
Reductive fractionation of woody biomass into lignin monomers and cellulose by tandem metal triflate and Pd/C catalysis†
Xiaoming
Huang
a,
Olivia M.
Morales Gonzalez
a,
Jiadong
Zhu
a,
Tamás I.
Korányi
a,
Michael D.
Boot
b and
Emiel J. M.
Hensen
 *a
*a
aSchuit Institute of Catalysis, Inorganic Materials Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.j.m.hensen@tue.nl
bCombustion Technology, Department of Mechanical Engineering, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 2nd December 2016
Abstract
A catalytic process for the upgrading of woody biomass into mono-aromatics, hemi-cellulose sugars and a solid cellulose-rich carbohydrate residue is presented. Lignin fragments are extracted from the lignocellulosic matrix by cleavage of ester and ether linkages between lignin and carbohydrates by the catalytic action of homogeneous Lewis acid metal triflates in methanol. The released lignin fragments are converted into lignin monomers by the combined catalytic action of Pd/C and metal triflates in hydrogen. The mechanism of ether bond cleavage is investigated by lignin dimer models (benzyl phenyl ether, guaiacylglycerol-β-guaiacyl ether, 2-phenylethyl phenyl ether and 2-phenoxy-1-phenylethanol). Metal triflates are involved in cleaving not only ester and ether linkages between lignin and the carbohydrates but also β-O-4 ether linkages within the aromatic lignin structure. Metal triflates are more active for β-O-4 ether bond cleavage than Pd/C. On the other hand, Pd/C is required for cleaving α-O-4, 4-O-5 and β–β linkages. Insight into the synergy between Pd/C and metal triflates allowed optimizing the reductive fractionation process. Under optimized conditions, 55 wt% mono-aromatics – mainly alkylmethoxyphenols – can be obtained from the lignin fraction (23.8 wt%) of birch wood in a reaction system comprising birch wood, methanol and small amounts of Pd/C and Al(III)-triflate as catalysts. The promise of scale-up of this process is demonstrated.
Introduction
Biorefining is gaining in importance as a technology to increase energy security, to mitigate emissions of carbon dioxide, and to increase rural development.1 Widespread production of biomass-derived fuels and chemicals will require cost-effective processes for breaking down the principle biomass constituents cellulose, hemicellulose and lignin into their monomers.2 One-pot conversion of whole biomass results in a complex mixture of aromatics, alkanes and alcohols, which limits applications as fuels and chemicals.3–5 In order to improve conversion efficiency and product selectivity, biomass is usually pre-treated by physical, chemical or biological methods. Pre-treatment of lignocellulosic biomass is one of the most costly operations in its valorization.6,7 Biological approaches are time-consuming, while mechanical operations are usually energy- and capital-intensive. Chemical pretreatment with alkali, ozone, peroxide or organic solvents is typically targeted at the extraction of lignin from the whole biomass with the purpose of enhancing enzymatic degradability of cellulose.8,9 Treatment with mineral acids, usually H2SO4 and HCl, is widely practiced to hydrolyze (hemi-)cellulose into fermentable sugars, leaving behind lignin as solid residue.6,9 A significant problem in converting technical lignin is its much lower reactivity as compared to the original lignin structure contained in the lignocellulose matrix.10–12 During pre-treatment, lignin's relatively labile β-O-4 ether linkages can be easily cleaved and the resulting fragments condense into more recalcitrant structures. As a consequence, technical lignin contains more C–C bonds than the original lignin. The condensation reactions typically involve phenolic fragments, formaldehyde13 and sugar-derived dehydration products such as 5-hydroxymethylfurfural and furfural.14,15 The problem of condensation of lignin fragments is more significant in the presence of acid or base catalysts. The low reactivity of technical lignin obtained by Kraft and soda pulping and organosolv methods has been emphasized in recent work. Detailed characterization revealed considerable condensation of lignin during extraction.16 As a consequence, the depolymerization of lignin requires high temperature (>250 °C) to increase the rate of (thermal) C–C bond cleavage.17–19Common approaches to depolymerize lignin such as pyrolysis, hydrocracking, hydrogenolysis, oxidation, and hydrolysis have been well documented in recent reviews.1,20–23 Most of such thermo-catalytic processes are non-selective and give rise to mixtures of tens or even hundreds of different compounds. The complexity of these product mixtures hinders application of lignin as feedstock for producing high-value chemicals. As an example, we refer to our own work in which technical lignin was catalytically converted in ethanol over a Cu–Mg–Al mixed oxide catalyst into lignin monomers in high yield (typically ∼60 wt% for soda and organosolv lignins and 86 wt% for Kraft lignin). High product yield was obtained under severe conditions (T = 380 °C, supercritical ethanol).13,24,25 Despite these and similar other efforts,10,26,27 efficient conversion of lignin into useful chemicals remains a significant challenge.
A recent major step forward has been the direct conversion of the lignin fragments removed from whole woody biomass before condensation. For instance, Yan et al. reported that birch wood lignin can be hydrogenated to alkylmethoxyphenols in 46 wt% yield in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) dioxane/water solvent mixture over carbon-supported noble metal (Pt, Ru, Pd and Rh) catalysts at 200 °C for 4 h.28 Li et al. reported the direct catalytic conversion of woody biomass into diols and alkylmethoxyphenols in water over a carbon-supported Ni–W2C catalyst at 235 °C for 4 h.5 Song et al. upgraded birch wood lignin into alkylmethoxyphenols in alcohols over nickel catalysts and reported a lignin conversion of about 50%.29 Ferrini et al. discussed a catalytic biorefining method that converts lignin from woody biomass into bio-oil rich in phenolic compounds and (hemi-)cellulose-rich pulp over a RANEY® Ni catalyst in a H2O/2-propanol mixture. The obtained bio-oil was further upgraded using the same catalyst in 2-propanol.18 The group of Abu-Omar used a bimetallic Zn/Pd/C catalyst to convert lignin in lignocellulosic biomass into two alkylmethoxyphenols in methanol. A yield of 52 wt% of lignin monomers was obtained from birch hardwood after reaction at 225 °C for 12 h.19,30 Sels and co-workers reported that birch wood sawdust was efficiently delignified through simultaneous solvolysis and catalytic hydrogenolysis in the presence of Ru/C or Pd/C in methanol under a H2 atmosphere at elevated temperature. The main products were a carbohydrate pulp and a lignin oil containing more than 50% phenolic monomers.6,31 The addition of acids such as H3PO4 improved the overall efficiency of the process.32
:1 (v/v) dioxane/water solvent mixture over carbon-supported noble metal (Pt, Ru, Pd and Rh) catalysts at 200 °C for 4 h.28 Li et al. reported the direct catalytic conversion of woody biomass into diols and alkylmethoxyphenols in water over a carbon-supported Ni–W2C catalyst at 235 °C for 4 h.5 Song et al. upgraded birch wood lignin into alkylmethoxyphenols in alcohols over nickel catalysts and reported a lignin conversion of about 50%.29 Ferrini et al. discussed a catalytic biorefining method that converts lignin from woody biomass into bio-oil rich in phenolic compounds and (hemi-)cellulose-rich pulp over a RANEY® Ni catalyst in a H2O/2-propanol mixture. The obtained bio-oil was further upgraded using the same catalyst in 2-propanol.18 The group of Abu-Omar used a bimetallic Zn/Pd/C catalyst to convert lignin in lignocellulosic biomass into two alkylmethoxyphenols in methanol. A yield of 52 wt% of lignin monomers was obtained from birch hardwood after reaction at 225 °C for 12 h.19,30 Sels and co-workers reported that birch wood sawdust was efficiently delignified through simultaneous solvolysis and catalytic hydrogenolysis in the presence of Ru/C or Pd/C in methanol under a H2 atmosphere at elevated temperature. The main products were a carbohydrate pulp and a lignin oil containing more than 50% phenolic monomers.6,31 The addition of acids such as H3PO4 improved the overall efficiency of the process.32
Earlier, we demonstrated that Lewis acids (metal acetates, metal chlorides and metal triflates) can be used as catalyst to depolymerize a soda lignin in supercritical ethanol. Metal triflates are promising water-tolerant Lewis acids catalysts for lignin conversion.33–35 A combination of a 10 mol% triflic acid with a heterogeneous Ru or homogeneous Ir catalyst is effective in depolymerizing organosolv lignin in 1,4-dioxane solvent.27 A similar catalytic system involving metal triflate and a homogeneous Rh catalyst was employed for the depolymerization of various organosolv lignins and wood sawdust in a 1,4-dioxane/H2O solvent mixture. Lignin monomer yields from these feedstocks were unsatisfactory.36 Related to these “lignin-first” approaches, we have recently demonstrated that metal triflates act as efficient co-catalysts in the delignification of biomass.37 In combination with Pd/C and methanol as the solvent a narrow stream (3–7 compounds) of lignin-derived alkylmethoxyphenols is obtained under mild conditions. Cellulose is retrieved as a separate fraction with a composition comparable to that of cellulose obtained by Kraft pulping of birch wood. We have already discussed mechanistic aspects about the role of metal triflates in liberating lignin fragments from lignocellulosic biomass. By use of model compounds, we demonstrated that these homogeneous catalysts are effective in catalysing cleavage of γ-ester and phenyl ether and glycoside bonds between lignin and (hemi-)cellulose. The resulting lignin fragments can then be disassembled into aromatic monomers by Pd-catalyzed hydrogenolysis reactions.
In this work, we focus on the synergistic effect of metal triflates in the Pd-catalyzed deconstruction of lignin fragments. For this purpose, we employ benzyl phenyl ether (BPE), guaiacylglycerol-β-guaiacyl ether (GG), 2-phenylethyl phenyl ether (PPE-H) and 2-phenoxy-1-phenylethanol (PPE-OH) as model reactants. Plausible mechanisms for the conversion of these model lignin compounds as well as the depolymerization of wood lignin will be discussed. We also varied the type of metal triflates and supported metal catalysts as well as the solvent to identify optimum conditions for woody biomass upgrading. In so doing, we were able to optimize the tandem process of birch wood towards high aromatic monomers yield under mild conditions (aromatic monomers yield of 55 wt% at T = 180 °C for t = 2 h). Finally, we also demonstrate that this process can be scaled-up to convert 100 g biomass with nearly similar mono-aromatics yield.
Results and discussion
The catalytic process
Fig. 1 illustrates the overall concept of the tandem catalytic system for the conversion of lignin from woody biomass. Metal triflates are effective in the extraction of lignin from the lignocellulosic matrix by cleaving ether and ester lignin–carbohydrate intralinkages. These lignin fragments are then cleaved by Pd-catalyzed reactions involving molecular hydrogen. In an experiment using Pd/C and Yb(III)-triflate at 180 °C for 2 h in 30 bar H2, the C9 lignin monomers yield was 43 wt% based on lignin content in birch wood. The main products were 4-n-propylsyringol (PS-H), 4-n-propanolsyringol (PS-OH) and 4-n-methoxy propyl syringol (PS-OCH3) and their corresponding guaiacyl counterparts, namely PG-H, PG-OH and PG-OCH3.37Table 1 shows a detailed analysis of the birch wood composition, the solid residue and product oil composition after upgrading by Pd/C only and by a combination of Pd/C and metal triflates. Cellulose remains nearly unconverted in these experiments as evident from the unchanged glucan content. Hemi-cellulose is hardly degraded by the action of Pd/C, but significantly removed from the solid matrix when metal triflates are present. Metal triflates strongly promote the reductive fractionation of lignocellulose. The lignin monomers yield with only Pd/C was limited to 14 wt%. Addition of Yb(III)- or Al(III)-triflate to the reaction strongly increased the removal of lignin from the biomass and, at the same time, increased the lignin monomers yield. The use of Al(III)-triflate led to a higher lignin monomers yield, but also degraded slightly more cellulose. Without Pd/C, mainly lignin fragments were obtained when Yb(III)-triflate was the catalyst with a low monomers yield of 7 wt%.37 These results demonstrate that metal triflates and Pd/C work in tandem in delignifying lignocellulosic biomass and converting the resulting fragments to monomers. We next demonstrate that metal triflates also affect the performance of Pd-catalyzed hydrogenolysis reactions.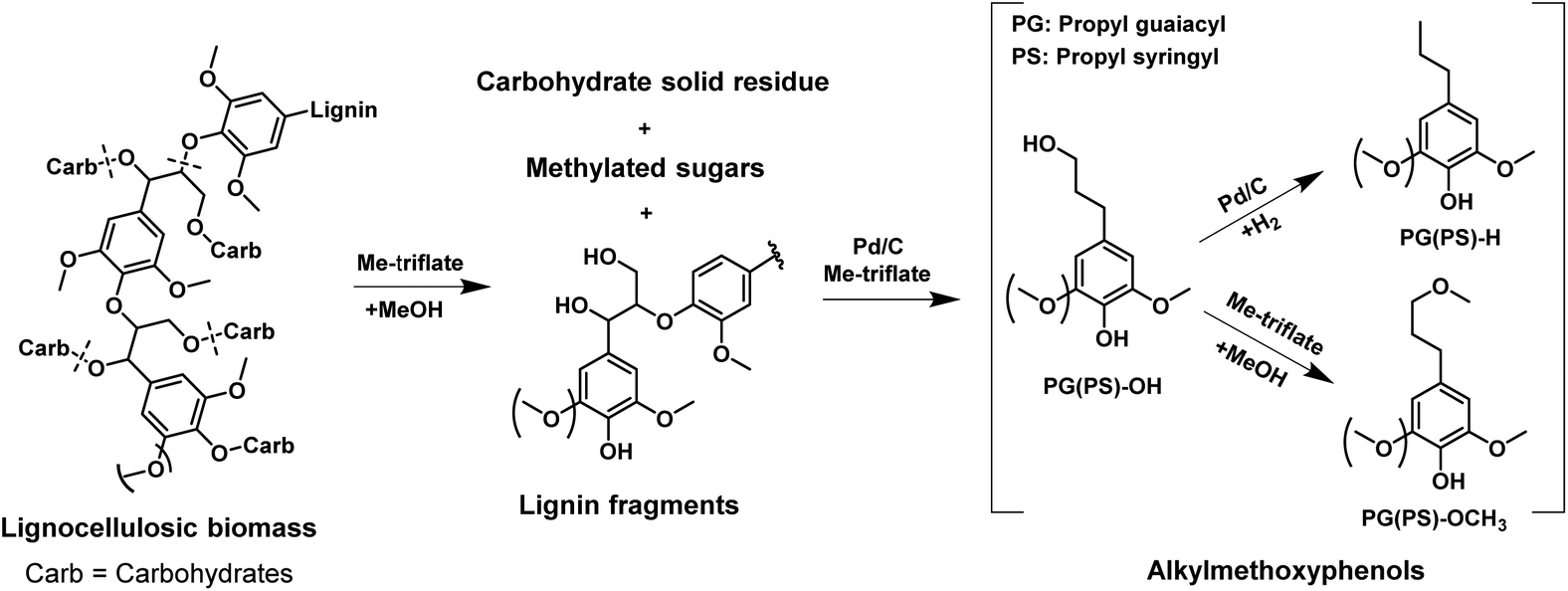 | ||
| Fig. 1 Tandem catalytic system for the reductive fractionation of woody biomass into alkylmethoxyphenols and sugars over Pd/C and metal triflate in methanol. | ||
| Catalyst | Birch wooda | Reductive fractionationb | ||
|---|---|---|---|---|
| — | Pd/C | Pd/C | Pd/C | |
| Yb(III)-triflate | Al(III)-triflate | |||
| Notes:a Birch wood sample subjected to Soxhlet extraction with water followed by ethanol.b Reaction conditions: 2000 mg extracted birch wood sawdust (125–300 μm), 200 mg 5 wt% Pd/C, 0.0322 mmol metal triflate, 40 ml methanol, 30 bar H2, stirring speed: 500 rpm, 2 h.c Solids analysis done by Celignis Limited Nexus Innovation Centre, University of Limerick, Ireland (http://www.celignis.com); mass of the residue excluding Pd/C catalyst and moisture.d Based on monomeric sugars detected after two-stage acid hydrolysis following a standard protocol.38e Based on the of acid-soluble and -insoluble (Klason) lignin.f Methylated sugars are detected and quantified by GC-FID.g Based on the total lignin content. | ||||
| Solidsc | 2000 | 1790 | 1360 | 1190 |
| Sugarsd | 1276 | 1249.4 | 1082.6 | 975.8 |
| Glucan | 800.0 | 801.9 | 817.4 | 772.8 |
| Xylan | 422.0 | 407.9 | 238.7 | 187.7 |
| Arabinan | 6.0 | 2.0 | 0.0 | 0.0 |
| Galactan | 14.0 | 9.9 | 1.6 | 1.4 |
| Mannan | 26.0 | 23.8 | 21.8 | 19.5 |
| Rhamnan | 8.0 | 4.0 | 0.0 | 0.0 |
| Lignine | 475 | 319.4 | 120.5 | 91.2 |
| Klason | 402.0 | 281.2 | 109.9 | 83.4 |
| Acid soluble | 73.0 | 38.2 | 10.6 | 7.8 |
| Ash | 5.5 | 17.2 | 22.1 | 14.4 |
| Unknown | 243.5 | 204.0 | 137.9 | 103.1 |
| Product oil | — | 210 | 640 | 810 |
| Methylated sugarsf | — | 6 | 205 | 352 |
| Phenolicsg | — | 64 | 203 | 212 |
| Unknown | — | 140 | 232 | 246 |
Model compound studies
With the purpose of understanding better the role of metal triflates in Pd-catalyzed deconstruction of lignin fragments, we employed benzyl phenyl ether (BPE) and guaiacylglycerol-β-guaiacyl ether (GG) as models for α-O-4 and β-O-4 ether lignin intralinkages in woody biomass, respectively. BPE is often used as model compound in screening studies.39–41 Although it does not contain hydroxyl and methoxy groups usually encountered in lignin, its benefit is the relative facile product identification and quantification in evaluating catalyst performance in ether linkage disruption and aromatic ring hydrogenation.20 GG is the most suitable model compound for the most abundant β-O-4 (40–65%) ether linkage in native lignin.23 Therefore and also because it is commercially available, GG has been frequently used in mechanistic studies of lignin deconstruction.10,26,27,42–44 Model reactions using these two dimer compounds were performed at 160 °C for 2 h in methanol and 30 bar H2. Blank experiments (without catalyst) were compared to catalytic experiments with either Pd/C or Yb(III)-triflate or a combination of the two. Fig. 2 shows the product yield obtained in these experiments. Without a catalyst, the ether bonds are relatively stable under the given reaction conditions. Pd/C is very active in cleaving the α-O-4 ether bond: complete ether bond conversion was achieved within 2 h. However, Pd is much less active in cleaving the β-O-4 ether bond in GG and the product of ether bond cleavage, guaiacol, is only obtained in 18% yield. The substantial activity difference stems from competitive adsorption between the ether bond and the α-OH and γ-OH groups of the reactant.19 Yb(III)-triflate is nearly inactive in cleaving the α-O-4 ether bond of BPE, yet very active in rupture of the β-O-4 ether bond in GG. This difference is caused by the strong activation of the ether bond upon coordination of a Lewis acid to oxygen atoms of OH groups in α- and γ-positions of the ether bond.19,43,45,46 This explains the higher activity of Yb(III)-triflate in cleaving the β-O-4 ether bond in GG as compared with the α-O-4 ether bond in BPE. A combination of Pd/C and Yb(III)-triflate results in similar BPE conversion and products as Pd/C, suggesting that in this case the metal triflate does not affect the performance of Pd/C. Fig. 2b shows the yield of guaiacol and thereof-derived products obtained by conversion of GG. Contrary to the results with BPE, the rate of ether bond conversion in GG is significantly lower in the tandem catalyst system compared with Yb(III)-triflate. Thus, the ether bond cleavage activity of Yb(III)-triflate is suppressed in the presence of Pd/C. The reason is that Pd/C is active in the removal of the α-OH group. This converts the β-O-4 ether bond into an ether bond which cannot be cleaved by the metal triflate. As we will show later, this result is important in further optimizing the tandem catalyst system towards high aromatic monomers yield.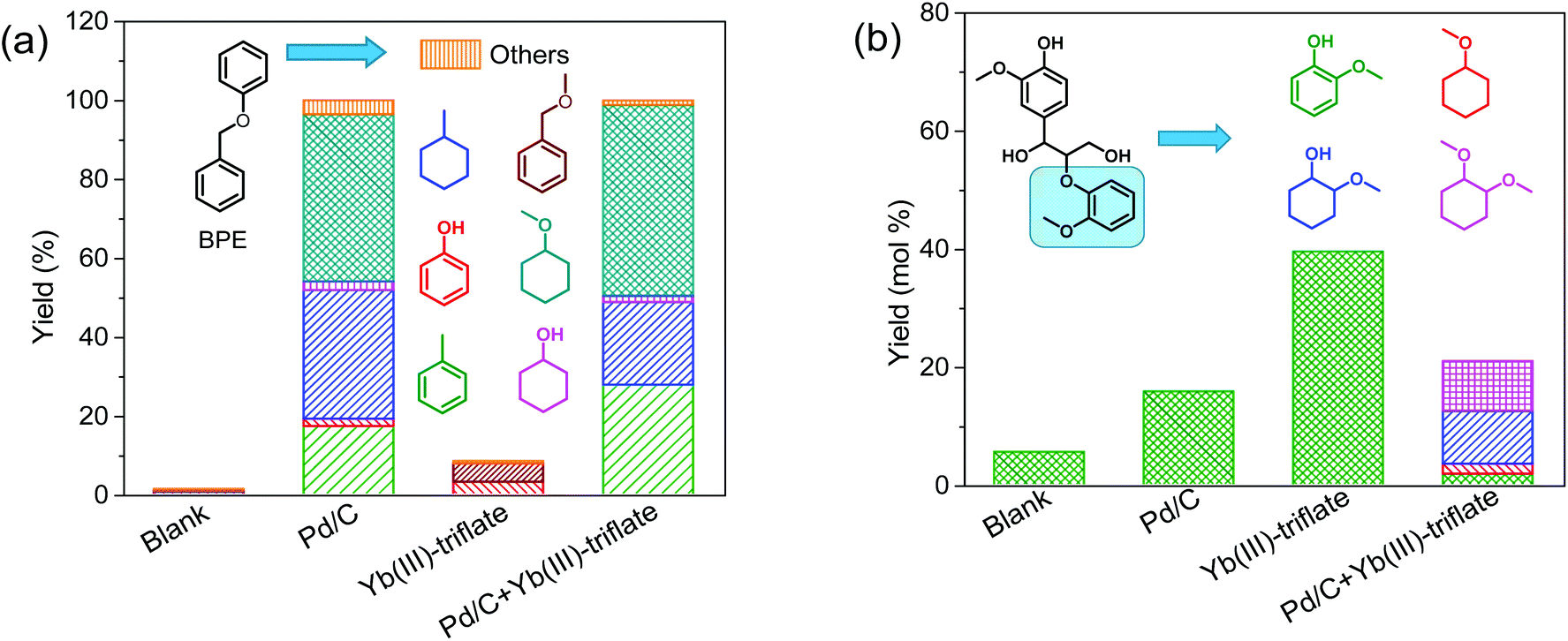 | ||
| Fig. 2 Reaction of (a) benzyl phenyl ether (BPE) and (b) guaiacylglycerol-β-guaiacyl ether (GG) in methanol over Pd/C and/or Yb(III)-triflate catalysts at 160 °C for 2 h in 30 bar H2. | ||
It is worthwhile to mention that in the tandem system ring-saturated products are obtained, very different from the experiments with only Pd/C or the metal triflates. The enhanced hydrogenation activity of Pd/C in the presence of Yb(III)-triflate is confirmed in an experiment with guaiacol as the reactant. The use of Pd/C results in a conversion of 61% to ring-saturated products. In the presence of Yb(III)-triflate, full conversion was achieved under the same conditions. Accordingly, we conclude that there is synergy between Pd/C and Yb(III)-triflate in aromatic ring hydrogenation. On the other hand, no synergy was observed with respect to ether bond cleavage. Synergy in aromatic ring hydrogenation between Lewis acids and Pd/C has been described in recent literature.45,46 For instance, Zhou and co-workers have shown that the presence of Lewis acids such as AlCl3 and ZnCl2 on Pd/C improve aromatic ring hydrogenation activity.45 Our own results evidenced that the presence of propanol side chains on aromatic rings hinders their hydrogenation, presumably due to competitive adsorption for Pd surface sites.37 The presence of a hydroxyl-group-rich substrate (i.e., the biomass itself and derived sugars) also appears to suppress hydrogenation of aromatic rings. This is demonstrated by comparing the hydrogenation of guaiacol in the presence and absence of glucose. Model reactions were performed at 160 °C for 2 h in methanol using Pd/C as the catalyst. The results attest to the lower rate of aromatic ring hydrogenation of guaiacol in the presence of glucose (Fig. S1†). These findings also explain the absence of ring hydrogenation of aromatics in the conversion of woody biomass. We speculate that the synergy between Pd/C and metal triflate in aromatic ring hydrogenation is due to the reduced amount of hydroxyl groups, owing to increased rate of etherification with methanol catalyzed by metal triflate.
The distinct difference observed in the way the two catalysts convert the model compounds prompted us to more systematically investigate the importance of aliphatic OH groups close to the ether bond linkage. This is cumbersome with GG as side-reactions such as etherification and hydrogenation occur. Thus, we carried out reactions with two other model compounds 2-phenoxy-1-phenylethanol (PPE-OH) and 2-phenylethyl phenyl ether (PPE-H). The former compound contains an OH group in the position β to the ether bond, while the latter does not. The reactions were conducted at 200 °C for 4 h in presence of 30 bar H2. Consistent with the BPE and GG results, Pd/C is much more active in cleaving the ether bond in PPE-H than in PPE-OH (Fig. 3).
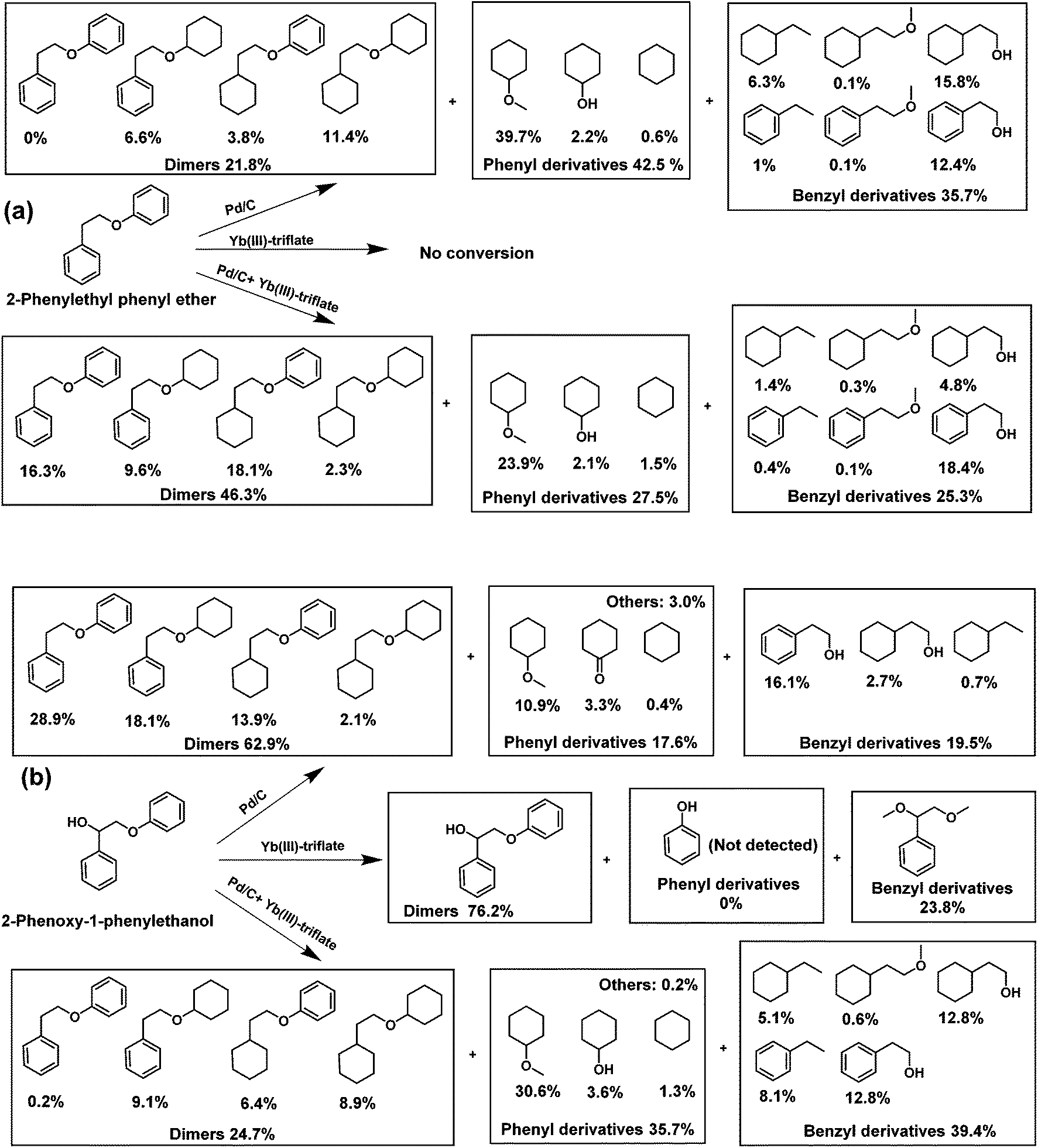 | ||
| Fig. 3 Catalytic results for the reaction of (a) 2-phenylethyl phenyl ether and (b) 2-phenoxy-1-phenylethanol in methanol solvent over Pd/C and/or Yb(III)-triflate at 200 °C for 4 h (the product quantification for the model lignin compound was based on the GC area normalization method). | ||
As expected, the amount of products derived from the phenyl and benzyl parts of the reactant were very similar. Yb(III)-triflate alone did not convert PPE-H, in keeping with the result obtained with BPE. On the other hand, the metal triflate is very active in ether bond cleavage of PPE-OH due to the presence of the α-OH group. We observed only one product in this case, namely 1-phenyl-1,2-dimethoxyethane deriving from the trans-etherification of the α-OH group and ether linkage by methanol. No products from the phenyl part were observed by GC-MS, indicating that phenolic intermediates rapidly underwent secondary reactions. When Pd/C was used in combination with Yb(III)-triflate, the conversion of the PPE-H ether linkage decreased. The opposite was observed when PPE-OH was used as substrate, that is, Yb(III)-triflate promoted the Pd-catalyzed ether bond cleavage.
Based on these model lignin compound investigations, we propose a reaction mechanism for the cleavage of the β-O-4 ether linkage as illustrated in Fig. 4. Pd/C is very active in cleaving the ether linkage in PPE-H by hydrogenolysis (Fig. 4a). However, in the presence of metal triflate, the Lewis acid will coordinate to the ether bond,47 preventing its cleavage as evident from experiments with BPE and PPE-H as the model reactant (Fig. 4b). The preference of ether linkages that coordinate to the metal triflates over the metal surface results in a decreased rate of ether bond cleavage. When PPE-OH is the reactant, the ether bond conversion rate is slower due to the competitive adsorption of the α-OH and ether bonds on the Pd active sites (Fig. 4c). We argue that this competitive adsorption for sites on the metal surface is partially suppressed by the strong coordination of the metal triflate to the α-OH group (Fig. 4d). This interaction increases the ether bond cleavage rate, as it contributes to weakening of the ether linkage. This aspect of the mechanism also explains why addition of metal triflate significantly increases the lignin monomers yield during sawdust wood upgrading.
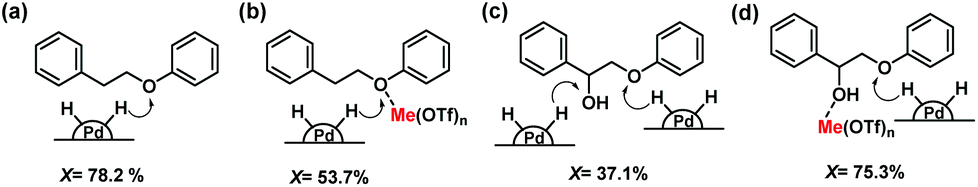 | ||
| Fig. 4 Proposed mechanism for the cleavage of β-O-4 ether linkage using (a) PPE-H by Pd/C; (b) PPE-H by Pd/C + metal triflate; (c) PPE-OH by Pd/C and (d) PPE-OH by Pd/C + metal triflate (X = ether bond conversion). | ||
Different metal triflates
In order to explore whether Yb can be replaced by the other less scarce metals, divalent (Ni and Cu), trivalent (Al, Sc and La) and tetravalent (Hf) triflates were evaluated for woody biomass conversion. All of the tested metal triflates significantly increase the C9 lignin monomers yield as compared with the Pd/C-only reference (Table 2 entry 1). The promoting effect is higher for tri- and tetravalent metal triflates than for divalent metals, which is most likely related to the increasing Lewis acidity with increasing cation charge of the transition metal. Quantitative determination of the Lewis acidity of metal triflates is challenging, especially when a coordinating solvent is involved. Lawton ranked the Lewis acidity of metal triflates on the basis of pyridine IR in an ionic liquid ([bmim][OTf]) and found that trivalent (Al, Sc, La, In) and tetravalent (Zr and Hf) metal triflates are much stronger Lewis acids than divalent metal triflates.48 These Lewis acidity differences qualitatively explain the activity differences in woody biomass conversion. Among the metal triflates tested here, Al(III)-triflate and Hf(IV)-triflate gave the highest lignin monomers yield, slightly higher than the monomers yield obtained with Yb(III)-triflate (Table 1). Concomitantly, the methylated C5 sugar yield with these two metal triflates was higher, showing a higher degree of removal of hemi-cellulose from the biomass matrix. In lignocellulose, lignin is typically bound to hemi-cellulose. High monomer yield was already obtained after 1 h reaction. Prolonging the reaction time to 2 h resulted only in a slightly increase of the monomer yield. We argue that this is due to rapid conversion of the predominant β-O-4 ether bonds, followed by slower cleavage of other ether bonds. As the β-O-4 ether bonds are the most abundant lignin intralinkages, the maximum lignin monomer yield should mainly relate to the β-O-4 ether bond content of the woody biomass. Conventional Lewis acids such as AlCl3 and ZnCl2 were much less effective in deconstructing lignocellulose. Hydrolysis of these Lewis acids by water generated in etherification and hydrogenolysis reactions will likely contribute to the lower efficiency of these Lewis acids. Based on these results, the preferred Lewis acid catalyst is Al(III)-triflate. Replacing the hydrogen atmosphere by nitrogen resulted in a much lower lignin monomer yield (Table 2 entry 11). This result shows that transfer hydrogenation reactions are not effective in the current process.| Entry | Catalyst | Co-catalyst | Residue massa (mg) | Lignin monomers yield (wt%) (mg) | Methylated C5 sugars (mg) | Total yield (%) | |
|---|---|---|---|---|---|---|---|
| 1 h | 2 h | ||||||
| Conditions: 2000 mg extracted birch wood sawdust (125–300 μm), 200 mg 5 wt% Pd/C, 0.0322 mmol co-catalyst, 40 ml methanol, 30 bar H2, stirring speed: 500 rpm.a Mass of the residue excluding Pd/C catalyst and moisture.b 30 bar N2 was added instead of H2. | |||||||
| 1 | 5 wt% Pd/C | — | 1790 | 10 | 14 (64) | 6 | 93 |
| 2 | 5 wt% Pd/C | Ni(II)-triflate | 1340 | 32 | 37 (176) | 194 | 86 |
| 3 | 5 wt% Pd/C | Cu(II)-triflate | 1360 | 33 | 34 (163) | 162 | 84 |
| 4 | 5 wt% Pd/C | Al(III)-triflate | 1190 | 42 | 45 (212) | 352 | 88 |
| 5 | 5 wt% Pd/C | Yb(III)-triflate | 1360 | 39 | 43 (203) | 205 | 88 |
| 6 | 5 wt% Pd/C | Sc(III)-triflate | 1250 | 37 | 39 (187) | 259 | 85 |
| 7 | 5 wt% Pd/C | La(III)-triflate | 1390 | 32 | 36 (171) | 190 | 88 |
| 8 | 5 wt% Pd/C | Hf(IV)-triflate | 1110 | 40 | 42 (200) | 345 | 83 |
| 9 | 5 wt% Pd/C | ZnCl2 | 1690 | 14 | 22 (104) | 31 | 91 |
| 10 | 5 wt% Pd/C | AlCl3 | 1540 | 20 | 28 (134) | 64 | 87 |
| 11 | 5 wt% Pd/C | Yb(III)-triflateb | 1470 | 9 | 17 (82) | 106 | 83 |
Optimization catalyst ratio in tandem Pd/C–Al(III)-triflate system
Using the optimal Al(III)-triflate catalyst, we then varied the ratio between the Pd/C metal and Lewis acid functions. In the first set of experiments, we fixed the amount of 5 wt% Pd/C to 200 mg (0.0940 mmol Pd) and varied the amount of Al(III)-triflate between 5 mg and 45 mg so as to obtain molar Pd/Al ratios of 1, 2, 3 and 9. At the highest Pd/Al ratio (5 mg Al(III)-triflate, Table 3, entry 2), the monomers yield is significantly higher than the Pd-only case. Increasing the amount of Al(III)-triflate to 15 mg (Pd/Al ratio = 3, Table 3 entry 3) results in a strong increase of the lignin monomers yield to 44 wt% after reaction at 180 °C for 2 h. At Pd/Al ratio = 2 (Table 3, entry 4), the same monomers yield was already reached upon heating to 180 °C. After 2 h, the monomers yield amounted to 48 wt%. At the highest Pd/Al ratio of 1 (Table 3, entry 5), the highest lignin monomers yield of 45 wt% was also reached during heating and it slightly decreased when prolonging the reaction time. For longer reaction times, a larger variety of aromatic products were obtained. By GC-MS, we could detect 12 monomeric compounds in amounts greater than 2 mg, whereas at a Pd/Al ratio of 3 only 6 compounds were observed in such amount. The larger number of products is the result of etherification of different hydroxyl groups, among which the phenolic OH groups, due to the high amount of metal triflate. In addition, some aliphatic ketones such as 2,5-hexanedione and esters such as the methyl ester of levulinic acid were observed in higher amounts than in the other experiments. This points to more extensive degradation of cellulose in this case at low Pd/Al ratio. Consistent with this, only 156 mg solid residue remained after 2 h reaction, showing that in this case not only hemi-cellulose but also a large part of cellulose was converted. Because the presence of these additional products is undesired as their separation is not straightforward, the amount of metal triflates should be limited during the reaction.| Entry | Pd/Al molar ratio | Al(III)-triflate (mg) | Residue mass (mg) | Lignin monomers yield (wt%) | ||
|---|---|---|---|---|---|---|
| 0 ha |
1 h | 2 h | ||||
| Note:a Heating time (30 min), the amount of 5 wt% Pd/C is fixed as 200 mg (0.0940 mmol Pd). | ||||||
| 1 | No triflates | 0 | 1790 | N/A | 10 | 14 |
| 2 | 9 | 5 | 1645 | 12 | 15 | 23 |
| 3 | 3 | 15 | 1180 | 23 | 39 | 44 |
| 4 | 1.5 | 30 | 828 | 43 | 46 | 48 |
| 5 | 1 | 45 | 156 | 45 | 42 | 41 |
Using an amount of 15 mg Al(III)-triflate (0.0316 mmol), which is sufficient to break the lignin–carbohydrate intralinkages but small enough to prevent release and degradation of glucose, we then varied the amount of Pd/C from 0 mg to 300 mg (Pd/Al ratios from 0 to 4.5). The product distribution and molecular weight distribution of the product mixture are shown in Fig. 5 and 6, respectively. Without Pd/C, the amount of lignin monomers is low (Fig. 5a) and the product mixture contains mainly dimers and oligomers, as evident from the gel permeation chromatogram shown in Fig. 6. Addition of a small amount of Pd/C (20 mg, Pd/Al ratio = 0.3) was already effective in converting most of these oligomers and a significant fraction of the dimers to monomers (Fig. 5b and 6). The lignin monomers yield for Pd/Al = 0.3 was 55 wt% after reaction at 180 °C for 2 h. Two products were obtained in high yield, namely 160 mg PS-OCH3 and 40 mg PG-OCH3 (the lignin content of the feedstock was 476 mg). Increasing the amount of Pd/C led to enhanced depolymerization of the lignin oligomers, as the broad peak due to higher-molecular-weight compounds decreased (Fig. 6). This confirms that Pd/C is active in cleaving ether bonds that cannot be cleaved by metal triflates. It is also seen that the amount of dimers does not change appreciably for Pd/Al > 0.3, indicative of the recalcitrance of the C–C intralinkages in these dimers. These C–C intralinkages are either from the native lignin structure, or retained after the cleavage of the recalcitrant ether linkages such as β-5 and β–β.20,29,49 and this does not contribute to monomers formation. Thus, the use of very small amounts of Pd/C and Al(III)-triflate is sufficient to reach maximum lignin monomers yield. With increasing Pd/Al ratio, the product distribution shifts from PS-OCH3 and PG-OCH3 to PS-OH, PG-OH, PS-H, and PG-H. Moreover, despite enhanced depolymerization of oligomers, the lignin monomers yield is seen to decrease with increasing Pd/Al ratio (Fig. 5). These two trends are caused by competitive reactions catalyzed by Pd/C and Al(III)-triflate as shown in Fig. 1. Pd/C catalyzes the removal of γ-OH groups by hydrogenolysis, while Al(III)-triflate catalyzes the etherification of OH groups by methanol solvent. Therefore, the increasing hydrodeoxygenation activity and associated removal of γ-OH groups when more Pd/C is added results in the different product mixture. Consistent with this, comparison of the data at short and long reaction times (Fig. 5a) shows that at the initial stages of the reaction a smaller amount of Pd/C is preferred. Using too much Pd/C appears to hinder the catalytic action of Al(III)-triflate, which can be explained in two manners. First, the high activity of Pd/C in the removal of α-OH and γ-OH groups close to ether bonds decreases the ability of metal triflates to cleave ether bonds. Second, part of the metal triflate might be immobilized on the activated carbon, for instance, by interaction with surface hydroxyl groups. Such interactions have also been proposed to be relevant in the ZnCl2 and Pd/C catalyzed depolymerization of lignocellulose.19,46
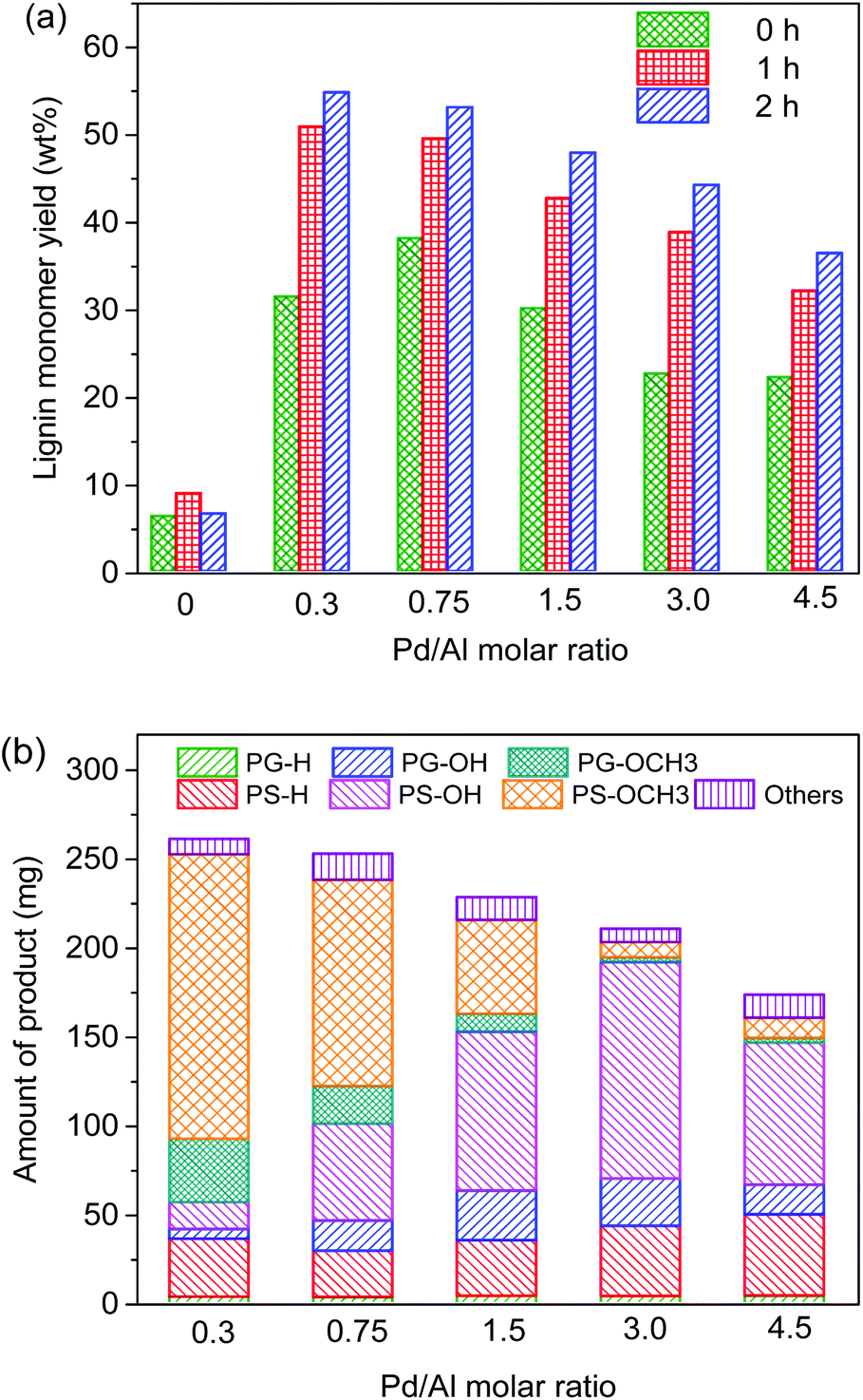 | ||
| Fig. 5 Influence of Pd/Al molar ratio on (a) lignin monomers yield (different reaction times) and (b) product distribution after reaction in methanol for 2 h. Conditions: 2000 mg birch wood, 15 mg (0.0316 mmol) Al(III)-triflate, 40 ml methanol, 180 °C, 30 bar H2. | ||
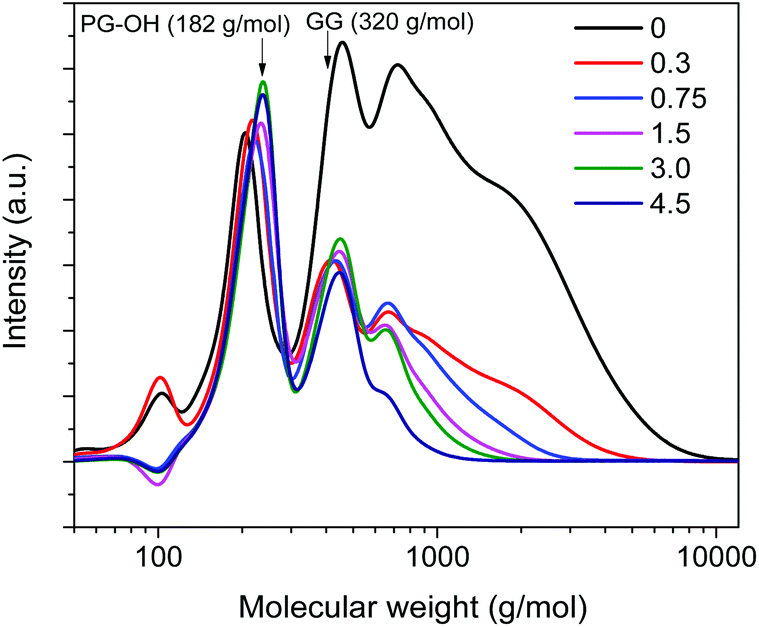 | ||
| Fig. 6 Influence of the Pd/Al molar ratio on the molecular weight distribution of the product mixture determined by GPC. Reaction conditions: 2000 mg birch wood, 15 mg (0.0316 mmol) Al(III)-triflate, 40 ml methanol, 180 °C, 30 bar H2. The maxima observed in GPC chromatograms of solutions of PG-OH (monomer model) and GG (dimer model) are indicated. | ||
Scheme 1 illustrates a plausible scenario for the influence of the ratio of the metal and Lewis acid functionalities in the tandem reductive depolymerization of wood lignin. Al(III)-triflate is active in catalysing the cleavage of ether and ester interlinkages between lignin and carbohydrates, facilitating the release of lignin from the lignocellulosic matrix. The metal triflate can also selectively cleave β-O-4 lignin ether bonds, which contain both α-OH and γ-OH groups. The β-O-4 ether linkage is the dominant lignin intralinkage, usually making up between ca. 43% and ca. 65% of all intralinkages in soft- and hardwood, respectively.23 Metal triflates are more active in cleavage of these bonds than Pd/C. On the other hand, metal triflates cannot cleave ether bonds in the absence of an α-OH group. Such ether linkages (α-O-4, 4-O-5 and β–β) are also common in lignin, accounting for ca. 12–16% and ca. 13–22% in soft- and hardwood, respectively.23 In the tandem system, Pd/C is able to cleave these bonds. This specificity for cleaving ether bonds explains the benefit of combining metal triflates and Pd/C in the reductive depolymerization of lignin fragments released from biomass. The amount of metal triflate should be kept relatively low to limit the release of glucose from cellulose and its further degradation. The results show that using a small amount of metal triflates does not impede its effectiveness in releasing lignin from biomass and aiding in the depolymerization of the released lignin fragments. The Pd/Al ratio is therefore a key parameter controlling the activity and product selectivity. When the Pd/Al ratio is too high (Pd/Al ratio > 3–4.5), the monomers yield is decreased, because Pd/C removes α-OH groups that enable the metal triflates to cleave ether bonds. The model compound results show how the positive effect of metal triflates in the presence of α-OH groups changes to a negative effect once these hydroxyl groups are removed. A time course study for the conversion of the PPE-OH model compound in the tandem Pd/C and Yb(III)-triflate system confirms that the α-OH group are completely removed during heating to 200 °C (t = 30 min) before ether bond cleavage commences (Fig. 7). Accordingly, the amount of Pd/C should also be limited. Optimum Pd/Al ratios are in the 0.3–3 range.
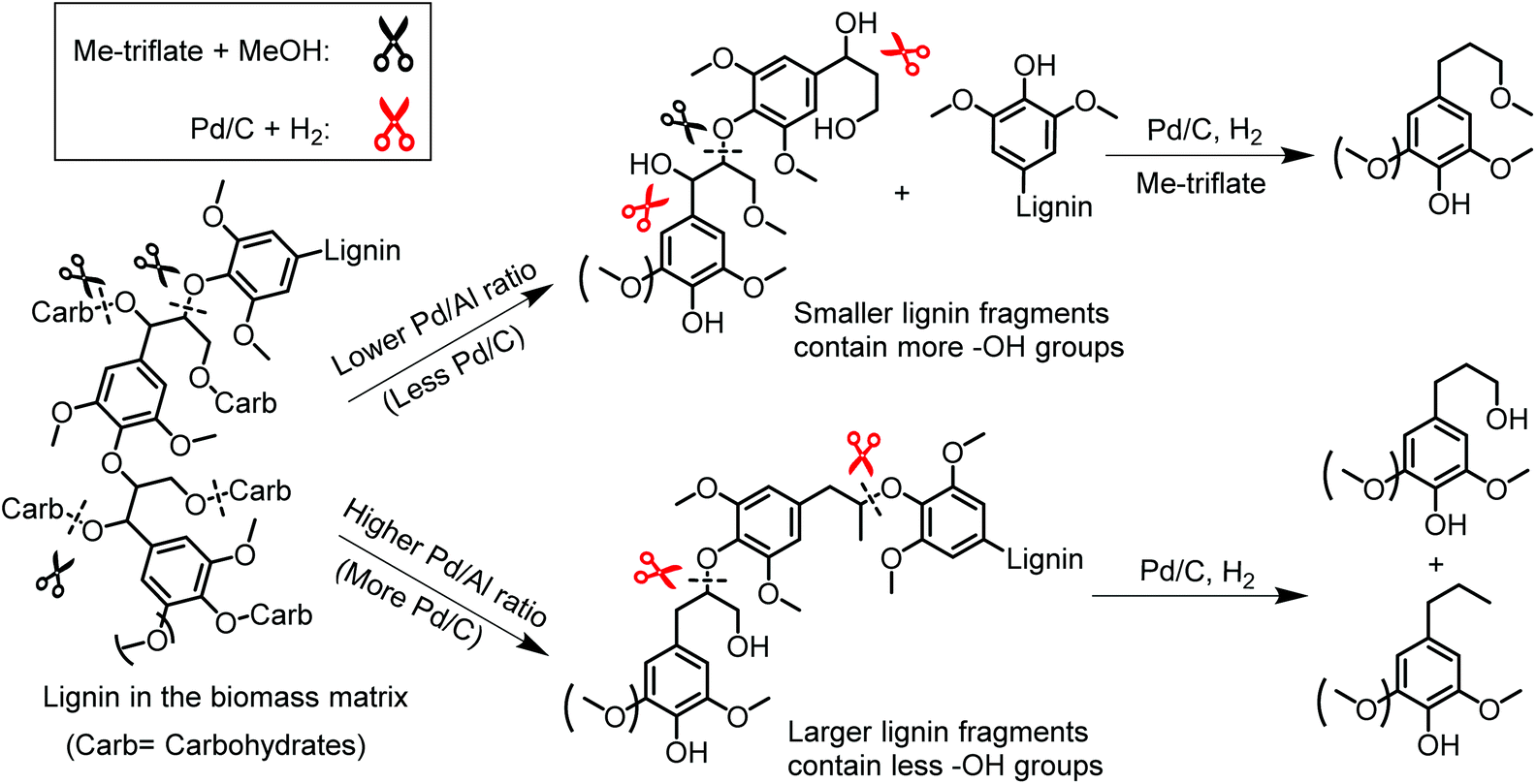 | ||
| Scheme 1 Plausible scenario for the tandem catalyzed reductive depolymerization of wood lignin into phenolic monomers over Pd/C and Al(III)-triflate at different Pd/Al ratios. | ||
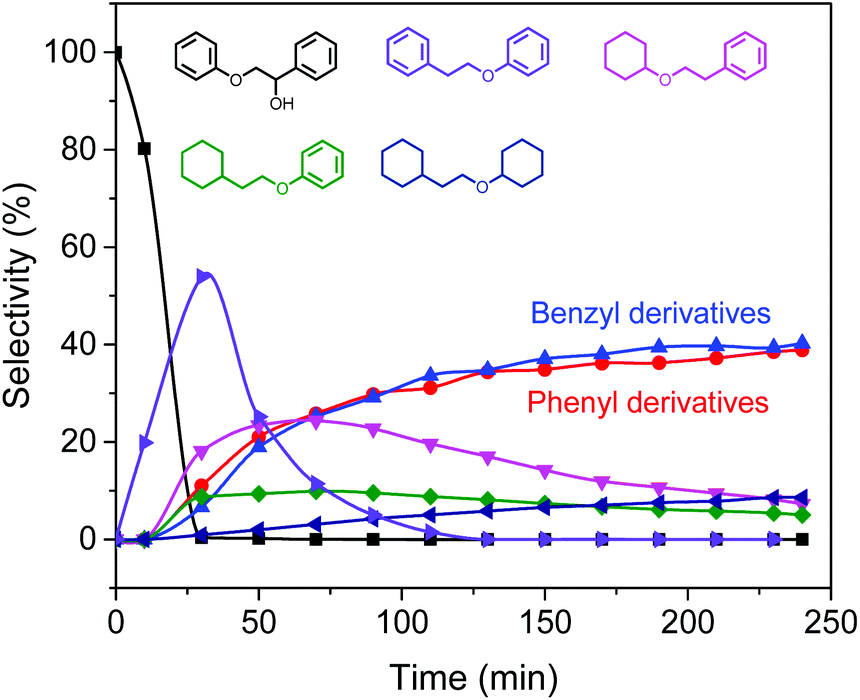 | ||
| Fig. 7 Time course study for the conversion of the PPE-OH model compound in the tandem Pd/C and Yb(III)-triflate system at 200 °C for 4 h. | ||
Thus, the role of the metal triflates as a homogeneous catalyst is two-fold. Metal triflates enhance delignification of the solid biomass as demonstrated before,31 and they rapidly decrease the molecular weight of the released lignin backbone by cleaving β-O-4 ether linkages. The smaller fragments are more accessible for Pd/C. Pd/C is also active in cleaving more recalcitrant ether linkages as well as removing α-OH and γ-OH groups. In this way, the ether linkages of the lignin structure can be effectively cleaved by metal triflate via trans-etherification, followed by Pd/C catalyzed hydrogenolysis reactions.
Recycling metal triflates
The literature contains many instances in which metal triflates could be recycled without loss of activity.50,51 We evaluated the possibility to recycle Al(III)-triflate in the reductive fractionation of woody biomass. A partitioning work-up using ethyl acetate and water solvent was applied in order to recover Al(III)-triflate from the liquid products. As metal triflates are better soluble in water than in common organic solvents, they can be extracted into the aqueous phase and recovered by evaporating water. Table 4 compares lignin monomer yields of a reference experiment and recycle tests in which Al(III)-triflate was recovered twice and then combined with fresh Pd/C catalyst, methanol and woody biomass. The amount of recovered Al(III)-triflate could not be determined as the solid also contained released sugars. The lignin monomer yield was 55 wt% after reaction at 180 °C for 2 h using the fresh catalysts. After recycling, the yield after 2 h decreases to 49 wt% and 37 wt%, indicating that not all Al(III)-triflate could be recovered by our work-up procedure. Incomplete recovery may be due to inefficient extraction into the aqueous phase and adsorption of metal triflate on activated carbon support of the Pd catalyst. To verify the latter, we determined by elemental analysis the uptake of Al(III)-triflate by Pd/C in methanol (15 mg in 40 mL). In this way, we found that that about 20–30% of the metal triflate was adsorbed by the Pd/C catalyst. Although these experiments show the principle possibility of recovering and reusing metal triflates, the interaction of the metal triflates with activated carbon leads to partial loss of the homogeneous co-catalyst.Recycling the Pd/C catalyst is a challenge because it ends up in the solid cellulose-rich residue. Possible solutions such as the use of a magnetic catalyst18,29 or embedding the metal function in a cage30 for separation have been proposed. An alternative solution is to separate the lignin release step and hydrogenolysis steps. Initial experiments show that nearly similar yield is obtained in this way (Table 5). We first treated birch wood with Yb(III)-triflate at 180 °C in methanol for 2 h. The C9 lignin monomer yield after 2 h was only 5 wt%. The solid residue was then removed by filtration and the filtrate was subjected to a second reaction in the presence of Pd/C and H2, leading to nearly similar C9 lignin monomer yield as in the one-step experiment (Table 5). These initial experiments suggest that a two-reactor approach is possible. In the first reactor, solid biomass is contacted with a solution of metal triflates in methanol. After addition of H2, the released fragments are then taken to a second reactor containing the Pd/C catalyst. The decreased contact time of the metal triflates with the Pd/C catalyst in the second reactor will also decrease the loss of metal triflate due to adsorption on the activated carbon surface.
| Entry | Catalyst | Lignin monomer yield (wt%) | |||
|---|---|---|---|---|---|
| 0 h | 0.5 h | 1 h | 2 h | ||
| a Reactions performed in two consecutive steps. In the first step, 20 mg Yb(III)-triflate was used as catalyst to convert 2000 mg birch wood. After reaction, the reactor mixture was filtered, and the filtrate was subjected to a second reaction in the presence of 200 mg Pd/C and pressurizing to 30 bar H2. | |||||
| One-pot | Yb(III)-triflate + Pd/C | 21 | 31 | 37 | 43 |
| Two-stepa | Yb(III)-triflate | n.a. | n.a. | n.a. | 5 |
| Pd/C | 37 | 36 | 37 | 36 | |
Scale-up of reduction fractionation process
As a first step towards scale-up of this process, we employed a 4 L high-pressure autoclave to convert a larger amount of biomass. For this purpose, we obtained from a local timber manufacturer sawdust from oak hardwood and Douglas fir softwood. The sawdust was dried, sieved to obtain particles in two size ranges (0–300 μm and 300–1000 μm), and used without pre-treatment in the reductive fractionation process. The autoclave was charged with 2 L methanol, 100 g sawdust, 1.5 g Pd/C and 0.75 g Al(III)-triflate and then pressurized to 30 bar with H2. The lignin monomer yields obtained after heating to 180 °C and prolonged reaction at this temperature are listed in Table 6 (detailed composition of oak and Douglas fir wood in the Experimental section, Table 7). For oak hardwood, the lignin monomer yield was already 40 wt% after heating to the reaction temperature and only increased slightly after 4 h. Nearly similar results were obtained with the smaller particles, demonstrating the effectiveness of the approach. Similar to the results obtained for Scotch pine softwood,37 conversion of Douglas fir softwood led to lower lignin monomer yield (17 wt%). The result shows that the lignin monomer yield did not increase much when the reaction was prolonged is likely due to the relatively long heating time (1 h) of the large autoclave. These initial results evidence that the woody biomass reductive fractionation process can be easily scaled up. An important aspect is that promising lignin monomer yield was obtained without fine-milling and extractive pre-treatment of the wood sawdust. This greatly contributes to reducing the number of steps and the overall cost of the process.| Entry | Feedstock | Lignin monomer yield (wt%) | |||
|---|---|---|---|---|---|
| 0 ha |
1 hb |
2 hb |
4 hc |
||
| Conditions: 100 g dry wood substrate, 1.5 g 5 wt% Pd/C, 0.75 g Al(III)-triflate, 2 L MeOH, 30 bar H2, 180 °C, stirring speed: 800 rpm.a Heating time (1 h).b Samples were taken during reaction via a sampling valve.c Samples were taken after the reactor was cooled down to room temperature. | |||||
| 1 | Oak (300–1000 μm) | 40 | 40 | 41 | 46 |
| 2 | Oak (0–300 μm) | 36 | 39 | 39 | 40 |
| 3 | Douglas fir (300–1000 μm) | 14 | 17 | 16 | 17 |
| Woody biomass | Total sugars | Glucose | Xylose | Mannose | Arabinose | Galactose | Rhamnose | Klason lignin | Acid soluble lignin | Extractives | Ash |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Oak | 56.75 | 37.55 | 15.65 | 1.83 | 0.42 | 0.98 | 0.32 | 23.30 | 2.98 | 8.80 | 0.32 |
| Douglas fir | 60.45 | 41.88 | 3.25 | 12.15 | 1.06 | 2.00 | 0.11 | 29.29 | 0.50 | 5.77 | 0.16 |
Conclusions
We describe a tandem catalyst system comprising Pd/C and metal triflates to remove lignin fragments from lignocellulosic biomass and convert these fragments in good yield to alkylmethoxyphenols, leaving cellulose as a solid residue. Under optimized conditions (20 mg Pd/C, 15 mg Al(III)-triflate, 180 °C, 2 h), the monomers yield obtainable from birch wood is 55 wt% (based on a lignin content of 23.8 wt% in the biomass). This optimized lignin monomers yield is close to the theoretical maximum value, which is based on the typical ether content of lignin in lignocellulosic biomass in the 67%–76%.20 The solid residue is mainly composed of cellulose with small amounts of hemi-cellulose and lignin (Table 1). The metal triflate is effective in breaking the bonds between lignin and the carbohydrate part of biomass, which are typically comprised of ether and ester linkages. Lignin dimer model compound experiments revealed that both metal triflates and Pd/C contribute to the further depolymerization of the resulting fragments into aromatic monomers. Metal triflates catalyst can cleave (β-O-4) ether linkages within the aromatic lignin structure due to their interaction with vicinal OH groups that active the ether bond. Metal triflates are more active for this reaction than Pd/C. Pd/C is able to cleave a wider range of ether linkages such as α-O-4, 4-O-5 and β–β. The Pd-to-metal triflate ratio is an important parameter to the overall efficiency of the reductive fraction process of woody biomass. Lower Pd/Al ratios resulted in PS/PG-OCH3 as dominant reaction products, while higher ratios led to formation of PS/PG-OH and PS/PG-H products. It is important to use a small amount of Pd/C, as otherwise rapid hydrodeoxygenation of aliphatic OH groups occurs, which limits the synergetic action of metal triflates. In addition to deoxygenation, Pd/C also saturates C–C double bonds of small lignin fragments, which are formed from fragmentation reactions catalyzed by metal triflate. These insights allowed decreasing the amount of metal and metal triflate catalysts and the reaction temperature and time. A range of metal triflates are effective with Al(III)-triflate providing the most promising results. We also demonstrated that the process can be easily scaled up to convert 100 g woody biomass with nearly similar lignin monomer yield as obtained in a small-scale experiments. The mild conditions, short reaction times and use of small amounts of catalyst make this tandem process very promising as a method to remove and upgrade lignin from second-generation biomass. Prospects of the resulting alkylmethoxyphenols are their use in the fragrance industry (e.g., dihydroeugenol), their upgrading to alkylbenzenes or alkylphenols by catalytic hydrodeoxygenation, or combining demethoxylation and dealkylation to obtain phenol, a valuable base chemical.Experimental
Chemicals and materials
Birch wood was obtained from the Energy Research Center of Netherlands (ECN). Oak and Douglas fir wood sawdust were obtained from houtzagerij Menten (http://www.houtzagerijmenten.nl). Detailed composition of these woods is provided in Table 7. 5 wt% Pd/C and all the metal triflates were purchased from Sigma Aldrich. 4-n-Propanolguaiacol (PG-OH) was purchased from TCI. 4-n-Propylsyringol (PS-H) and 4-n-methoxy propyl syringol (PS-OCH3) were recovered from the reaction mixture (T = 180 °C, t = 2 h over Pd/C and Yb(III)-triflate catalyst using birch wood sawdust). The reaction mixture was first subjected to a partitioning work-up37 to removed sugars and then separated using column chromatography by choosing 1:9 (v/v) hexane/diethyl ether as eluent. These phenolic compounds were used to determine the weight response factors relative to n-dodecane. Extra-dry absolute methanol was purchased from Biosolve. Benzyl phenyl ether and 2-phenoxy-1-phenylethanol were purchased from Sigma Aldrich. Guaiacylglycerol-β-guaiacyl ether (>97%) was purchased from TCI. 2-Phenylethyl phenyl ether was purchased from Santa Cruz Biotechnology. All the other commercial chemicals are analytical grade and used without further purification.
Feedstock pretreatment
Birch wood was first ball milled to small particles and then sieved to obtain particle with sizes between 150 μm and 300 μm. The wood sawdust was subjected to Soxhlet extraction with water followed by ethanol to remove extractives. After extraction, the feedstock particles were dried at 105 °C overnight for catalytic reaction.Catalytic activity measurements
In a typical reaction experiment, the autoclave was charged with a suspension of 2.0 g feedstock, 200 mg Pd/C (5 wt% Pd loading), 15 mg Al(III)-triflate and 30 μl n-dodecane internal standard in 40 ml methanol. The reactor was sealed and purged with nitrogen and hydrogen several times sequentially. After leak testing, the pressure brought to 30 bar with hydrogen and the reaction mixture was heated to reaction temperature under continuous stirring at 500 rpm within 0.5 h. A sampling valve was installed, which allows taking liquid samples during the reaction. In some cases, about 0.4 ml solutions were taken out from autoclave during reaction and analysed directly by GC-MS. After reaction, the heating oven was removed and the reactor was cooled to room temperature in an ice-water bath. After releasing the pressure, the autoclave was opened and the mixture was collected and combined with the solution obtained from washing the autoclave with methanol. The collected reaction mixture was filtered by using a filter crucible under vacuum. The carbohydrate and Pd/C solid residue was left in air to dry for one day to determine the amount of all solid residue. In some cases, the solid residue was further subjected to a two-stage acid hydrolysis to determine the (hemi-)cellulose, lignin, and ash composition.A mini-autoclave with a volume of 12 ml was used for reactions with model compound. In a typical run 50 mg model compound (BPE and GG), 10 mg Pd/C, 5 mg Yb(III)-triflate and 30 μl n-dodecane internal standard were added to 5 ml methanol. After sealing, purging and checking for leaks, the autoclave was heated to 160 °C and maintained at that temperature for 2 h. After reaction, an aliquot of 1 ml was taken from the reaction mixture and directly analysed by GC-MS without dilution following filtration with a 0.45 μm syringe filter. In experiments involving PPE-H and PPE-OH as model compounds, 30 mg reactant was used, and the reaction temperature and time were 200 °C and 4 h, respectively.
Metal-triflate recycling
The reaction mixture was treated by the work-up procedure mentioned in our previous work (ref. 37). From the liquid fraction, methanol was removed from the liquid products (methanol-soluble lignin and sugar products) by evaporation. The residue was separated by extraction with water and ethyl acetate. The water soluble fraction will contain most of the initial metal triflate. Water was removed from this solution by rotary evaporation under vacuum. The resulting product was re-dissolved in 40 ml methanol and used in a new reaction under standard conditions.Scale-up of reductive fractionation
Oak and Douglas fir wood sawdust were used to explore conversion of larger amounts of woody biomass. The sawdust samples were dried overnight in an oven at 105 °C and sieved to obtain particles with sizes between 0–300 μm and 300–1000 μm for oak, and between 300–1000 μm for Douglas fir. No extraction treatment was employed for these samples.Product analysis and characterization
The liquid phase products were analysed by a Shimadzu 2000 GC-MS system equipped with a RTX-1701 column (60 m × 0.25 mm × 0.25 μm) and a flame ionization detector (FID) together with a mass spectrometer (MS) detector. For lignin monomers, the product peaks were identified using MS and by authentication with reference compounds. All the quantitative analyses of liquid phase products were based on GC-FID. Experimentally determined weight response factors (RF) were used for calculation relative to n-dodecane, which was the internal standard. For some of the commercially unavailable compounds (side-products obtained in small amounts), the RF of PS-OCH3 was used for quantification. The yield of lignin monomers was calculated using following equation: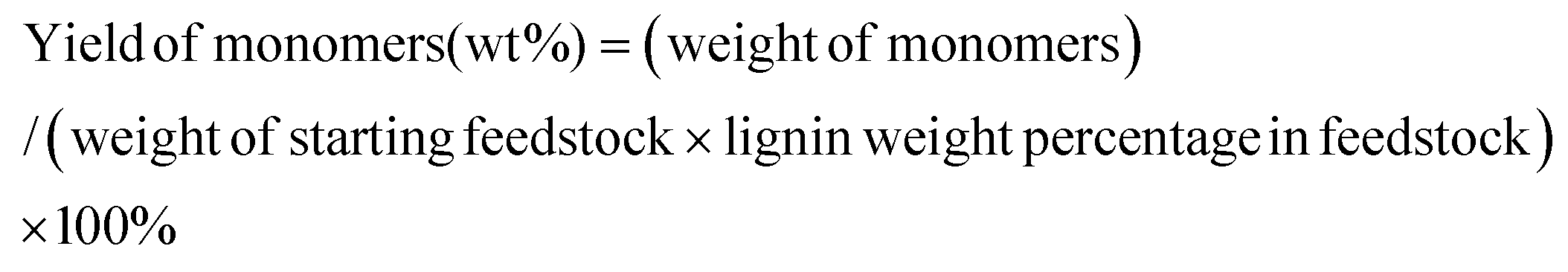
The product quantification for the model lignin compound reactions (BPE, PPE-H and PPE-OH) was based on the GC area normalization method. In the case of GG model lignin compound reaction, the experimentally determined weight response factor for guaiacol relative to n-dodecane was used for product quantification.
Lignocellulosic composition analysis
All the wood samples were delivered to Celignis Limited Nexus Innovation Centre, University of Limerick, Ireland (http://www.celignis.com) for analysis. All the samples were analysed in duplicate and take the mean value. The detailed analysis procedure is shown as follows.Acid insoluble lignin (Klason lignin)
Weight 300 mg of the sample into a pressure tube, then adding 72% H2SO4 (3 ml) by means of an automatic titrator. The sample and acid were then mixed thoroughly using a glass rod and the pressure tube was transferred to a water bath, maintained at 30 °C, for a period of 1 h. The sample/acid mixture was stirred every 5 min. Subsequently 84 ml of water was added to achieve a 4% acid concentration and the pressure tube sealed. All of the pressure tubes of an analytical batch (between 16 and 20) were then transferred to an autoclave along with two/three pressure tubes each containing 10 ml of a solution of a known sugar composition to which 348 μl of 72% H2SO4 was added. These additional tubes are referred to as the sugar recovery solutions and were used to determine the sugar losses associated with this secondary hydrolysis step (121 °C for 60 min). The pressure tubes were removed from the autoclave once the temperature fell below 80 °C and allowed to cool to room temperature at which point the hydrolysates were filtered (using vacuum suction), through filter crucibles of known weight, and the resulting filtrate was stored. Residual solids were washed from the tube using deionised water until all the residue resided on the filter crucible. This was then dried overnight at 105 °C and weighed to determine the Acid Insoluble Residue (AIR) content. The filter crucible was then ashed to determine the acid-insoluble ash (AIA) content. The Klason lignin content was determined as AIR minus AIA. The sum of Klason and acid soluble lignin was used for lignin monomer yield calculation.Acid soluble lignin (ASL)
The hydrolysate was placed in a 1 cm path-length (3 ml volume) quartz cuvette and diluted with water until the UV-absorbance was within a linear region. The spectrum of the sample was collected in transmission mode using a HP Agilent 8452A diode array spectrophotometer. The absorbance at 205 nm was used to determine the ASL content using an absorptivity constant of 110 M−1 cm−1.Sugar analysis
The hydrolysates obtained from the two-stage hydrolysis of the carbohydrate residue were diluted 5 times with a solution containing a known concentration of the internal standard fucose. A series of sugars including arabinose, galactose, rhamnose, glucose, xylose, and mannose were used for sugar quantification. Relative response factors were determined via sugar standard samples injected at regular intervals in the analytical sequence. Detailed description of the analysis procedure can be found in ref. 38 and http://www.celignis.com/package.php?value=1.Gel permeation chromatography (GPC)
GPC analyses were performed using a Shimadzu apparatus equipped with two columns in series (Mixed-C and Mixed-D, polymer laboratories) and a UV-Vis detector at 254 nm. The column was calibrated with polystyrene standards. Analyses were carried out at 25 °C using THF as eluent at a flow rate of 1 ml min−1. For the lignin residue analysis, the sample was prepared at a concentration of 2 mg ml−1. All samples were filtered using 0.45 μm filter membrane prior to injection.Acknowledgements
This work was funded by the “New Energy House” project of the Eindhoven Energy Institute in collaboration with the Knowledge and Innovation Community InnoEnergy of the European Institute of Innovations and Technology.References
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS PubMed.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47, 1503–1512 CrossRef CAS PubMed.
- C. Z. Li, M. Y. Zheng, A. Q. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383–6390 CAS.
- P. F. H. Harmsen, W. J. J. Huijgen, L. M. B. López and R. R. C. Bakker, Literature Review of Physical and Chemical Pretreatment Processes for Lignocellulosic Biomass, ECN-E–10-013, Food & Biobased Research, Wageningen UR, 2010 Search PubMed.
- A. T. W. M. Hendriks and G. Zeeman, Bioresour. Technol., 2009, 100, 10–18 CrossRef CAS PubMed.
- J. L. Xu, X. M. Zhang and J. J. Cheng, Bioresour. Technol., 2012, 111, 255–260 CrossRef CAS PubMed.
- V. Chaturvedi and P. Verma, Biotechnology, 2013, 3, 415–431 Search PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- F. P. Bouxin, A. McVeigh, F. Tran, N. J. Westwood, M. C. Jarvis and S. D. Jackson, Green Chem., 2015, 17, 1235–1242 RSC.
- J. L. Wen, B. L. Xue, S. L. Sun and R. C. Sun, J. Chem. Technol. Biotechnol., 2013, 88, 1663–1671 CrossRef CAS.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, Green Chem., 2015, 17, 4941–4950 RSC.
- X. M. Zhang, P. Murria, Y. Jiang, W. H. Xiao, H. I. Kenttamaa, M. M. Abu-Omar and N. S. Mosier, Green Chem., 2016, 18, 5219–5229 RSC.
- P. Sannigrahi, A. J. Ragauskas and S. J. Miller, BioEnergy Res., 2008, 1, 205–214 CrossRef.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. D. Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- C. R. Kumar, N. Anand, A. Kloekhorst, C. Cannilla, G. Bonura, F. Frusteri, K. Barta and H. J. Heeres, Green Chem., 2015, 17, 4921–4930 RSC.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- I. Klein, C. Marcum, H. Kenttämaa and M. M. Abu-Omar, Green Chem., 2016, 18, 2399–2405 RSC.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- B. Joffres, D. Laurenti, N. Charon, A. Daudin, A. Quignard and C. Geantet, Oil Gas Sci. Technol., 2013, 68, 753–763 CrossRef CAS.
- C. P. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- C. Z. Li, X. C. Zhao, A. Q. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- X. Huang, C. Atay, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ACS Catal., 2015, 5, 7359–7370 CrossRef CAS.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. T. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed.
- Q. Song, F. Wang, J. Y. Cai, Y. H. Wang, J. J. Zhang, W. Q. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttamaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- T. Renders, W. Schutyser, S. Van den Bosch, S. F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS.
- B. Guvenatam, E. H. J. Heeres, E. A. Pidko and E. J. M. Hensen, J. Mol. Catal. A: Chem., 2015, 410, 89–99 CrossRef.
- B. Guvenatam, E. H. J. Heeres, E. A. Pidko and E. J. M. Hensen, Catal. Today, 2016, 269, 9–20 CrossRef.
- B. Guvenatam, E. H. J. Heeres, E. A. Pidko and E. J. M. Hensen, Catal. Today, 2016, 259, 460–466 CrossRef.
- R. Jastrzebski, S. Constant, C. S. Lancefield, N. J. Westwood, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2016 DOI:10.1002/cssc.201600683.
- X. Huang, Z. Jiadong, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2016 DOI:10.1002/cssc.201601252.
- D. J. M. Hayes, Bioresour. Technol., 2012, 119, 393–405 CrossRef CAS PubMed.
- B. Guvenatam, O. Kursun, E. H. J. Heeres, E. A. Pidko and E. J. M. Hensen, Catal. Today, 2014, 233, 83–91 CrossRef.
- J. Y. He, L. Lu, C. Zhao, D. H. Mei and J. A. Lercher, J. Catal., 2014, 311, 41–51 CrossRef CAS.
- H. Fan, Y. Yang, J. Song, Q. Meng, T. Jiang, G. Yang and B. Han, Green Chem., 2015, 17, 4452–4458 RSC.
- J. Tsujino, H. Kawamoto and S. Saka, Wood Sci. Technol., 2003, 37, 299–307 CrossRef CAS.
- H. L. Wang, L. B. Zhang, T. S. Deng, H. Ruan, X. L. Hou, J. R. Cort and B. Yang, Green Chem., 2016, 18, 2802–2810 RSC.
- M. Zaheer and R. Kempe, ACS Catal., 2015, 5, 1675–1684 CrossRef CAS.
- H. Z. Liu, T. Jiang, B. X. Han, S. G. Liang and Y. X. Zhou, Science, 2009, 326, 1250–1252 CrossRef CAS PubMed.
- T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttamaa, F. Ribeiro, J. T. Miller and M. M. Abu-Omar, Chem. Sci., 2013, 4, 806–813 RSC.
- Z. Li, R. S. Assary, A. C. Atesin, L. A. Curtiss and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 104–107 CrossRef CAS PubMed.
- M. C. Lawton, PhD Thesis, University of Johannesburg, 2009.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS.
- A. G. M. Barrett and D. C. Braddock, Chem. Commun., 1997, 351–352 RSC.
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W. L. Lam, Chem. Rev., 2002, 102, 2227–2302 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02962k |
| This journal is © The Royal Society of Chemistry 2017 |