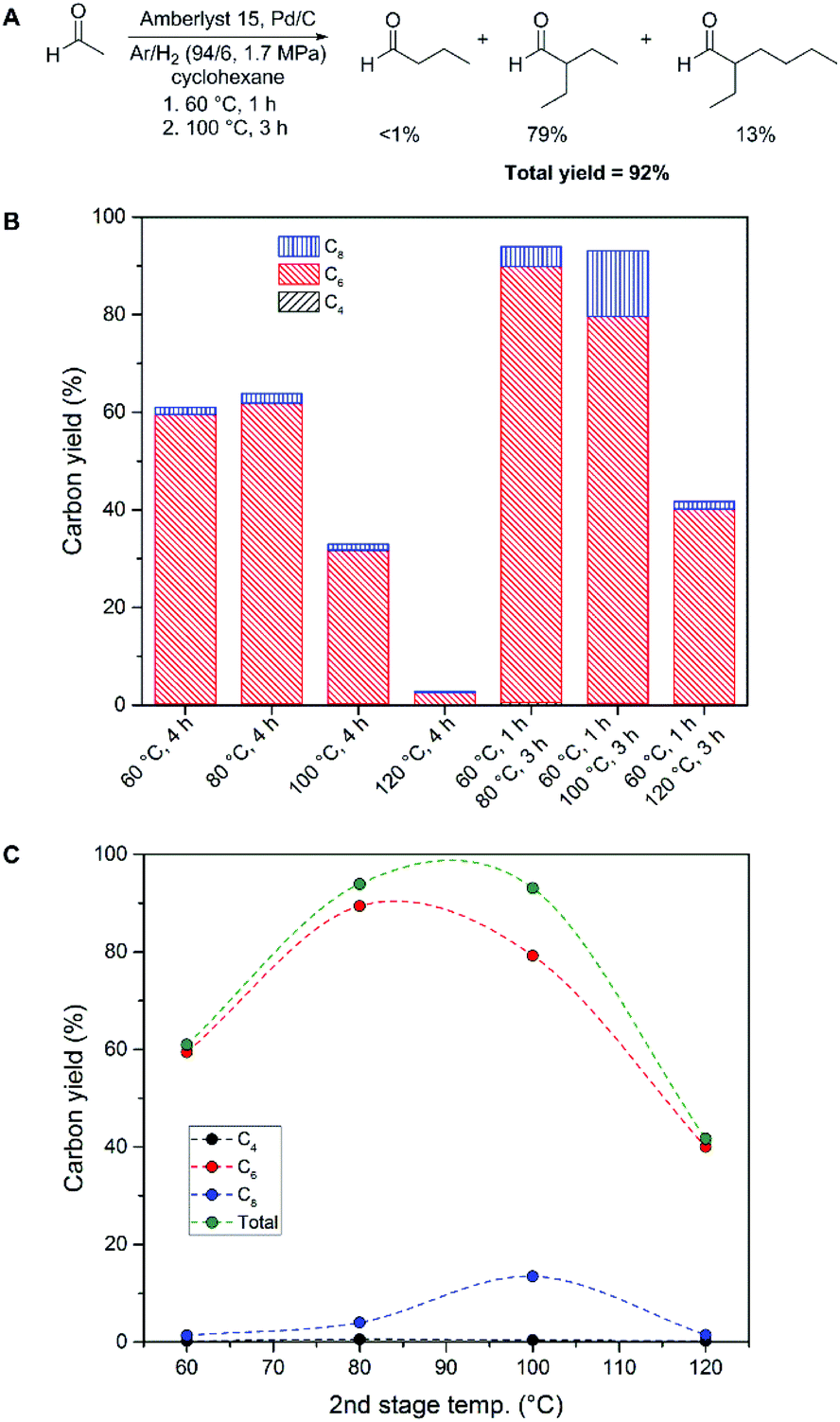 | ||
| Fig. 2 Optimized conditions for poly-aldol condensation of acetaldehyde in cyclohexane solution (A), effects of reaction temperatures and time on condensation yields (B) and comparison of second stage reaction temperature and condensation yields (C; 1 h at 60 °C, 3 h at specified temperature, lines are drawn to guide the eye). Acetaldehyde (4.43 mmol), Amberlyst 15 (0.30 g), Pd/C (10 wt%; 0.05 g), cyclohexane (4.0 mL). | ||
The effect of the two-stage heating scheme was further evaluated by comparison to heating solutions at fixed temperatures for a total of four hours. As can be seen in Fig. 2B, the overall yields of acetaldehyde condensation products are increased by performing two heating steps with an initial condensation at 60 °C, as compared to holding a constant temperature over the same time period. We attribute this behaviour to the formation of butyraldehyde at 60 °C, followed by further condensation at an elevated temperature. As an example, the initial formation of butyraldehyde from acetaldehyde at 60 °C is maximum after 30 minutes, and beyond this time point the butyraldehyde concentration begins to diminish (Fig. S3†). Futhermore, the effect of the two-stage heating scheme is most prominent with reactions at 120 °C, where a 3% overall yield was observed with a constant temperature, as opposed to 42% overall yield using an initial 60 °C reaction, followed by heating to 120 °C (Fig. 2B). Increasing the second stage temperature beyond 100 °C was also found to have a detrimental effect on the overall yield of the poly-aldol condensation process (Fig. 2C). Optimum yields of 93 and 92% were obtained when the second stage heating was performed at 80 and 100 °C, respectively.
As a further demonstration of the efficiency of our system for aldol condensations of aldehydes, we examined the self-condensation of butyraldehyde. We performed the condensation reaction at 100 °C using dilute H2 in Ar with Pd/C and Amberlyst 15 as catalysts. Under these conditions, we observed a near quantitative yield of 2-ethylhexanal (Fig. 3A). The observed selectivity is superior to a related palladium-impregnated Amberlyst resin that promotes the condensation of crotonaldehyde to 2-ethylhexanal (67% selectivity, 98% conversion) in supercritical carbon dioxide30 and a Ce-modified γ-Al2O3 for the condensation of butyraldehyde to 2-ethyl-2-hexenal (91% selectivity, 91% conversion).31 This result further demonstrates the importance of the reaction temperature and subsequent efficiency of the acetaldehyde poly-aldol condensation reaction in the present system. Using Ni/SiO2–Al2O3, 2-ethylhexanal can be readily hydrogenated to provide 2-ethyl-1-hexanol in excellent selectivity (Fig. 3A). The commercial importance of this alcohol is illustrated in Fig. 3B, where a variety of chemical products manufactured using this alcohol are highlighted. Currently, approximately 3 × 106 Mt of 2-ethyl-1-hexanol are produced annually; the synthesis begins with the homogeneously-catalysed hydroformylation of propene yielding butyraldehyde, then subsequent condensation using NaOH and finally hydrogenation.32 Alternatively, this alcohol could be produced by dehydrogenation of bio-derived 1-butanol to yield butyraldehyde,33 followed by condensation and hydrogenation. Futhermore, recent analyses have demonstrated that co-production of bioproducts can enable low cost biofuels by offsetting production expenses.34,35 In line with this strategy, the poly-aldol condensation process utilises a single chemical input and is capable of producing three distinct product streams: valuable chemicals (such as 2-ethyl-1-hexanol) and various biofuel molecules (vide infra) which are potentially suitable for gasoline or diesel application.
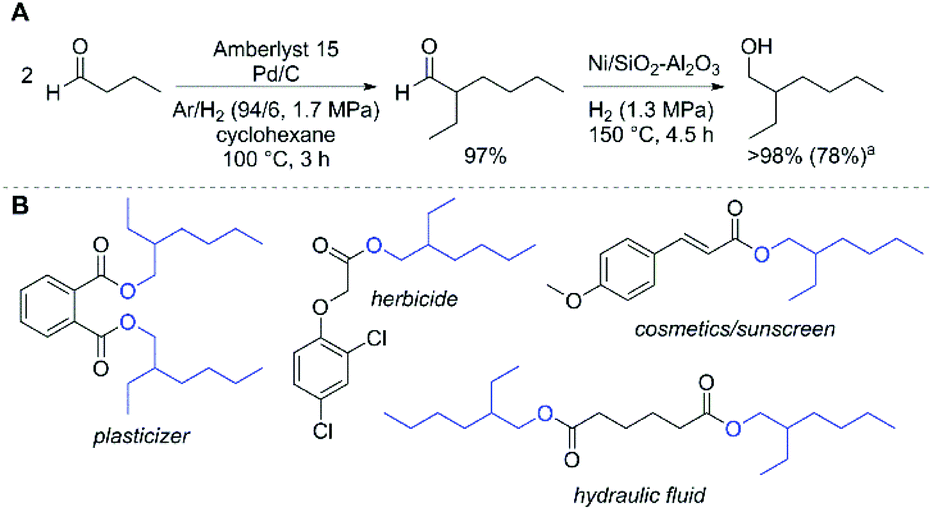 | ||
Fig. 3 Route to produce 2-ethyl-1-hexanol from butyraldehyde (A; a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) isolated yield, see ESI† for full reaction conditions), and select molecules derived from 2-ethyl-1-hexanol and their applications (B). isolated yield, see ESI† for full reaction conditions), and select molecules derived from 2-ethyl-1-hexanol and their applications (B). | ||
To determine the potential suitability of these products to be employed as potential drop-in fuel replacements, we predicted their corresponding derived cetane number (DCN) and research octane number (RON). These two measures of fuel performance are dimensionless numbers, usually within 0–100, and are derived from the autoignition of reference standards to provide a basis for determining the application of fuels for either spark ignition (gasoline, high RON) or compression ignition (diesel, high cetane number, or CN) engines.36 A group contribution method based on functional group additivity was employed for DCN prediction,37 in combination with a relationship between RON and DCN.38
While this method provides solely predictions, it yields a first approximation of the possibility of a given product as a gasoline or diesel fuel. The results of this analysis are presented in Fig. 4 and demonstrate that the majority of the products described herein are potentially suitable for diesel applications, based on their respective predicted DCN's.39 It should be noted that more extensive fuel property testing is required to deem these molecules as suitable blendstocks or drop-in replacements. In the case of the alcohols, the predicted RON values (or measured in the case of 1-butanol) are near the minimum values required for gasoline fuels. From a fuel combustion perspective, there is no advantage to performing hydrodeoxygenation of these molecules to obtain branched alkanes. This notion is in contrast to the large majority of synthetic efforts to fully deoxygenate bio-derived molecules to produce fuels. Specifically, 2-ethylbutane and 2-ethylhexane (derived from the hydrodeoxygenation of 2-ethylbutyraldehye and 2-ethylhexanal, respectively) have measured cetane numbers and octane numbers that fall outside of the range for both diesel and gasoline, respectively. This is an important distinction given that hydrodeoxygenation of these, and any other oxygenates would cause a negative impact on the economics of an industrial process since this transformation requires added H2. This fact further demonstrates the benefit to performing preliminary fuel property analyses, given that alkanes are not always ideal fuel targets for bio-derived inputs,36 and the isolation of oxygenates requires less processing steps, separations and overall less energy input when compared to alkanes. Additionally, the formation of acetals incorporates additional ethanol equivalents into a single molecule while increasing the predicted DCN when compared to the alcohols (Fig. 4B), and accordingly these molecules are ideal future fuel targets.
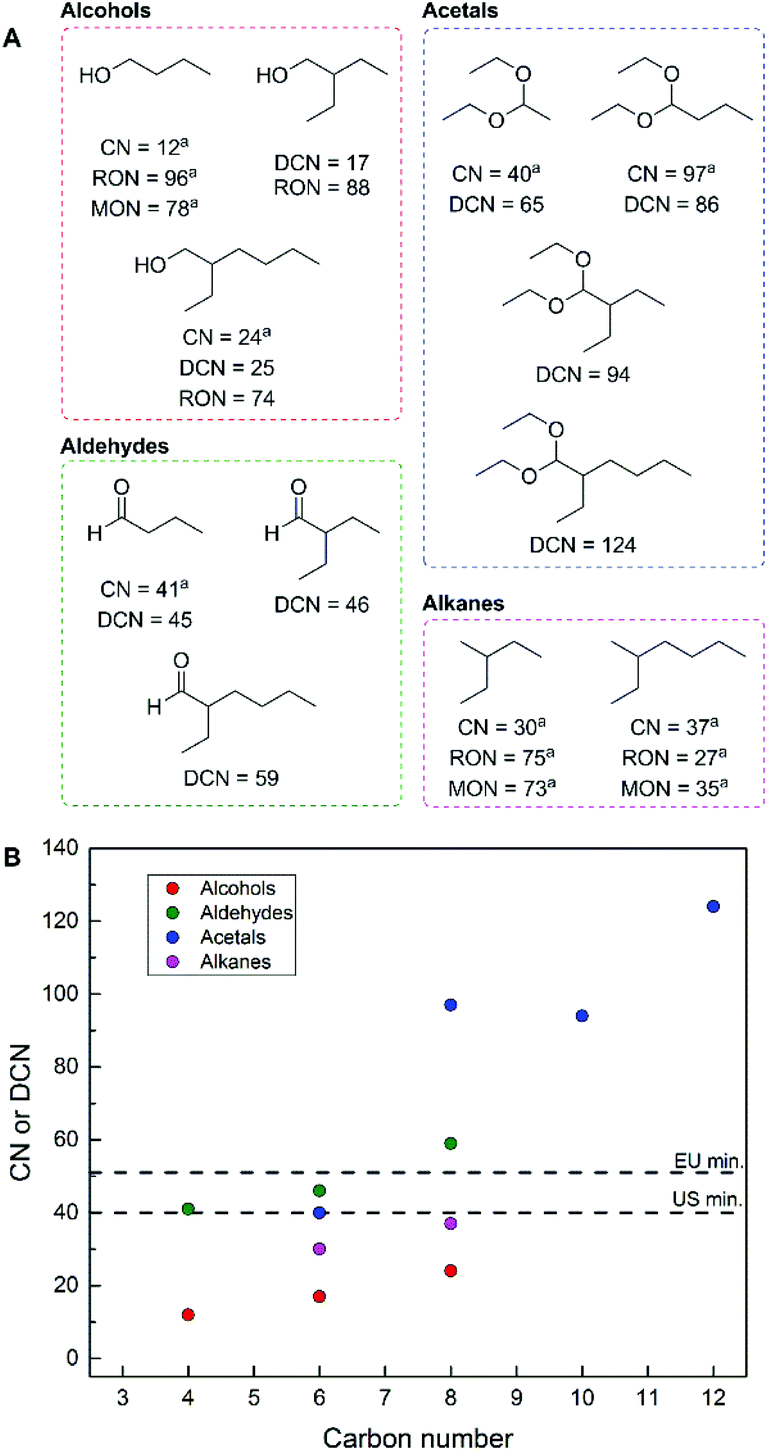 | ||
| Fig. 4 Predicted derived cetane number (DCN) and research octane number (RON, where applicable) for select molecules that can be produced using this process (A; areported experimental values, CN = cetane number) and graphical representation of cetane data from A with respect to minimum allowed fuel values for the EU and US (B). | ||
Conclusions
In conclusion, we have demonstrated a fully heterogeneous catalyst system (Amberlyst 15 and Pd/C) capable of efficiently upgrading acetaldehyde to fuels and chemicals. When combined with known ethanol dehydrogenation, this process represents an alternative upgrading approach to Guerbet chemistry by separating the reaction steps proposed within the Guerbet pathway. Yields of C4+ products using this methodology can be >93% with high selectivity for C6 products. Precise selection of reaction temperature plays a key role in the efficiency of the process and a two-stage heating protocol was found to provide the highest yields of condensation products. Preliminary fuel performance analyses demonstrate that the products derived from the poly-aldol condensation of acetaldehyde could be competent drop-in replacements for diesel fuel. Future efforts will be focused on developing this chemistry in flow, along with full fuel property testing, and will be described in due course.Acknowledgements
We thank the LANL Laboratory Directed Research and Development (LDRD) program (LDRD20160095ER) and the Office of Energy Efficiency & Renewable Energy (EERE) Bioenergy Technology Office (BETO) for financial support. Additionally we thank the LANL LDRD program for a Director's Postdoctoral Fellowship to CMM. Los Alamos National Laboratory is operated by Los Alamos National Security, LLC, for the National Nuclear Security Administration of the U.S. Department of Energy under contract DE-AC5206NA25396.Notes and references
- Fueling a High Octane Future: 2016 Ethanol Industry Outlook, Renewable Fuels Association, 2016, http://www.ethanolrfa.org/wpcontent/uploads/2016/02/Ethanol-Industry-Outlook-2016.pdf Search PubMed.
- Renewable Fuel Standard, US Department of Energy Alternative Fuels Data Center; http://www.afdc.energy.gov/laws/RFS, accessed August 2016.
- M. Balat, Energy Sources, Part A, 2009, 31, 1242–1255 CrossRef CAS.
- Flexible Fuel Vehicles, US Department of Energy Alternative Fuels Data Center; http://www.afdc.energy.gov/vehicles/flexible_fuel.html, accessed August 2016.
- M. Guerbet, C. R. Acad. Sci. Paris, 1899, 128, 511 Search PubMed.
- D. Gabriels, W. Y. Hernandez, B. Sels, P. Van Der Voort and A. Verberckmoes, Catal. Sci. Technol., 2015, 5, 3876–3902 CAS.
- J. T. Kozlowski and R. J. Davis, ACS Catal., 2013, 3, 1588–1600 CrossRef CAS.
- T. Moteki and D. W. Flaherty, ACS Catal., 2016, 6, 4170–4183 CrossRef CAS.
- K.-N. T. Tseng, S. Lin, J. W. Kampf and N. K. Szymczak, Chem. Commun., 2016, 52, 2901–2904 RSC.
- S. Chakraborty, P. E. Piszel, C. E. Hayes, R. T. Baker and W. D. Jones, J. Am. Chem. Soc., 2015, 137, 14264–14267 CrossRef CAS PubMed.
- R. L. Wingad, P. J. Gates, S. T. G. Street and D. F. Wass, ACS Catal., 2015, 5, 5822–5826 CrossRef CAS.
- B. G. Harvey and H. A. Meylemans, Green Chem., 2014, 16, 770–776 RSC.
- B. G. Harvey, W. W. Merriman and T. A. Koontz, Energy Fuels, 2015, 29, 2431–2436 CrossRef CAS.
- C. K. Narula, Z. Li, E. M. Casbeer, R. A. Geiger, M. Moses-Debusk, M. Keller, M. V. Buchanan and B. H. Davison, Sci. Rep., 2015, 5, 16039 CrossRef CAS PubMed.
- P. Anbarasan, Z. C. Baer, S. Sreekumar, E. Gross, J. B. Binder, H. W. Blanch, D. S. Clark and F. D. Toste, Nature, 2012, 491, 235–239 CrossRef CAS PubMed.
- S. Sreekumar, Z. C. Baer, E. Gross, S. Padmanaban, K. Goulas, G. Gunbas, S. Alayoglu, H. W. Blanch, D. S. Clark and F. D. Toste, ChemSusChem, 2014, 7, 2445–2448 CrossRef CAS PubMed.
- Y. Guan and E. J. M. Hensen, Appl. Catal., A, 2009, 361, 49–56 CrossRef CAS.
- F.-W. Chang, H.-C. Yang, L. S. Roselin and W.-Y. Kuo, Appl. Catal., A, 2006, 304, 30–39 CrossRef CAS.
- T. Lu, X. Li, L. Gu and Y. Zhang, ChemSusChem, 2014, 7, 2423–2426 CrossRef CAS PubMed.
- H. Zhu, R. Gonzalez and T. A. Bobik, Appl. Environ. Microbiol., 2011, 77, 6441–6450 CrossRef CAS PubMed.
- J. Blumenstein, J. Albert, R. P. Schulz and C. Kohlpaintner, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- W. Kagunya and W. Jones, Appl. Clay Sci., 1995, 10, 95–102 CrossRef CAS.
- L. M. Baigrie, R. A. Cox, H. Slebocka-Tilk, M. Tencer and T. T. Tidwell, J. Am. Chem. Soc., 1985, 107, 3640–3645 CrossRef CAS.
- Z. D. Young, S. Hanspal and R. J. Davis, ACS Catal., 2016, 6, 3193–3202 CrossRef CAS.
- J. E. Rekoske and M. A. Barteau, Ind. Eng. Chem. Res., 2011, 50, 41–51 CrossRef CAS.
- S. Luo and J. L. Falconer, Catal. Lett., 1999, 57, 89–93 CrossRef CAS.
- A. D. Sutton, C. M. Moore, R. W. Jenkins, O. Staples and T. J. Brooks, US Pat, 62/199,669, 2016 Search PubMed.
- Note that acetaldehyde polymers may be formed under these reaction conditions and may contribute to the observed carbon loss.
- T. L. Jordison, C. T. Lira and D. J. Miller, Ind. Eng. Chem. Res., 2015, 54, 10991–11000 CrossRef CAS.
- T. Seki, J.-D. Grunwaldt, N. van Vegten and A. Baiker, Adv. Synth. Catal., 2008, 350, 691–705 CrossRef CAS.
- C. Xiong, N. Liang, H. An, X. Zhao and Y. Wang, RSC Adv., 2015, 5, 103523–103533 RSC.
- H. Bahrmann, H.-D. Hahn, D. Mayer and G. D. Frey, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- P. Suchorska-Woźniak, O. Rac, R. Klimkiewicz, M. Fiedot and H. Teterycz, Appl. Catal., A, 2016, 514, 135–145 CrossRef.
- M. J. Biddy, R. Davis, D. Humbird, L. Tao, N. Dowe, M. T. Guarnieri, J. G. Linger, E. M. Karp, D. Salvachúa, D. R. Vardon and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 3196–3211 CrossRef CAS.
- Bioproducts to Enable Biofuels Workshop Summary Report, US Department of Energy, Office of Energy Efficiency & Renewable Energy, Bioenergy Technologies Office, 2015.
- R. W. Jenkins, C. M. Moore, T. A. Semelsberger, C. J. Chuck, J. C. Gordon and A. D. Sutton, ChemSusChem, 2016, 9, 922–931 CrossRef CAS PubMed.
- M. Dahmen and W. Marquardt, Energy Fuels, 2015, 29, 5781–5801 CrossRef CAS.
- G. T. Kalghatgi, Soc. Automot. Eng., [Spec. Publ.] SP, 2005, 1972, 203–220 Search PubMed.
- We note that the limited stability of aldehydes may prohibit them from being employed as alternative fuels, however little information has been reported to date on this issue (see ref. 36).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02507b |
| This journal is © The Royal Society of Chemistry 2017 |