
Liquid biopsy on chip: a paradigm shift towards the understanding of cancer metastasis
Amogha
Tadimety†
a,
Abeer
Syed†
a,
Yuan
Nie
a,
Christina R.
Long
a,
Kasia M.
Kready
a and
John X. J.
Zhang
*ab
aThayer School of Engineering at Dartmouth College, Hanover NH, 03755, USA. E-mail: John.Zhang@dartmouth.edu
bDartmouth-Hitchcock Norris Cotton Cancer Center, Lebanon NH, 03766, USA
First published on 1st December 2016
Abstract
This comprehensive review serves as a guide for developing scalable and robust liquid biopsies on chip for capture, detection, and analysis of circulating tumor cells (CTCs). Liquid biopsy, the detection of biomarkers from body fluids, has proven challenging because of CTC rarity and the heterogeneity of CTCs shed from tumors. The review starts with the underlying biological mechanisms that make liquid biopsy a challenge before moving into an evaluation of current technological progress. Then, a framework for evaluation of the technologies is presented with special attention to throughput, capture rate, and cell viability for analysis. Technologies for CTC capture, detection, and analysis will be evaluated based on these criteria, with a focus on current approaches, limitations and future directions. The paper provides a critical review for microchip developers as well as clinical investigators to build upon the existing progress towards the goal of designing CTC capture, detection, and analysis platforms.
 Amogha Tadimety | Amogha Tadimety completed her BSE at Princeton University in 2014, with a major in Chemical and Biological Engineering and certificates in Engineering Biology and Values & Public Life. During that time she completed research internships at the Weatherall Institute of Molecular Medicine at Oxford University and the Wyss Institute at Harvard. Amogha is currently pursuing her PhD at Dartmouth College's Thayer School of Engineering in the Zhang Research Group. Her research focuses on the development of nanoplasmonic sensors for capture, enrichment, and detection of circulating biomarkers, with a focus on low-cost nanostructure fabrication. |
 Abeer Syed | Abeer Syed received her PhD in Biomedical Engineering from the University of Glasgow, UK in 2013 followed by postdoctoral research at New York University Abu Dhabi, UAE and the Dartmouth College, USA. Her research focuses on design, fabrication and use of microfluidic systems for low cost, point-of-care diagnostics. |
 Yuan Nie | Yuan Nie received her BS degree in Mechanical Engineering from Huazhong University of Science and Technology, Wuhan, China, in 2011, and her MS degree in Mechanical Engineering from Hong Kong University of Science and Technology, Hong Kong, in 2014. She is currently a PhD student in Thayer School of Engineering, Dartmouth College, NH, USA. Her main research is focused on the development of the immunomagnetic microfluidic platform for the capture and analysis of rare cancer biomarkers. She is also interested in the finite volume based simulation of fluidic dynamics at micro/nano-scale. |
 Christina R. Long | Christina Long is a Dartmouth undergraduate studying engineering modified with computer science. She has been working as an assistant to the Zhang lab group since summer of 2015. Her work focuses on improving methods for the immunomagnetic capture of circulating tumor cells. |
 Kasia M. Kready | Kasia Kready, currently an undergraduate at Dartmouth College, studies engineering with a focus in biotechnology at the Thayer school of Engineering. Broadly, she is interested in ‘gaps’ in women's healthcare, nanotechnology, and bioengineering. Kasia has been an undergraduate research assistant in John Zhang's lab since 2014. Her work involves fabricating nanoplasmonic arrays for biosensing applications. Additionally, she studies Arabic, French, and Greek. She is expected to graduate in June 2017. |
 John X. J. Zhang | John X. J. Zhang is a Professor at Thayer School of Engineering, Dartmouth College, and an Investigator of Dartmouth-Hitchcock Medical Center. He received his PhD from Stanford University, and was a Research Scientist at Massachusetts Institute of Technology. Dr Zhang is a Fellow of American Institute for Medical and Biological Engineering, and recipient of NIH Director's Transformative Research Award, NSF CAREER Award, DARPA Young Faculty Award and many other recognitions. Zhang's research focuses on exploring bio-inspired nanomaterials, scale-dependent biophysics, and nanofabrication technology, towards developing new diagnostic devices and methods on probing complex cellular processes and biological networks critical to development and diseases. |
Insight, innovation, integrationDetection of rare biomarkers, such as circulating tumor cells (CTCs), has proven challenging because of the rarity and heterogeneity of targets in samples. Quantitative detection and downstream analyses of biomarkers have become increasingly feasible with advances in micro/nanoscale engineered systems. Emerging microchip based technologies enable high throughput screening with single cell resolution, which helps understand tumor heterogeneity and the metastatic process by performing “omics” (genomics, proteomics) studies and by characterizing cell phenotypes based on surface protein expression and deformability. In this review, the significance of throughput, accuracy, and efficiency is detailed for capture, detection, and analysis of cancer cells. This review provides bioengineers and clinical investigators a framework for developing simple, sample-to-answer diagnostic platforms for initial diagnosis and continued monitoring. |
1. Introduction
Although researchers, medical caregivers, and informed citizens have curbed cancer's death rate by 22% from 1991 to 2011 in the USA, cancer remains the greatest cause of non-accidental deaths globally.1 The Cancer Moonshot Initiative encourages scientists to build upon these advances through better predictive screening, diagnostics, prognostics, targeted therapies, and monitoring. Cancer diagnostic tools sustainably contribute to this success, especially excisional biopsies.Once the presence of abnormal tissue or fluid is discerned, medical professionals have invasive and minimally invasive procedures to remove, or ‘biopsy’, a portion of it in order to perform diagnostic tests.2 These procedures prevail as the sole diagnostic means of determining whether a suspicious area is cancerous. Unfortunately in the case of breast biopsies, many women must undergo these procedures only to reveal the news that these suspicious regions are benign.3,4 In certain cases, the suspicious tissues are located in regions of the body that are inaccessible for biopsies. Additionally, excisional biopsies only provide a time dependent snapshot of the dynamic genetic fluctuations of a tumor. An invasive or even minimally invasive biopsy is risky to spatially and temporally perform on a patient to monitor disease progression. The aforementioned information presents an outstanding opportunity to improve upon the state of the art methods of biopsies, especially when rates of survival correlate with earlier detection.
The burgeoning field of microfluidics is producing innovative liquid biopsies on chip as a non-invasive cancer diagnostic tool. Liquid biopsies on chip consist of this general scheme: liquid from a patient (i.e. blood or serum) is loaded into a microfluidic device that performs a combination of capturing, detecting, and or analyzing tumor derived material, such as circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), or exosomes, that are shed by a tumor into the circulatory system (Fig. 1). In addition to being a novel non-invasive cancer biopsy, liquid biopsies on chip are low cost, less exhaustive of resources, superior at reproducing results, conducive to sequential sampling, and allow automation. However, microchip developers encounter numerous challenges in developing functional devices. One of the greatest challenges is that the tactics employed by cancer to evade the immune system create sophisticated hurdles that researchers must overcome to integrate cancer biology into chips to capture, detect, and analyze cancer biomarkers. Even within the same type of cancer, there are stratifications consisting of different biological signatures, or ‘biomarkers’ to capture the CTCs, ctDNA, or tumor-derived exosomes. A major design challenge is integrating the biomarkers of cancer into microchip design.
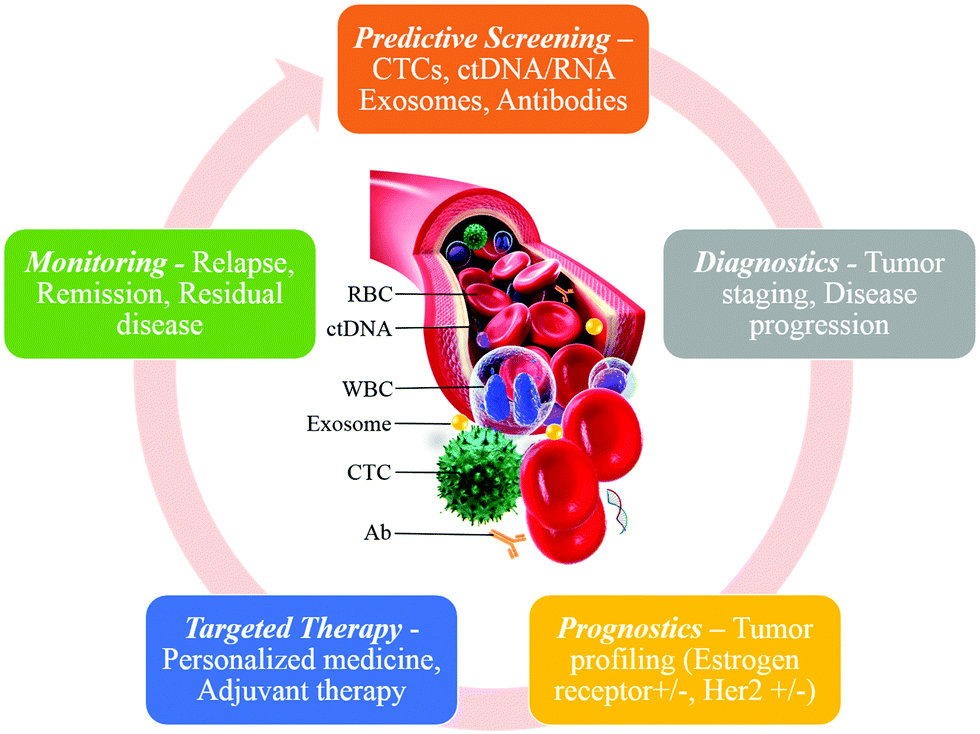 | ||
| Fig. 1 Liquid biopsy as a diagnostic and prognostic tool. The information derived from liquid biopsy can be used for continuous monitoring of the patients, from initial screening to personalized treatment. | ||
CTCs remain the biomarkers with the greatest wealth of research, analysis, and validation, although there have been many recent strides in ctDNA5 and extracellular vesicle analysis.6–9 There are over 3500 papers from the last ten years on PubMed relating to CTC analysis, including papers relating to their impact on therapeutics,10–12 capture,13–15 and underlying biology.16–18 CTCs are larger than the other biomarkers and their molecular contents are protected inside intact cells by a bilayer. Furthermore, validation of capture is easier using CTCs than it is for ctDNA and vesicles due to their larger size and well-characterized surface markers. CTCs provide rich data about the current tumor status of the patient, and provide a large enough sample for a variety of molecular analyses. The integrated findings from CTC capture (count, molecular contents, surface expression) provide information for patient diagnosis, monitoring, drug response, and prognosis. Clinically relevant analysis of CTCs majorly factors into the design of future liquid biopsies on chip.
Beyond molecular characterization of a population of cells, there have also been a number of recent studies conducted at the single cell level.19–22 Transcriptional profiling, surface marker analysis, and deformability all vary greatly between CTCs within the same patient. This heterogeneity is characterized in our current gold standard biopsy through mutation analysis within tumor tissue; similarly, a recent phenomenon has been shown with CTCs.23 This type of analysis can provide clues as to the status of the full tumor because CTCs may shed from different sites within the tumor. Single cell molecular analysis also has the potential to prevent leukocyte contamination through an additional check on molecular contents. Thus, the capture and analysis of single CTCs on chip is valuable to the determination of prognosis and therapy from liquid biopsies. Because of this, the review will outline technologies with special attention to platforms that are compatible with a single-cell analysis.
In order to facilitate the design process of liquid biopsies on chip, this review functions as a guide for microchip development. The review is structured to explain and evaluate existing technologies in the context of cancer biology, throughput, and ease of analysis. We begin this review with the relevant background knowledge of invasive and minimally invasive biopsies, and then delve into the perspectives and limitations of liquid CTC biopsies on chips. We then present a framework with key parameters for liquid CTC biopsies on chip based upon state of the art technology and clinically significant data. We then quickly overview the mechanisms of cancer and the theorized journey CTCs, so that we can critically analyze methods and existing technologies integrating biomarkers of CTCs into microchip designs for capture, detection, and analysis. Where appropriate, this review focuses on breast cancer, not only because of its impact on healthcare, but because of power and depth of existing knowledge to illustrate the opportunities for liquid biopsies on chip.
2. Current biopsy gold standards and liquid biopsy
2.1 Surgical biopsy
Early in the 10th century, Al Zahrawi, an Arab surgeon, invented a hollow needle to examine abnormal tissues in thyroid glands.24 Since Al Zahrawi's first recorded biopsy, major foundations of biopsies have not changed. Today, the existing categories of biopsies, surgical, core-needle, and fine-needle, still require the excision of suspicious tissues by way of an invasive or minimally invasive procedure (Table 1).| Advantages | Disadvantages |
|---|---|
| Surgical | |
| ✓ Most data retrieved (type, grade, receptor status, margins) | – Requires local anesthetic |
| ✓ Removes part or all of tumor | – Requires image guidance |
| – Invasive and uncomfortable for patient | |
| – No real-time monitoring | |
| Minimally invasive | |
| ✓ Greater patient comfort | – Requires local anesthetic |
| ✓ Better cosmetic results | – Requires image guidance |
| ✓ Can fully excise small lesions | – No real-time monitoring |
| Liquid biopsy | |
| ✓ Does not require appreciable symptoms for diagnosis | – Collecting rare biomarkers is challenging |
| ✓ Far less invasive | – Does not provide a treatment |
| ✓ Enables real-time monitoring | – Very few FDA approved technologies |
| ✓ Does not require pathologist | – Location of tumor unknown |
| – Size of tumor unknown | |
| – Margins unknown | |
Surgical biopsies are the most invasive; the patient receives a local anesthetic, allowing the surgeon to remove all or part of the abnormal tissue, nearby normal tissue, and implant a small piece of metal to mark the location of the tumor in case further surgery is required.25 The removal of part or all of the abnormal tissue conveniently decreases the amount of treatment necessary for remission.26 Once excised, the biopsied material is sent to pathologists, who then produce a report with clinically relevant information indicating the presence of cancerous cells. If cancerous cells are discovered, the type, the grade, and the receptor status of the tumor are reported along with the location of the tumor, the distance of the tumor from normal tissue, and the margins.27 Margins are a measure of how well the tumor was removed, and they are broken down into three different types. A positive margin signifies that cancer cells are at the boundary of the tumor and suggest that the tumor has advanced beyond the immediate area.26 A negative margin indicates that cancer cells are not found at the boundary of the tumor, and that the cancer is localized to that specific region. A close margin means that the distance between cancerous tissue and normal tissue is less than 3 millimeters. Surgical biopsies grant the most data to be gleaned from the tumor and the most of the tumor removed.26 However, this data is only a snapshot of the dynamic genetic fluctuations of the tumor, and repeated surgical biopsies cannot be performed to monitor patients in real-time because of surgical risks and patient discomfort.
2.2 Minimally invasive biopsy
The focus of minimally invasive techniques is obtaining the diagnosis, not complete excision or treatment of cancer. In addition to reducing morbidity and mortality, minimally invasive techniques offer unparalleled cosmetic results and a higher degree of patient comfort.2 These techniques have allowed for cost-effective and holistic medical care by focusing surgery on negative margins.28–31 Core-needle biopsy (CNB),32,33 fine-needle aspiration cytology (FNAC),34,35 large core biopsy,33,36 and vacuum-assisted core biopsy35,37 are the cutting-edge procedures.During a CNB, a hollow needle is inserted into a locally anesthetized region above the abnormal tissue and a ‘core’ of the abnormal tissue is excised. Large core biopsies remove the entirety of small abnormal lesions through the hollow core of a large needle.38 Fine-needle aspirations are performed when an abnormal tissue is expected to be fluid filled or when an abnormal tissue is superficial. Fine-needle aspirations are used to drain the fluid and remove cells for testing from suspected fluid filled abnormal tissues.38 FNACs excise abnormal tissue from superficial regions through minimally invasive fine-needles.34 Vacuum-assisted core biopsies apply a vacuum to a hollow needle, pulling the tissue into the center of the needle where it is cut and retrieved after the surgery. All of these procedures are guided by imaging techniques such as mammograms, magnetic resonance imaging (MRI), or ultrasounds. These image guidance techniques allow the visualization of the location of the abnormal tissue without physically viewing it.
Similar to surgical biopsies, minimally invasive biopsies do not support real-time monitoring of the patient's response to therapies. Although minimally invasive surgeries are more cosmetically appealing and diminish discomfort in comparison to surgical biopsies, they require local anesthetics, imaging guidance, and only remove small abnormal lesions. Invasive and minimally invasive biopsies suffer from a reliance on appreciable symptoms of tumors to signal that the operation is required.39 They also require a highly skilled pathologist who cannot robotically reproduce results, and surgeons who could potentially perturb the CTCs into proliferation. In some cases, regardless of the surgeon's and pathologist's expertise, the tumor could be anatomically challenging to biopsy with substantial risks.39 Even with these results, they do not represent a tumor's heterogeneity or its evolving resistance mechanisms. Without these, cancers cannot be precisely stratified, which is imperative for personalized treatments.
Innovative, non-invasive biopsies must be developed to revamp outdated procedures that rely less upon perceivable symptoms and image guidance. By the nature of biopsy, symptoms need to be present before a biopsy can be performed and definitive diagnosis formed. There is some debate over whether improved life expectancy of cancer patients is due to earlier detection or better treatment methods. A study of life expectancy by Chie et al. found that for breast, cervical, colorectal, gastric, and liver cancer, early detection was responsible for a larger share of improved life expectancy than improved treatment methods.40 This result shows great promise for the impact of early detection on patient prognosis.
2.3 Liquid biopsy
Liquid biopsies are a non-invasive means of testing the patient's blood to identify CTCs and circulating tumor DNA (ctDNA) cast out by the primary tumor and metastatic sites.41 As such, liquid biopsies are appealing because they offer an up-to-date insight into the patient's condition and allow for situation-specific responses and informed treatment choices.42 A number of samples, including blood, plasma, urine, CSF, and breast milk are being studied for liquid biopsy platform development.41 Apart from providing accurate information regarding the cancer's advancement and intensity, liquid biopsies can effectively gauge the body's responses to the chosen method of treatment, and they can detect early recurrence and identify possible causes behind resistance to treatment.43 Liquid biopsy analysis can combine with ongoing treatment to map molecular disease changes, allowing clinicians to chart the growth of secondary resistance and isolate heterogeneous subclonal populations of tumor cells that continue to grow.44Liquid biopsy enables real-time monitoring of ongoing changes in the tumor at the molecular level, allowing for more extensive insights into disease state and progression.44 The use of liquid biopsies holds significant potential and it is likely to replace extensive imaging and invasive tissue biopsies. Likely, liquid biopsies will frequently monitor and inform cancer treatment choices. With technological advances, liquid biopsy may conceivably detect early tumors yet invisible on conventional imaging.
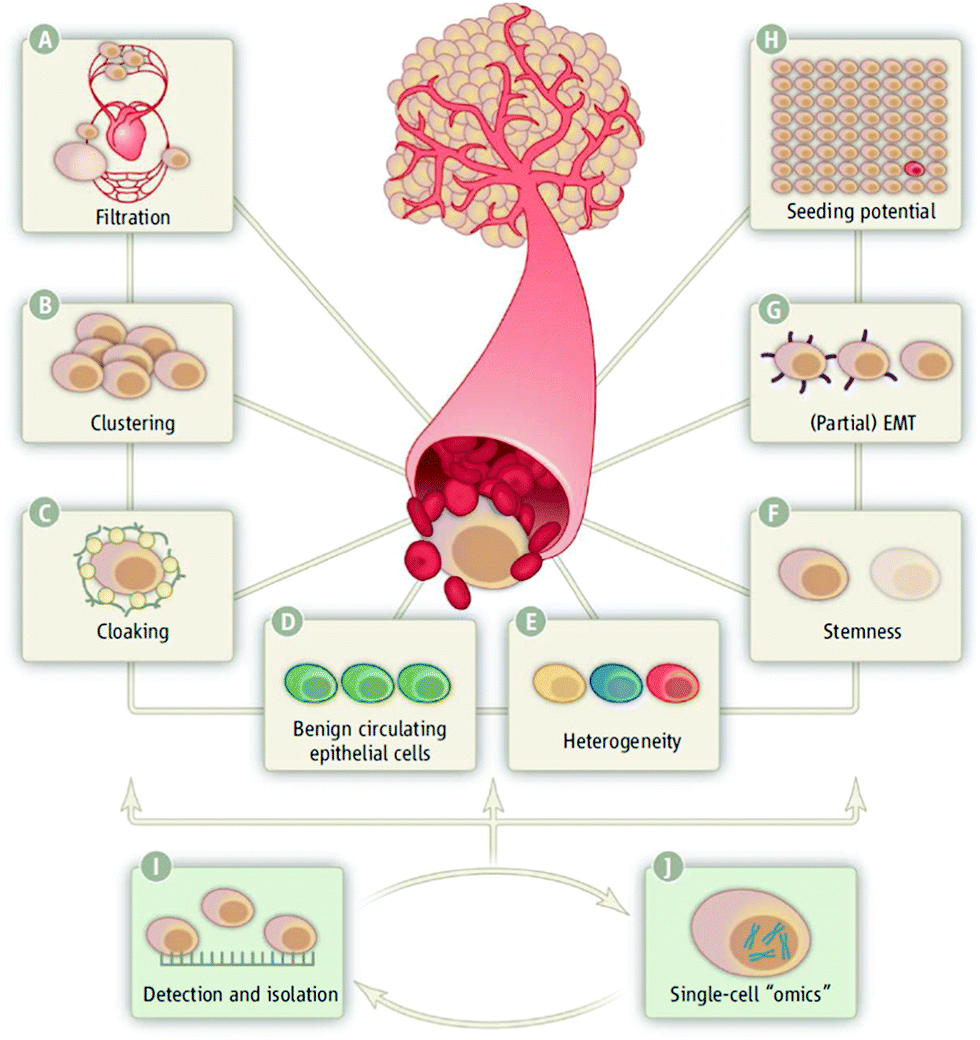 | ||
| Fig. 2 A schematic representation of challenges in CTC research. CTCs are difficult to detect because they may (A) undergo filtration in smaller capillaries, (B) form clusters likely to lodge in capillaries, (C) become cloaked by platelets and coagulation factors, thereby escaping the immune response, (D) may not provide useful information, (E) are heterogeneous, (F) show stemness, (G) show partial EMT, and (H) unclear metastatic seeding potential. Reprinted from Science, 341, Plaks, Circulating tumor cells, 1186–1188, Copyright (2013), with permission from AAAS. | ||
3. CTC liquid biopsy framework
Information from CTC capture can aid in predictive screening, diagnostics, prognostics, targeted therapies, and monitoring. For predictive screening and diagnostics, just the presence and enumeration of CTCs is valuable, and the analysis may include validation that the captured cells are CTCs and whether the cell is metastatic. A more advanced diagnostic may measure the surface markers, secreted proteins, or mutations inherent within the CTCs genome. For determining patient prognosis, enumeration is very important, as number of captured CTCs has been shown to correlate with overall survival rate. A mutational or epigenetic analysis may also be done because certain mutations result in a more aggressive cancer presentation. Similarly for monitoring, the number of captured CTCs and whether they decrease as a patient is being treated can give the clinician valuable information about patient status and recurrence of disease. In terms of targeted therapies, a more in-depth level of genomic analysis may be done or drug response can be evaluated by CTC culture. Through this method clinicians can learn which treatment the CTCs respond to most readily, which could help them prescribe the correct therapy from the start. Some advantages of liquid biopsy include the possibility for serial sampling, automation, and reproducible results. For each of the applications discussed, including diagnostics, monitoring, and prognosis, the ability to serially sample and automate the testing allows for validation and confirmation of the result (Table 2).| Capture | Detection | Analysis |
|---|---|---|
| ✓ High throughput | ✓ High throughput | ✓ Effective enumeration |
| ✓ High capture efficiency | ✓ High detection efficiency | ✓ Accuracy |
| ✓ Low contamination | ✓ Minimal damage to cells | ✓ Repeatability |
| ✓ Minimal damage to cells | ✓ Contrast with background | ✓ Comparison to clinical Gold standard |
| ✓ Contrast with contaminant |
When designing liquid biopsy chips, it is ideal to integrate capture, detection, and analysis into simple platforms. Capture is the first step, and involves separating out CTCs from the starting sample, most commonly whole blood. A number of techniques exist for CTC capture and involve separating CTCs based on distinctions in physical or chemical characteristics from other cells. This can involve binding the antigens, separating them by size or optical properties, or by mechanical properties. The ideal capture technology would have a high capture rate (above 90% effective), high throughput (milliliters of sample ran in a few hours), low contamination rate, and do minimal damage to the cells (above 90% cell viability). This would mean that the technology quickly picks out the most CTCs while leaving behind other types of cells, and does this quickly and leaves the cells viable. The capture technologies discussed later in the paper will be evaluated based on these criteria.
A very similar set of criteria can be used to evaluate the quality of a method of detection, or confirming the presence of the captured CTCs. Among available methods include immunofluorescent staining, light microscope imaging, nanoparticle based methods, and label-free detection. A clinically translatable detection method would allow for high throughput (milliliters of sample ran in a few hours), enumeration and high detection efficiency (above 90% effective), and significant contrast between the CTC and both the background and other cells. Ideally, an advanced detection method would maintain cell viability for analysis (above 90% cell viability).
Finally, the analysis step may be as simple as enumeration but often involves a molecular analysis of the captured cells. First, the effectiveness of the enumeration through analysis should be confirmed because of the importance of CTC count. This analysis step is key to benchmarking the liquid biopsy technology against current gold standard. Because of this, the accuracy of the analysis, repeatability, and comparison to current clinical standard are some of the criteria for evaluation of a method of CTC analysis. Based on the kind of molecular analysis that is typically done on a tumor biopsy, the analysis of the CTCs should be able to provide similar data. The best way to do this kind of validation is to run, in parallel and on the same patients, the liquid biopsy assay and a tissue biopsy analysis. An analytic validation of the diagnostic capabilities required for FDA approval of a liquid biopsy chip.47 The analysis methods reviewed in this paper will be described in terms of their accuracy, repeatability, and in the context of the current gold standards.
Oftentimes it makes the most sense to work backwards from the clinical need to determine the best capture and detection technology. For example, immunofluorescence staining and fixation can damage the cells, so it would not be an apt technology to use if you wanted to culture or do a genomic analysis on the cells afterwards. In this case, you may want to use a label free method of capture and detection, such as optical detection or mechanical method, in order to minimize the potential harm to the cells. On the other hand, if the goal is simply to detect whether CTCs are present and to count them, antibody–antigen binding followed by immunofluorescence staining may be the most successful method because of its demonstrated capture efficiency and ease of detection. Where appropriate, this paper will describe these design decisions and the possible technologies depending on desired analysis.
4. Biological background: mechanisms, EMT, & biomarkers
Integrating biology on chip for CTC capture, detection, and analysis requires an understanding of the underlying biological workings of cancer. In this section we discuss the mechanisms of cancer, the epithelial mesenchymal transition (EMT), biomarkers, and corresponding molecular analyses. The mechanism of cancer explains how six features of cancer are necessary for malignant growth and how tumor cells evade the host immune system, and many of these features underlie the challenges associated with CTC capture, detection, and analysis. Presumably, EMT boosts the challenges posed to CTC capture because the process involves additional heterogeneity during the process of cancer metastasis. Finally, some of the molecular studies discussed show this variability among CTC populations are the basis for single cell capture and analysis technologies. This biology section serves as a background for understanding relevant biomarkers and optimizing liquid biopsy chip design.4.1 Mechanisms of cancer
The unified efforts of researchers to understand the mechanisms of cancer have assembled an abounding body of knowledge that has elicited a well-accepted theory regarding the genesis of cancer. Hanahan and Weinberg listed six hallmarks of cancer (Fig. 3), and later other critical features have been used as the starting point for cancer research.48,49 Any combination of three of these six features are necessarily characteristic of any malignant growth: sustaining proliferative signaling, evading growth suppressors, activating invasion and metastasis, enabling replicative immortality, inducing angiogenesis, and resisting cell death.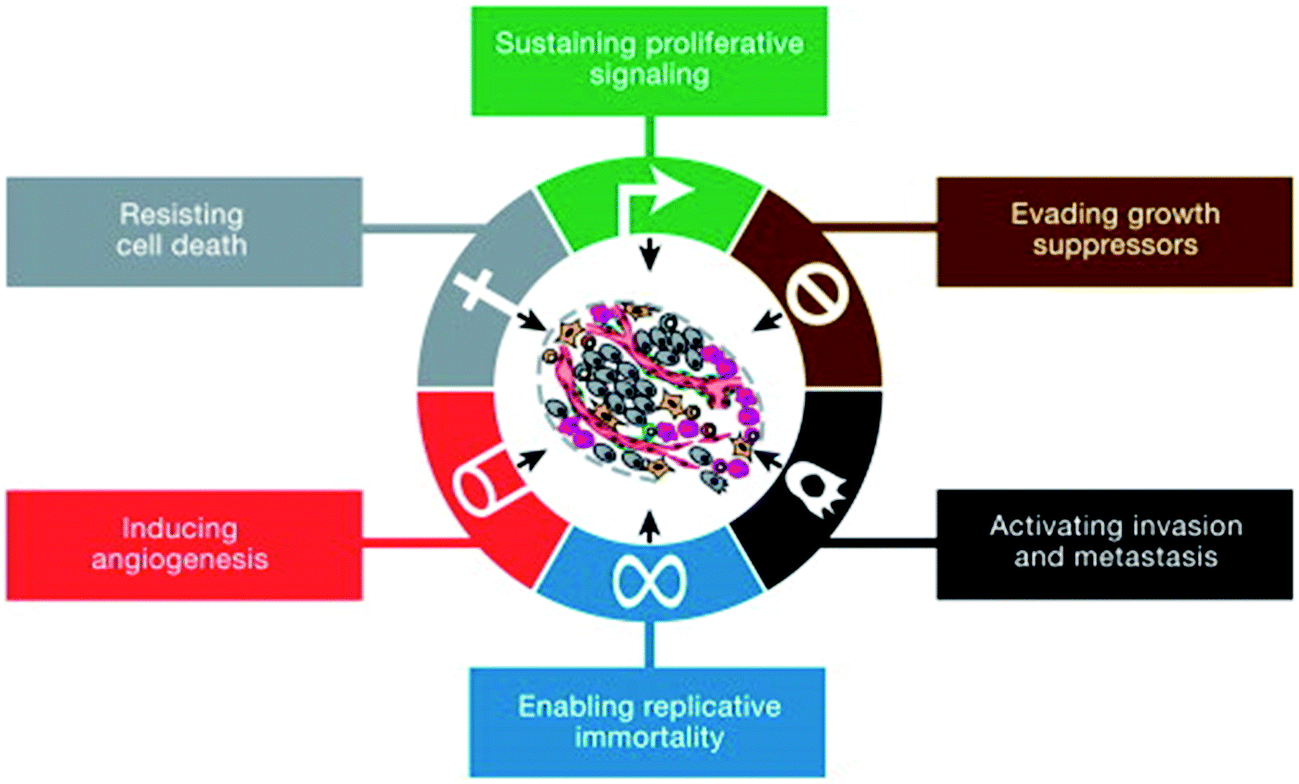 | ||
| Fig. 3 Schematic representation of six hallmarks of cancer. The cancerous cell produces sufficient growth signals while blocking the antigrowth signals to proliferate uncontrollably. Cells evade apoptosis, angiogenesis and activate metastasis. Reprinted from Cell, 14(5), Hanahan and Weinberg, Hallmarks of cancer: the next generation, 646–674, Copyright (2011), with permission from Elsevier. | ||
Since it is believed that at least three of these hallmarks are mandatory for malignancy, different categories of cancers arise; each one possibly shedding off a variety of heterogeneous strata of CTCs with contrasting phenotypic and functional characteristics.
4.2 Epithelial mesenchymal transition
In addition to the heterogeneity of cancer developed during its genesis, a second tier of heterogeneity is layered on through the hypothesized, and not well understood, process of epithelial mesenchymal transition (EMT). EMT is believed to simulate a spatial and temporal flux of CTCs between different disease states (epithelial, stemness, and mesenchymal) during primary tumor metastasis.57 First off, EMT enables an apical-basal polarized epithelial cell to transition through a number of biochemical changes to become a mesenchymal cell, leading to improved migratory capacity, invasiveness, improved resistance to apoptosis, and highly elevated extracellular matrix (ECM) component production (Fig. 4).58 Numerous molecular processes such as specific cell-surface protein expression, cytoskeletal protein rearrangement, synthesis of ECM-degrading enzymes, and changes in specific microRNA expression are involved in initiating the EMT process and facilitating its completion.59,60 Finally, the completion of this biological process is marked by the destruction of the underlying basement membrane, giving the cell, now classified as mesenchymal, the ability to escape the epithelial layer in which it was formed.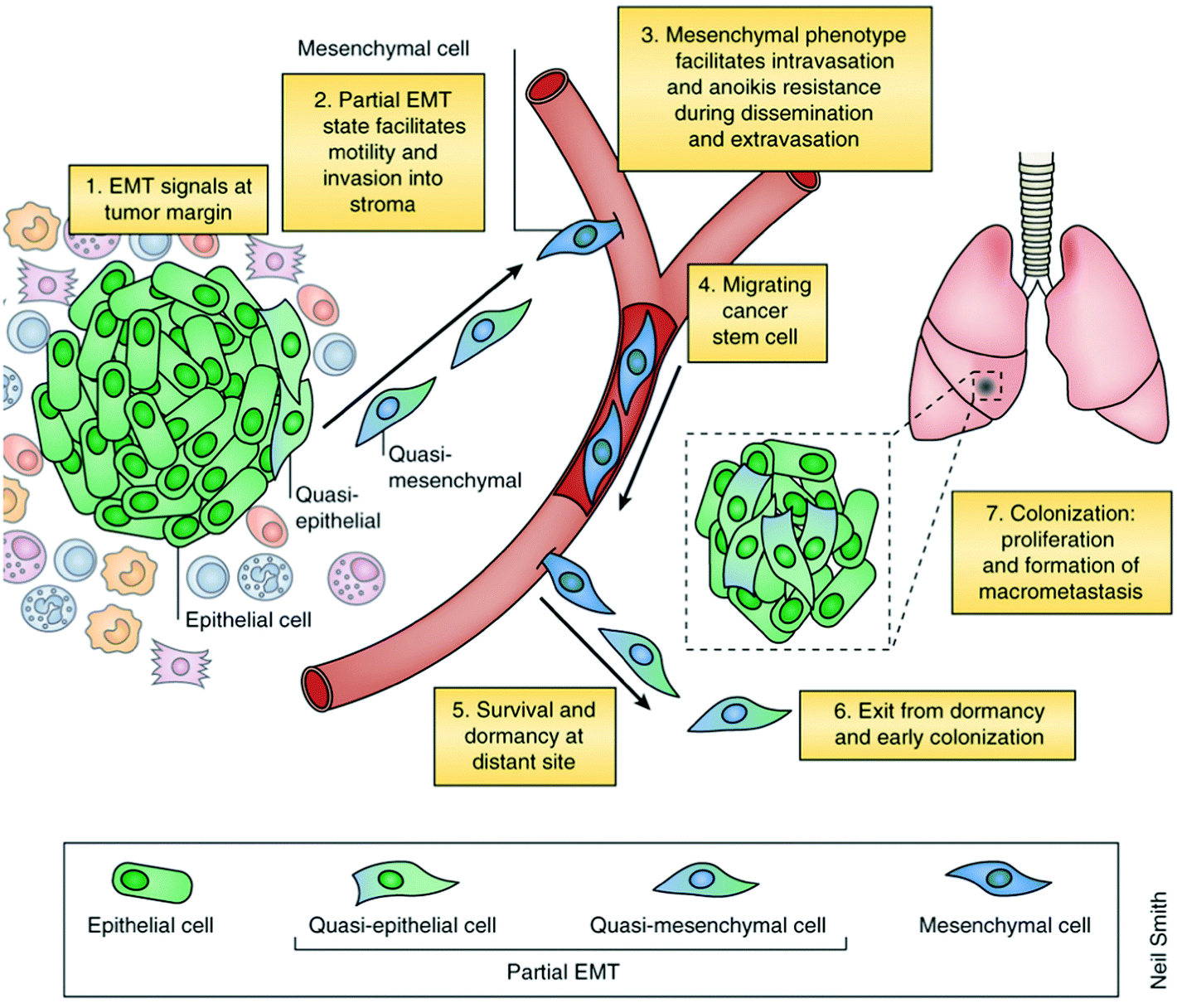 | ||
| Fig. 4 Schematic of EMT. During EMT, epithelial cells undergo cytoskeletal rearrangements, synthesize ECM, lose cell–cell junctions and show increased motility. The increased motility enables them to migrate to other organs through the blood vessels where they proliferate to initiate metastases. “Reprinted from Nature Medicine, 19, Tam and Weinberg, The epigenetics of epithelial-mesenchymal plasticity in cancer, 1438–1449, Copyright (2013), with permission from NPG. | ||
Another type of cancer cell that supposedly emerges during EMT is a tumorigenic circulating cancer stem cell (CSC). CSCs are often seen as the most dangerous because they mimic physiological cells, maintain multipotency, sustained self-renewal abilities and capabilities to initiate tumors.61 Researchers offer two hypotheses on the origin of CSCs. The first hypothesis proposes that during EMT cancerous somatic stem cells move from the primary tumor into the blood and become mesenchymal CSCs. The second hypothesis claims that circulating CSCs may originate from fully differentiated cancer cells and then adopt migratory properties during EMT as new pathways are developed. A number of studies have indicated that CSCs develop from differentiated progenitor cells or somatic stem cells. Sun and Qui (2013) claimed that only this particular subpopulation is responsible for relapse or metastasis in cancer patients.62 If it is the case that CSCs are the major cause of metastasis, present and future CTC research should focus on recognizing this specific circulating-CSC population.
There is a notable difference between normal and pathological EMT. EMT is a process to disperse cells in embryos and cause mesenchymal cells in injured tissues to induce invasive/metastatic behavior of epithelial cancers.59 All the steps involved in EMT during the embryological process are not crucial for establishing invasive phenotypes, and partial epithelial mesenchymal transition is sufficient to initiate tumor cells.63 Researchers must improve upon the theory of EMT to fully understand how pathological EMT differs from normal EMT and the essential signals necessary for transitioning between the stages of EMT.
4.3 Molecular heterogeneity
Research on CTCs is in the process of transitioning from capture and enumeration to molecular characterization of the cell's surface and contents.64 Significant variability has been found from transcriptional profiling and surface marker analysis, leading to an understanding that CTCs are highly heterogeneous.65 The heterogeneity of cells in primary breast cancer is visible in distinct mutations within the same slice of tumor tissue, but this principle was only recently shown in CTCs.65 In 2012, it was shown that CTC variability is highly consistent with tumor tissue variability, meaning that phenotyping of CTCs will bring to light many of the characteristics present in the tumor itself.65Recently, Ni et al. developed a method for whole genome amplification from single lung cancer CTCs to capture information for individual therapy and drug resistance.23 After capturing CTCs, a method of multiple annealing and looping was used to determine variations and deletions in the CTC exomes. It was found that individual patients had reproducible patterns of copy number variation, and that these variations differed between metastatic and non-metastatic tumors. Additionally, single nucleotide variations on a specific genome can strongly indicate metastasis. This variability shows the additional layer of heterogeneity due to specific genomic mutations that can be found in CTCs.23
The two major classes of single cell molecular profiling include mutational profiling studies and gene expression profiling studies.39 Both of these can elucidate the regulatory status of the tumor cells because they will shed light on the mutations or the over- and under-regulation of specific tumor-related genes. Among the molecular techniques used for mutational profiling are Sanger sequencing21,66 and comparative genomic hybridization (CGH).20,67 These studies show a high level of copy number variation and heterogeneity in expression-related mutations in a variety of cancers including breast, prostate, and colorectal cancer. For the expression-related studies, RNA analysis is typically performed and can be done through qRT-PCR microarray or RNA sequencing panels.65,68 These experiments can show profiles distinct from white blood cells, but also unique to specific cancer types. These studies also show a wide range of regulation of tumor and growth factor related genes.
These types of molecular analyses performed on populations of CTCs demonstrate the heterogeneity of CTCs. An understanding of the molecular heterogeneity of CTCs is particularly useful because it will inform development of targeted therapeutics for personalized medicine. Thus, the capture and analysis of single CTCs on chip is hugely valuable to the determination of prognosis and therapy from liquid biopsy. As it stands, there are very few technologies that employ novel, highly efficient capture technologies with single-cell level analysis. Some examples of hybrid devices to be developed include a monitoring device that also measures drug response at the single cell level or a diagnostic device that measures surface marker expression.
4.4 Biomarkers
Biomarkers are commonly used in research, both clinical and discovery-based; therefore, many disease-state specific biomarkers have been defined and characterized.69 Biomarkers can include clinical signs such as pulse or blood pressure, or biochemical signs such as surface markers and metabolites.69 A working definition according to the World Health Organization and the UN is “any substance, structure, or process that can be measured in the body or its products and influence or predict the incidence of outcome or disease”.69In the case of liquid biopsy of CTCs, there are two main classes of biomarkers that this review covers in detail. The first is surface protein biomarkers, mostly antigens that can be exploited for the capture and detection of CTCs. There are a number of heterogeneous sub-populations of CTCs, and the surface antigens of CTCs vary widely with function and state throughout EMT.70 The epithelial, mesenchymal and stemness markers are the most recognized families of antigens found on the circulating tumor cells and will be discussed in more detail in this section.70
There are also some studies that infer the presence of CTCs through expression-level analysis by capture of DNA or mRNA.39 These studies either analyze specific genes (such as cytokeratin-19 and cytokeratin-20)71 or detect epigenetic patterns such as methylation.72 Such analysis determines the specific mechanisms of aberrant behavior and similar genetic analysis provides clues as to the best method of treatment (Table 3).
| Biomarkers for CTC capture, detection, and analysis | |||
|---|---|---|---|
| Description | Application | Ref. | |
| Epithelial | |||
| EpCAM | Surface glycoprotein, downregulated in EMT | Capture, detection, enumeration | 70, 73 and 74 |
| E-cadherin | Component of adherens junctions, involved in cell architecture | Capture, detection, enumeration | 75 and 76 |
| Cytokeratins | Cytoskeletal filaments, downregulated in EMT | Capture, detection, enumeration, analysis | 71, 77 and 78 |
| Mesenchymal | |||
| N-cadherin | Adherens junctions, upregulated in EMT | Capture, detection, enumeration | 79–81 |
| Vimentin | Mesenchymal phenotype, enhances mobility | Capture, detection, enumeration | 80 |
| Plastin-3 | Induces EMT, activates mesenchymal genes | Capture, detection, enumeration | 82 and 83 |
| Stemness | |||
| ALDH-1 | Correlates to stemness characteristics and patient outcome | Capture, detection, enumeration | 84 |
| CD44 | Cell surface glycoprotein, cell migration and metastasis | Capture, detection, enumeration | 89 |
| Genes | |||
| KRAS | Commonly mutated in many cancers, especially colorectal | Analysis | 39 |
| VIM | EMT associated gene, often used for qPCR | Analysis | 65 |
| BRMS1 | Breast cancer regulator, epigenetic analysis | Analysis | 72 |
Epithelial CTC markers. Post CTC enrichment, if evidence of two or more of the markers – EpCAM, E-cadherin, cytokeratins – is found at single cell level, a particular CTC can be marked as a differentiated epithelial cancer cell; however, it should be confirmed that stem and mesenchymal markers are not observed.
Epithelial cellular adhesion molecule. Epithelial cellular adhesion molecule (EpCAM) is a cell surface glycoprotein found in epithelial cells. Its expression in epithelial cancer cells is higher than in normal epithelial cells, so it differentiates cancer cells from healthy epithelial cells.73 The development of EpCAM as a biomarker was mainly carried out by CellSearch system, and was used for the enumeration and detection of the CTCs (Veridex, Warren, New Jersey, USA).74 However, the expression of EpCAM depends on the stage of EMT, which when regulated downwards, is expressed in combination with N-cadherin and Vimentin and can eventually disappear from cell surfaces.70 This means that EpCAM alone as a surface biomarker will not capture all CTCs at various stages of EMT.
E-cadherin. As a component of adherens junctions (which maintain cell–cell interactions), E-cadherin is a hallmark of epithelial cells and functions closely with the actin cytoskeleton to provide resistance against cell detachment. Like some other proteins, E-cadherin is also involved in supporting the epithelial tissue architecture and its regulation plays an important role in the progression of epithelial mesenchymal transition: the expression decreases as the cell approaches mesenchymal state.75 E-cadherin may be inactivated or down-regulated by initiation of processes such as hypermethylation, histone deacetylation, and transcriptional repression once the cancer cells manage to get out of the primary tumor.76
Cytokeratins. Cytokeratins (CKs) are the largest class of intermediate filaments of the cytoskeleton and are the biomarkers of regular epithelial differentiation. CKs are used as a diagnostic tool for the detection of circulating carcinoma cells. To enable the detection, antibodies have to recognize most epitopes displayed by cancer cells. Epithelial cancer cells, besides expressing CKs, also express K8, K18 and K19. These specific markers are expressed by cells with epithelial phenotype; however, their expression is down-regulated during epithelial mesenchymal transition.77 Enabling the use of anti-CK antibodies in combination with others helps in tracking cells that lose the partial or whole epithelial phenotype.78
Mesenchymal-like CTC markers. Varying degrees of EMT take place in individual cells, making it hard to define a mesenchymal CTC subpopulation in pure state as they evolve from epithelial status to a mesenchymal state. The identification of mesenchymal biomarkers only shows the active status of an EMT process. When EMT is over and before the transition to the next stage, mesenchymal epithelial transition (MET), these markers can aid in distinguishing a pure population of cells with mixed phenotype (both epithelial and mesenchymal features). A cell can be defined to have a mesenchymal character when two mesenchymal markers are connected to epithelial or stem markers.70
N-cadherin. E-cadherin and N-cadherin subfamilies constitute the proteins of adherens junctions and N-cadherin is expressed in numerous cells, including the mesenchymal cells, making it a potential biomarker of EMT. While EMT is occurring, N-cadherin is active while E-cadherin is inactive. Upregulation of N-cadherin is considered a hallmark of mesenchymal characterized CTCs.79
Vimentin. Vimentin is a protein expressed in mesenchymal cells that induces a mesenchymal phenotype and enhances mobility.80 Vimentin has also been found in normal blood cells, however, Lustberg et al., who conducted this research, never found a cell that was positive for both CD45 and Vimentin. This finding indicates that Vimentin cannot mark hematopoietic cells. Vimentin expression alone cannot differentiate a mesenchymal phenotype from an epithelial phenotype, and evidence supporting the efficiency of this biomarker in identifying mesenchymal cells needs to be demonstrated.81
Plastin-3. It has been shown that Plastin-3 (PLS3) has the ability to induce EMT in colorectal cancer cells.82 While this study focuses on the colorectal cancer cells, one can designate this factor as a biomarker of mesenchymal CTCs because it activates a high level of mesenchymal genes.67 Plastin-3 can also be considered a putative clue to differentiate CTCs with both epithelial and mesenchymal traits.83
Stemness-like CTCs eiomarkers. This strata of CTCs, CTCs with stemness phenotypes and CSCs, are thought to be responsible for relapse or metastasis in cancer patients.30 Differentiating and isolating CSCs among CTCs will allow a fuller understanding of prognosis and treatment plans. The following markers may be used to identify CSCs in blood.
Aldehyde dehydrogenase-1 (ALDH1). ALDH1 is expressed in 1–2% of breast epithelial cells and was first described in a study by Ginestier et al.84 The group was able to demonstrate ALDH1 as a biomarker for increased risk of breast cancer using immunostaining. Although a number of cells have this characteristic, only a few are able to grow into differentiated solid tumor in NOD/SCID mice.84 Barriere et al. conducted a clinical trial with the aim of detecting CTCs in early breast cancer diagnosis and found that of the 130 patients in question, 49 were CTC positive and 17 of those were ALDH1 positive. These results show the presence of stemness CTCs in blood samples of cancer patients without metastasis.85 This level of biological understanding can inform specific personalized treatments.
CD44. CD44, a cell surface glycoprotein, has been implicated in cell migration and metastasis.86 Breast CSCs have the ability to intravasate into the blood; their characteristics are defined by the expression of CD44+/CD24low/−/Lin – cell surface markers. The involvement of CSCs in tumorigenesis was shown by Al-Hajj et al. who demonstrated that the transplantation of as few as a hundred CD44 CD24-breast carcinoma cells was enough to initiate differentiated solid tumor growth in NOD/SCID mice. On the other hand, even 105 CD44−/CD24+ tumor cells did not initiate the growth of tumors in animal models. It is worth remarking that cells populations with CD44+/CD24low/− may not necessarily coincide with cell populations consisting of ALDH1 positive cells.87
This evidence suggests that these proteins may be helpful in differentiating between epithelial and mesenchymal characteristics of stem cells.88 Alternative splicing of CD44 can result in a number of isoforms. A few of these isoforms can act as highly specific markers of stemness and need to be extensively studied. Similarly, CD44 is encoded by 20 exons, and 10 of them can be regulated by alternative splicing; these are named variants of v exons. The isoforms obtained are expressed exclusively in proliferating cells, cancer cells in particular. CSCs express CD44v6 in colorectal cancer and this expression gives the CSCs metastatic ability. When it comes to the detection of CSCs, CD44v6 appears to be one of the best markers.89
Another group used qPCR on whole genome amplified DNA to determine specific mutations in the KRAS gene in patients with colorectal cancer.90 They studied a number of clinically relevant mutations, including c.34G > T, c.34G > A, c.34G > C, c.35G > T, c.35G > A, c.35G > C, and c.38G > A in genomic DNA samples in order to quantify the heterogeneity of mutations among the CTC genome.90 Different primers were used for each of these point mutations in order to ensure single nucleotide specificity.
Another method of analysis determines epigenetic and regulatory changes to the genome by measuring sequence-specific methylation.72 They specifically studied the methylation of the breast cancer metastasis suppressor-1 (BRMS1) promoter. They found that methylation was not detected in noncancerous tissue, but in CTCs and breast cancer tissue more than one third of samples had methylated promoters.72 This study shows another biomarker, the BRMS1 promoter, which can be used for analysis of CTC heterogeneity and expression.
Methods of CTC analysis attempting to understand gene expression and regulation use a number of molecular biology techniques including amplification and epigenetic monitoring. Some of the relevant genes include VIM, CK, the KRAS gene, and, specific to breast cancer, the BRMS1 promoter. Any of these genes can be targeted and studied after CTC separation and detection in order to monitor regulatory changes in the tumor environment.
5. Macroscale technologies and challenges
As a precursor to liquid biopsy on chip, macroscale liquid biopsies such as ISET,91,92 Magnetic Activated Cell Sorting systems (MACs),93 CellSearch,93 MagSweeper,93 and GILUPI Nanomedizin,94 strive to make the detriments of surgical biopsies and minimally invasive biopsies obsolete. These devices strength is the capture of CTCs in the pursuit of a snapshot of the global genomic fluctuations of a tumor. Some devices, such as ISET, are better adapted for downstream analysis of captured CTCs. With the exception of GILUPI Nanomedizin, these devices require a simple blood draw, afflicting the patient less than minimally invasive biopsies. The scrutiny of liquid biopsy on chip against these technologies at each iteration of the design process enables microchip developers to hone in on an elegant platform.Sized-based filtration is a rudimentary macroscale method of CTC separation. One of the most commonly used techniques includes a physical filtration based ISET array developed by Vona et al.91 Using as little as 1 mL of blood, ISET can detect a single micropipetted CTC; additionally, ISET enumerates, characterizes immunomorphologically, and lends itself well to molecular analysis, such as Fluorescence in situ Hybridization (FISH) and real-time PCR (RT-PCR) of CTCs.91 ISET essentially filters CTCs that are larger than leukocytes; however, leukocytes range in sizes comparable to CTCs, which commonly contaminates and impedes the effectiveness of size-based enrichment (Fig. 5a). Furthermore, smaller CTCs are not captured in the filtration system.
 | ||
| Fig. 5 (a) Physical separation based ISET. CTCs are captured by a filter that allows red blood cells and leukocytes to pass through. The first two panels show individual isolated CTCs, the third shows a cluster. Reprinted from British Journal of Cancer, 105, Farace, A direct comparison of CellSearch and ISET for circulating tumour-cell detection in patients with metastatic carcinomas, 847–853, Copyright (2011), with permission from NPG. (b) Schematic of magnetic activated cell sorting systems (MACS). CTCS are captured by immunomagnetic particles that cling to the side of a tube in the presence of a magnetic field, allowing red blood cells and leukocytes to elute away. Reprinted from Lab on a Chip, 14(3), Chen, Multiscale immunomagnetic enrichment of circulating tumor cells: from tubes to microchips, Copyright (2013), with permission from RSC. (c) CellSearch. An FDA two part separation and enumeration platform that captures CTCs with epithelial phenotypes. Reprinted from Lab on a Chip, 14(3), Chen, Multiscale immunomagnetic enrichment of circulating tumor cells: from tubes to microchips, Copyright (2013), with permission from RSC. (d) MagSweeper. A magnetic rod functionalized with EpCAM antibodies swirls above the bottom of a dish containing whole blood, capturing CTCs. Reprinted from Lab on a Chip, 14(3), Chen, Multiscale immunomagnetic enrichment of circulating tumor cells: from tubes to microchips, Copyright (2013), with permission from RSC. (e) GILUPI Nanomedizin. An antibody conjugated medical wire is inserted into the vein for 30 min, capturing CTCs circulating in the blood of the patient. Reprinted with permission from authors. | ||
More predominant and mainstream approaches build upon Magnet Activated Cell Sorting Systems, MACS (Fig. 5b). Typically, MACS systems magnetically force CTCs labeled with magnetic particles to the side of conical tubes in which the samples are stored. Unlabeled cells are flushed, and the magnetically labeled CTCs are removed from the tube after withdrawing the external magnetic field.93 Manual MACS systems, such as those developed by Miltenyi Biotech, offer low throughputs, prolonged separation times, and weak external fields generated by bulky magnets for the enrichment of rare cells.93
The only FDA approved and commercial system for the separation of breast, colorectal, and lung CTCs is CellSearch (Fig. 5c). CellSearch is a two-part system for the capture and enumeration of EpCAM and Cytokeratin positive CTCs labeled with ferrofluids. CellSearch is limited to capturing CTCs with epithelial phenotypes, and possibly neglects CTCs with mesenchymal and stemness phenotypes. Furthermore, after fluorescence imaging for enumeration, captured CTCs are re-suspended in a solution. The troublesome recovery of these CTCs for further analyses hinders the effectiveness of CellSearch.93
Another device-based macroscopic system for the capture of CTCs is MagSweeper (Fig. 5d). A magnetic rod whirls above the bottom of a well containing a sample of CTCs labeled with magnetic particles. Weakly bound cells are flushed from the rod, and the strongly bound CTCs remain. This system can accommodate multiplex assays and screen multiple samples simultaneously. However, the magnetic rods are covered in a sheath for easy separation of the CTCs from the magnetic rod that consequently weakens the magnetic force and thwarts some of its capture efficiency.93
GILUPI Nanomedizin (Germany) developed a minimally invasive liquid biopsy, in which a biocompatible structured medical Seldinger guidewire (FSMW) functionalized with anti-EpCAM antibodies is inserted into the cubital vein for half an hour (Fig. 5e). After removal, the wire is rinsed, and the attached CTCs counted by immunocytochemically staining with EpCAM and/or cytokeratins.94 This device can sample large volumes of blood with minimal distress to the patient. Unfortunately, the hydrodynamic forces acting on the target CTCs cannot be controlled, which could interfere the devices capturing capabilities.
The popularity and success of immunomagnetic separation technologies is a sound takeaway from this brief review of macroscale liquid biopsy technologies. However, there is compelling evidence to suggest that it is advantageous to incorporate multiple separation methods into a simple platform. Additionally, it is imperative to juxtapose future microscale technologies in the pipeline towards clinical adoption; not only with industrial gold standards, but also with macroscale equivalents to fully assure that what is being developed is of clinical importance and practicality.
6. Microscale technologies
There are many advantages of using microfluidics in cancer cell enrichment and isolation. Microfluidics can minimize the total steps involved in cell pre-treatment and reduce lost or damaged cells.95 Additionally, microscale technologies consume small sample and reagent volumes, are low cost, and support higher throughput, sensitivity, resolution, portability, and automation.96 At the microscale, physical forces like laminar flow, diffusion, fluidic resistance, and surface tension become dominant.97 Shifting to a microscale also leads to the increase in relative amount of surface area exposure that a volume of fluid experiences. When the surface area contact increases, it leads to quicker diffusion and elevated heat conduction. Laminar flow enables free-flowing adjacent perfusion of various reagents that are useful in the localized treatment of cells, and when used in combination with controlled diffusion, it can aid the construction of chemical gradients with subcellular resolution.98The main strategies at the microscale include positive enrichment, negative enrichment, and label-free techniques. Positive enrichment typically refers to a process that selects for the CTCs while leaving the other particles behind.99 This typically has a very high accuracy, and one of the most common methods involves antibody tagging surface antigens on the CTC. Another strategy is negative enrichment, which involves targeting and removing other types of cells, in this case white and red blood cells.95 This generally leads to lower purity but does not have the challenge of removal of binding probes from the surface of the CTC, and is often able to bypass the challenge of CTC heterogeneity.99 A label-free technique is one that avoids biochemically tagging the desired molecule.100 This means that rather than using immune affinity for capture or sample cleaning, another method that does not involve labeling cells is used such as size-based, mechanical property based, acoustic, or optical. The following are methods employed in the field of microfluidics to capture and separate CTCs (Fig. 6).
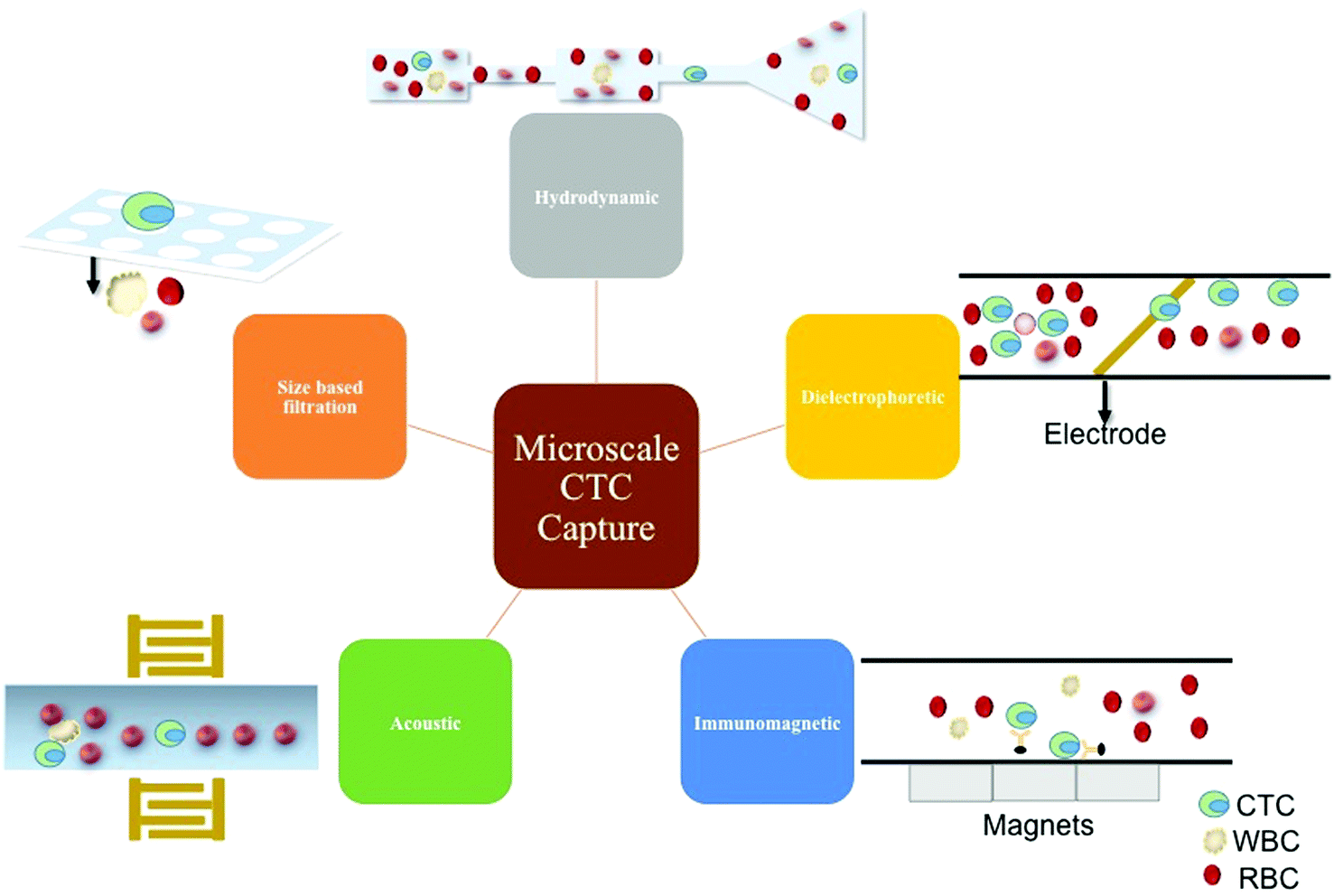 | ||
| Fig. 6 Microscale technologies for CTC capture. Overview of microscale CTC capture mechanisms. | ||
6.1 Hydrodynamic
The hydrodynamic method of CTC separation, deterministic lateral displacement (DLD), operates by causing the small particles to follow the laminar flow streams whereas large particles are continuously forced to change their laminar flow streams causing them to be displaced laterally.101 This method is well suited for label-free and negative enrichment of CTCs without significantly harming them. However, since leukocytes and CTCs are comparable sizes, this method suffers from significant leukocyte contamination and low throughput.6.2 Dielectrophoretic
When subjected to an inhomogeneous electric field, a cell becomes polarized, but the electrostatic forces on the two ends of the dipole are not equal inducing dielectrophoretic (DEP) based cell sorting.102–104 This method is label-free, has moderate to high capture efficiencies, and high throughput, but more needs to be understood about how a CTC charges change throughout the metastatic journey.6.3 Acoustic
An external transducer is used to generate a standing ultrasonic wave across a microchannel. When the cells experience this force, they either move towards the pressure node or the anti-node.105 This method is label-free, offers high throughput, and has demonstrated detection of pigmented cancer but still suffers from leukocyte contamination and limited number of detectable cancers.6.4 Size-based filtration
Size based filtration involves fabricating pores of appropriate size so that the RBCs can pass through whereas the WBCs and CTCs can be collected from the filter.106,107 This method has the potential to be used for label-free, positive, or negative enrichment of CTCs with high capture efficiencies while minimally damaging them. However, leukocyte contamination and moderate throughput are hindrances. Sized based filtration tends to allow more deformable CTCs with a greater metastatic potential to pass through the filter.6.5 Immunomagnetic
Scientists widely study immunomagnetic separation methods, and they have developed numerous capture platforms. Simply, this method involves conjugating an antibody to a magnetic particle, incubating the functionalized magnetic particle with a solution containing cells to capture them, and applying a magnetic field to separate them from the bulk solution.67,108 Positive immunomagnetic capture permits integration of cancer biology into the capture mechanism, and generally demonstrates high throughput, high capture efficiency, and low leukocyte contamination. Negative immunomagnetic enrichment has been employed to improve the diversity and heterogeneity of the enriched CTC population.Each of these methods is widely used at the microscale to capture CTCs from either an enriched or unprocessed sample. When considering these techniques for liquid biopsy, it is important to consider ease of detection, confirmation, and further analysis. Because immunomagnetic capture involves immunoaffinity, the success of the technology is highly dependent on the choice of biomarker. Another important aspect given our understanding of the biological mechanisms governing cancer is the ability to perform single cell tests, which will be discussed later in more detail. Thus, this review will focus on some immunomagnetic and microfluidic technologies that enable single cell molecular analysis (Table 4).
| Evaluation of CTC capture technologies | ||||||
|---|---|---|---|---|---|---|
| Technology | Biomarker | Capture efficiency | Throughput | Normal cell contamination | Cell viability | Ref. |
| Immunomagnetic | ||||||
| Permanent magnet | EpCAM | Moderate | Moderate | Low | High | 110 |
| Magnetic sifter | EpCAM | High | High | Moderate | High | 111 |
| CTC-iChip | Biomarker for WBCs | High | High | Moderate to high | Moderate | 109 |
| Array of micromagnets | EpCAM | High | High | Moderate | High | 15, 112 and 113 |
| Capture & release | ||||||
| Lasers & microfilms | EpCAM | High | Moderate | Low | High | 113 |
| Thermoresponsive | EpCAM | High | Moderate | — | High | 115 |
| pH & glucose | — | Moderate | Moderate | — | High | 116 |
| Aptamers | Protein tyrosine kinase 7 (PTK7) | Moderate | Moderate | Low | Moderate | 117 |
| Single cell capture | ||||||
| Microwells | — | Moderate | High | — | High | 118 |
| Programmable valves & channels | — | — | High | — | High | 119 |
| Capture & culture | — | — | High | — | High | 120 |
7. Capture technologies
According to the presented framework, optimal chips for the capture of CTCs should feature: a high capture rate, high throughput, low contamination rate, and high cell viability. The following immunomagnetic, capture and release, and single cell capture technologies are critically reviewed in order to illustrate the current optimization of these key parameters. A focus will be given to immunomagnetic technologies because of the numerous advantages for rare CTC capture, and the recent trend towards immunomagnetic microchip development.93 Some of the advantages of immunomagnetic chips include their sensitivity and specificity due to their use of antigen–antibody capture, and their high throughput and tunability due to their power over a larger area than some other capture methods which require direct input.93 For these reasons, immunomagnetic chips do well with our framework based on capture rate, throughput, contamination, and viability. For completeness, we also review other methods as discussed, including capture and release and studies at the single cell level.7.1 Immunomagnetic
Immunomagnetic microchips provide a versatile platform for CTC enrichment and isolation. In the future, a combination of both negative and positive enrichment, or highly integrated devices that take advantage of other techniques should be developed to increase capture efficiency and purity and better distribution of captured CTCs. Combining more than one technique to increase the capture efficiency of CTCs has recently been gaining considerable popularity.109 Apart from the design, other important feature to consider for next generation techniques is the diversity of surface biomarkers and how to exploit them to achieve higher sensitivity and specificity. This section will detail a number of immunomagnetic microscale technologies.Kang et al., designed a PDMS based device with one main channel and two dead-end side chambers for capture and culture of CTCs (Fig. 7a).110 The breast cancer cells were labeled with 2.8 μm magnetic beads conjugated to EpCAM. The CTCs were collected in the side chambers that had an NdFeB N52 permanent magnet beneath it. The microchannel at the inlet was oriented at 60 degrees to main channel to ensure cells would be closer to the magnetic field. The flow rate was optimized to be 1.2 mL h−1 based on the residence time of the bead-bound cells and time required to attract cells into the dead-end chambers. A capture efficiency of 90% was reported for breast cancer cells spiked in 1 mL of blood, and the captured cells were cultured and harvested for further molecular analysis.110 This device has great potential to impact the field of targeted therapies because it cultures CTCs from the patient, and showcases a moderate throughput and capture efficiency geared towards early detection, and monitoring. However, the biomarker of choice, EpCAM, is known to not capture the full heterogeneous population of CTCs.
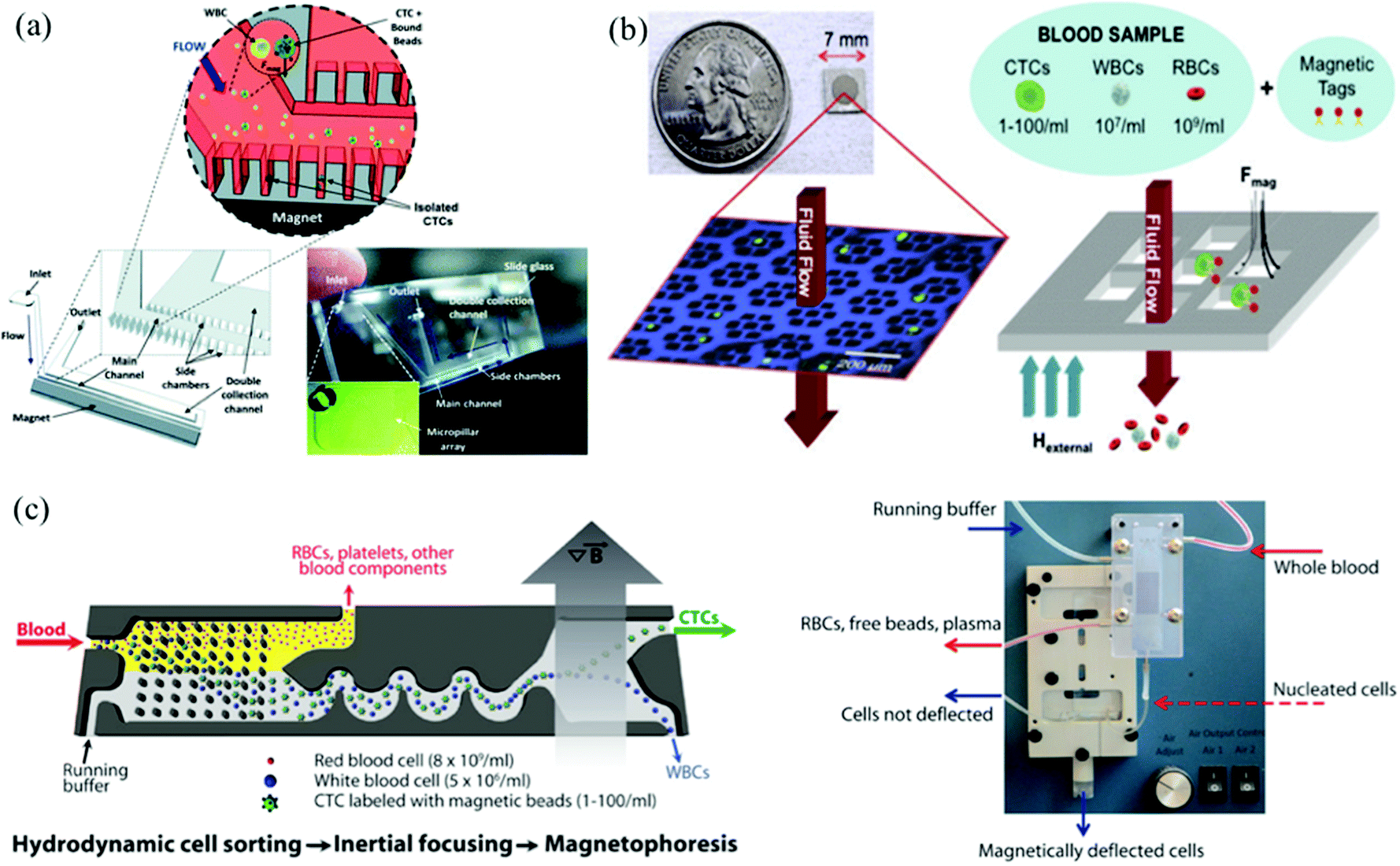 | ||
| Fig. 7 (a) Microchannels with dead-end side chambers. Side chambers were designed for immunomagnetic capture of CTCs Reprinted from Lab on a Chip, 12, Kang et al., A combined micromagnetic-microfluidic device for rapid capture and culture of rare circulating tumor cells, 2175–2181, Copyright (2012), with permission from RSC. (b) Micro sifter Biochip. CTCs were conjugated with magnetic tags and under the influence of magnetic forces, CTCs stay on the substrate while all the other blood cells are flushed away through the holes. Reprinted from Lab on a Chip, 14, Earhart et al., Isolation and mutational analysis of circulating tumor cells from lung cancer patients with magnetic sifters and biochips, 78–88, Copyright (2014), with permission from RSC. (c) CTC-iChip. A positive isolation method is shown here: CTCs are labeled with magnetic beads, and by hydrodynamic cell sorting, only nucleated cells like WBCs and CTCs are retained in the sample for the next stage of isolation. Then inertial focusing is applied to order the rest cells so that a better separation of CTCs and WBCs into different outlets is obtained. Reprinted from Science Translational Medicine, 5(179), Ozkumur et al., Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells, 179ra47, Copyright (2013), with permission from AAAS. | ||
Another platform that uses positive selection of CTCs was developed by Earhart et al.111 The magnetic sifter biochip (Fig. 7b) had a dense array of magnetic pores with high magnetic field gradients existing along the pore edges. When CTCs bound to magnetic beads labeled with anti-EpCAM antibodies are injected through the pores, they are captured at the pore edges under the influence of the external magnetic field whereas the unlabeled cells pass through the pores unhindered. The capture efficiencies for high EpCAM expressing CTCs was ∼90% compared to the low expressing CTCs which ranged from 17.7% to 48%. In addition to a high throughput and capture efficiency of only high EpCAM expressing CTCs, the simple and quick release of the captured CTCs from the sifter allows for enumeration and advanced genomic analysis appropriate for early detection, monitoring, prognosis, diagnosis, and targeted therapy.
The CTC–iChip combines immunomagnetic capture with DLD and inertial focusing (Fig. 7c).109 In the first stage, DLD was used to achieve hydrodynamic size based sorting by removing non-nucleated cells such as RBCs and platelets from the whole blood. The next stages used inertial microfluidics to focus the CTCs and WBCs for magnetophoretic sorting. Once the cells enter the magnetic field, the magnetically labeled CTCs are deflected and separated from the WBCs.109 The apt combination of DLD and immunomagnetic capture, in addition to negative enrichment of CTCs, grasps the most diverse population of CTCs. The platform primarily enumerates CTCs through fluorescence-based detection methods, which can damage the cells, preventing further genomic downstream analysis. One disadvantage is that this device has been demonstrated at spiking concentrations much higher than typical CTC concentrations, at about 200–1000 CTCs per mL but has also shown lower detection concentrations. This device does not satisfactorily meet the requirements for discovering targeted therapies, but it has demonstrated its early detection, prognosis, diagnosis, and monitoring capabilities.
Similarly, Zhang et al., developed immunomagnetic microchips for capture and release of CTCs at single cell level.15,112,113Fig. 8a and b show the original design of the microchip. The first generation device was PDMS based hexagonal microchannel with permanent magnets placed below the PDMS device. Magnetic nanoparticles conjugated to anti-EpCAM were added to the whole blood sample spiked with cancer cells and then pumped into the microchip. The CTCs were captured on the glass slides and the waste blood cells were eluted. This simple design enabled a higher flow rate of 10 mL h−1 compared with the commercially available CellSearch system and demonstrated considerably high capture rates of 90% and 86% for COLO205 and SKBR3 cells, respectively, with a very low tumor to blood cell ratios (about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, including red blood cells).15 The next generation microchip designs from Chen et al., had an array of ultra-thin micromagnets of sub-micron height with a spacing of 100 μm fabricated on the glass slides (Fig. 8c), to increase the magnetic attraction force by creating areas where the magnetic field intensity is locally increased. The micromagnet array both uniformly distributes the intensity throughout the glass slide and efficiently captures CTCs labeled with magnetic nanoparticles.112 In comparison to the CTC-iChip, this one has a higher throughput, demonstrated low normal cell contamination, and analogous capture efficiency. The positive selection of EpCAM phenotypic CTCs captures a uniform population of CTCs, but has the same disadvantages that we mentioned earlier due to the heterogeneity of the CTC population. This device allows for the quick enumeration of CTCs, ideal for early detection, prognosis, and basic diagnosis, but it is not suitable for an advanced analysis for target therapies.
:10, including red blood cells).15 The next generation microchip designs from Chen et al., had an array of ultra-thin micromagnets of sub-micron height with a spacing of 100 μm fabricated on the glass slides (Fig. 8c), to increase the magnetic attraction force by creating areas where the magnetic field intensity is locally increased. The micromagnet array both uniformly distributes the intensity throughout the glass slide and efficiently captures CTCs labeled with magnetic nanoparticles.112 In comparison to the CTC-iChip, this one has a higher throughput, demonstrated low normal cell contamination, and analogous capture efficiency. The positive selection of EpCAM phenotypic CTCs captures a uniform population of CTCs, but has the same disadvantages that we mentioned earlier due to the heterogeneity of the CTC population. This device allows for the quick enumeration of CTCs, ideal for early detection, prognosis, and basic diagnosis, but it is not suitable for an advanced analysis for target therapies.
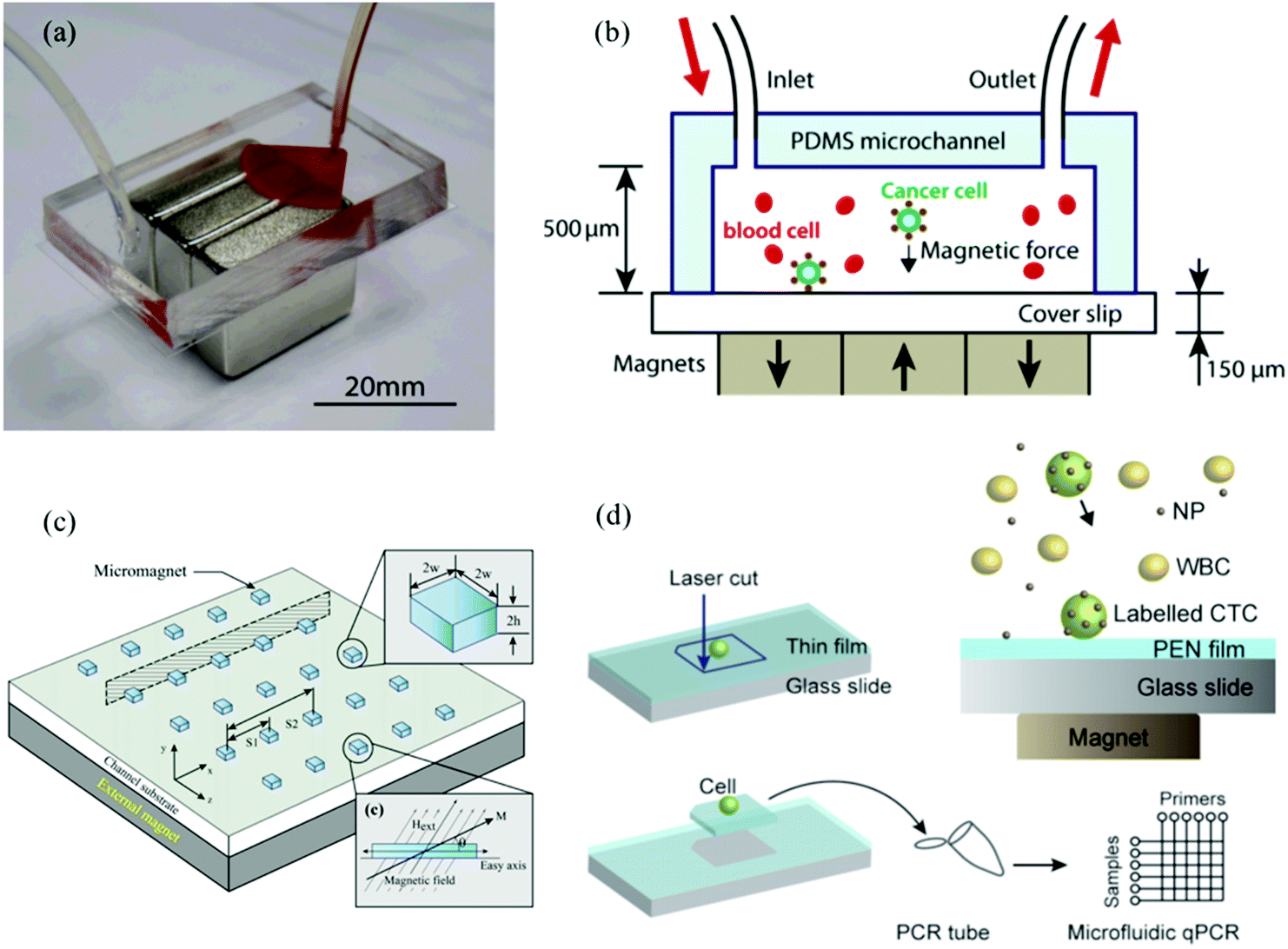 | ||
| Fig. 8 (a) and (b) An immunomagnetic microchip and its schematic. Whole blood sample with spiked cancer cells is flowed into the microchip and targeted cancer cells are captured under magnetic field because of antibody–antigen reaction with antibody conjugated micro magnetic beads. Reprinted from Lab on a Chip, 11, Hoshino et al. Microchip-based immunomagnetic detection of circulating tumor cells, 3449–3457, Copyright (2011), with permission from RSC. (c) Local micro magnets for better distribution of captured CTCs. Patterned thin-film micromagnets were fabricated on the substrate to increase local magnetic field gradients for better distribution of captured CTCs. Reprinted from Scientific Reports, 5, Chen et al. Microscale magnetic field modulation for enhanced capture and distribution of rare circulating tumor cells, 8745, Copyright (2015), with permission from NPG. (d) Single-cell-level capture and analysis of CTCs. PEN film was formed on top of glass slide so that after capture of CTCs, single CTC could be isolated and qPCR could be conducted. Reprinted from Journal of Circulating Biomarkers, Hoshino et al. An Immunofluorescence-assisted Microfluidic Single Cell Quantitative Reverse Transcription Polymerase Chain Reaction Analysis of Tumor Cells Separated from Blood, ISSN 1849-4544, Copyright (2015), under a Creative Commons License. | ||
All of the immunomagnetic techniques described above have all included a step that employs positive enrichment by selectively targeting markers on the surface of CTCs. Using negative enrichment alone provides a number of advantages because of its label-free nature. Karabacak et al. investigated a modification of the iChip to improve the removal of red blood cells for higher sensitivity negative enrichment of CTCs.114 This technology first separated nucleated cells and then used antibodies to deplete leukocytes and achieved a 97% yield of CTCs. This negative enrichment improvement is highly promising, but has not yet been demonstrated at a physiological concentration of CTCs.
7.2 Capture and release
As discussed previously, the capture and release of CTCs is optimal for single cell analysis. By allowing selective release, cells can be analyzed on an individual basis at favorable temporal and spatial separations. Lasers and microfilms, thermoresponsive polymers, pH and glucose sensitive polymers, and aptamers are methods of capture and release reviewed below.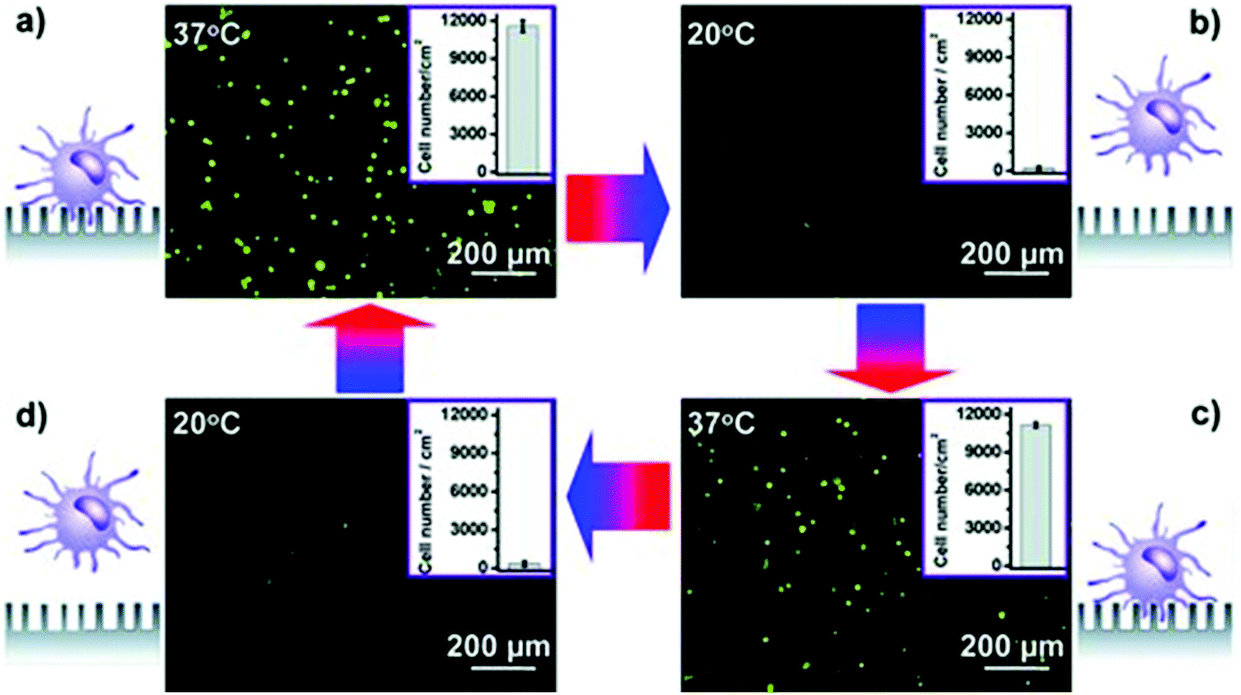 | ||
| Fig. 9 Thermoresponsive cell capture and release. Targeted cancer cells are reversibly captured and released using biotin-BSA and PNIPAAm-modified SINP through temperature changes between 37 °C and 20 °C. Reprinted from Advanced Materials, 25, Liu, Hydrophobic interaction-mediated capture and release of cancer cells on thermoresponsive nanostructured surfaces, 922–927, Copyright (2013), with permission from Wiley-VCH. | ||
7.3 Single cell and cluster capture
On the premise that a small population of cancer cells drives drug resistance, Lin et al. recently investigated a platform for microwell cell loading.118 The technology involves a dual-well design and has 77% single-cell loading. The goal of this research is to trap single cells through a small microwell and into a large microwell to allow for cell culture. The device took advantage of gravitational force and low flow rates to allow cells to settle into the capture wells before washing away uncaptured cells. The cells then drop from these capture wells into larger wells that cultures the cells over the course of several days. A number of flow rates were iterated through until a “cell sweeping” procedure was developed to maintain a high capture efficiency, and a flow rate of 3 μL min−1 was found to be best for allowing cells to move through the device while also settling within capture channels.118 While this sort of platform is less applicable to CTC separation, it is a feasible last step after CTCs have been separated to isolate individual cells for single-cell downstream analysis. It is also relatively low throughput, resulting in a very long timespan required for analysis.Over ten years ago Wheeler et al. developed microfluidic devices for single cell analysis for a number of applications, including testing cell viability and intracellular ion concentration.119 The technology builds upon the development of multilayer soft lithography with a series of valves at channel intersections (Fig. 10). These digitally controlled valves can close and open channels in a programmable method to separate out single cells from a solution. The device is designed to both separate an individual cell and provide it with small volumes of reagents, a function that relies on laminar flow. The use of this system to measure intracellular calcium ion concentration was investigated, and showed that this device can be used for a number of single cell assays.119 This has many applications to downstream analyses of individual CTCs after capture.
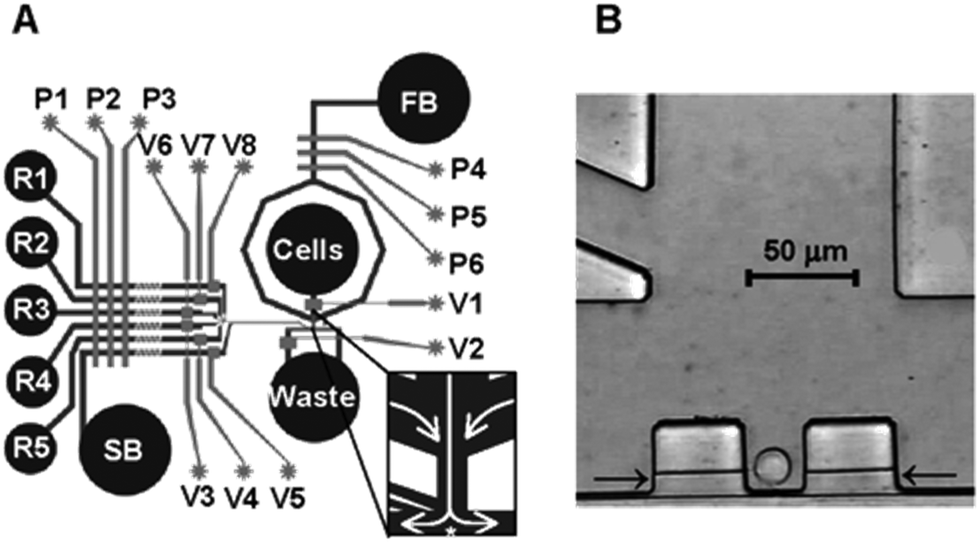 | ||
| Fig. 10 Single cell capture device. (A) A schematic of the multilayer microfluidic device with digitally controlled valves and control channels. V1–V8 control valves, and P1–P6 activate pumps. (B) A single cell captured in a junction. Reprinted from Analytical Chemistry, 75, Wheeler, Microfluidic Device for single-cell analysis, 3581–3586, Copyright (2003), with permission from ACS. | ||
One device that is able to separate out cells and successfully culture them for evaluation of therapeutic response has recently been developed by Khoo et al.120 This technology requires minimal reagents, is able to simulate in vivo conditions, and is simple and robust. Using label-free technology the device is able to capture CTC clusters and culture them directly from the blood in order to monitor the drug responses of patients.120 One particular weakness of this technology is that it may select for more aggressive and proliferative subtypes of CTCs within the patient because the selection is based on growth through culture. This, however, may also be an advantage because these are the more clinically relevant subtypes for understanding of patient prognosis. This technology shows a method of single or clustered CTC capture that allows for prognostic and therapeutic monitoring.
These reviewed devices are the vanguards of liquid biopsy on chip for the capture of CTCs; each one optimizing a different set of the key parameters listed in the framework for developing such capture devices. Partly due to the biology of cancer and to the infancy of this technology, no impeccable platform for CTC capture has emerged. More must be researched in order to capture the full heterogeneous population of CTCs to usher microscale technologies for early detection, monitoring, prognosis, diagnosis, and targeted treatment into mainstream clinical toolkits.
8. Detection technologies
Once the CTCs are captured from the sample, their presence must be detected. This involves confirmation of the cells location and verification that it is indeed a CTC. This can be done through fluorescence staining, either manual or automated, or through optical, photoacoustic, or electrochemical detection. While fluorescence tagging is very good for validation and enumeration, it is not label free and minimizes the opportunities for analysis afterwards. Other technologies for CTC detection have the advantage of allowing for an array of analysis possibilities.8.1 Fluorescence tagging
Most CTC isolation platforms focus on fluorescence-based detection of CTCs. Though these platforms are effective in CTCs enrichment and detection, isolation and downstream analysis of CTCs at single cell level is essential because the genetic and molecular analysis of the captured cells can provide more accurate information about tumor-CTC relationship, the disease state, and progression. Fluorescence staining methods, such as fluorescent microscopy, FACs, and image analysis are delicately affected by fluorescence intensity, which is affected by the biomarker chosen to tag the CTC with the fluorescent molecule. However, fluorescence-based detection methods of CTCs are optimal for early detection, monitoring, prognosis, and diagnosis.As a lower-throughput fluorescence-based method, Hoshino et al. described a fluorescent cell staining procedure used to detect, image, and enumerate captured CTCs using magnetic microchip immunoisolation.15 After cells were separated onto a glass coverslip, a cocktail of fluorescent dyes stained the captured cells and detected individual components within captured CTCs. These fluorescent dyes include DAPI, anti-cytokeratin, and anti-CD45; each targets individual components of the cells, and are positive and negative stains for CTCs.112 CTCs are positive for DAPI and cytokeratin, differentiating them from the white blood cells, which are positive for DAPI and CD45. Cells are visualized and imaged under a bright field microscope before observation of the fluorescence (Fig. 11).112 This technique displays captured CTCs and executes cell counting independent of trained scientists by automating thresholding and contrast analysis to detect and count cells.15
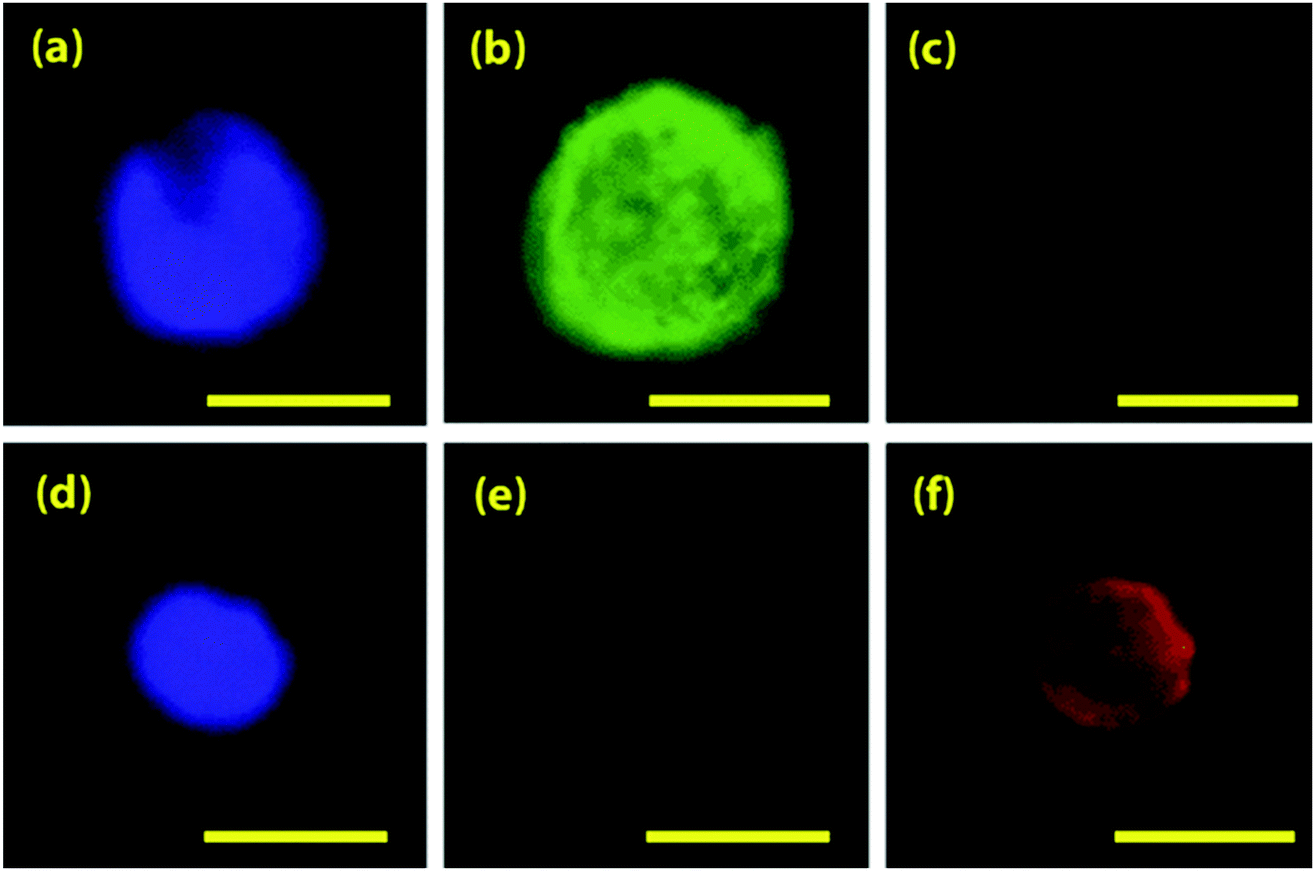 | ||
| Fig. 11 Immunofluorescent staining of a captured COLO205 cell and a leukocyte. COLO205 cancer cell stained for DAPI (a), anti-CK (b), and anti-CD45 (c). Leukocyte stained for the same 3 markers (d–f). Reprinted from Lab on a Chip, 11, Hoshino, Microchip-based immunomagnetic detection of circulating tumor cells, 3449–3457, Copyright (2011), with permission from RSC. | ||
Tan et al. immunofluorescently stained within a microdevice to limit the use of expensive dye reagents.121 They stained a number of cancer cell lines, including breast cancer and gastric carcinoma cell lines, with a similar series of dyes: EpCAM, CD45, and DAPI. In order to stain within the microdevice, a series of pressure differentials induced flow of low microliter volumes of staining reagents.122 This method miniaturizes the immunofluorescent staining process and permits a greater level of portability and potential automation.
Another automated method of cell enumeration and separation is fluorescence-activated cell sorting (FACS), which combines flow cytometry with sorting. This technique is used in combination with immunofluorescence staining and uses a focused laser beam to scatter light into a detector.123 After the fluorescent signal is analyzed, each cell is encapsulated in a droplet and sorted. FACS can gate cells based on the scatter and the fluorescence intensity.124 This technique is valuable because it can detect and sort cells into individual well plates, which can then be used for molecular characterization or culture.118 Some technologies are capable of detecting multiplexed suspensions of cells and can sort up to 50000 cells per second.123 This system can collect a valuable data set including size, gene expression, and fluorescence of each of the capture cells that can be used to explore the heterogeneity of detected cells.100 This sort of system can be used with an enriched sample of CTCs or with labeled whole blood to try to detect low-concentration CTCs. The FACS process can often damage cells due to the high shear forces required for operation118 and can be prohibitively expensive because of the large machines and costs to operate.
Once example of a cytometry based approach is the multicolor flow cytometry protocol developed by Hristozova et al. for studying squamous cell carcinoma of the head and neck (SCCHN).125 They used this protocol to detect and characterize CTCs in the blood after red blood cell lysis and fluorescence staining. This methodology was able to use an electronic threshold for fluorescence signals to accurately enumerate the CTCs and their epidermal growth factor (EGFR) expression. This procedure was able to detect a low number of CTCs, with an average of 1.7 CTCs per 3.75 mL of blood.125 This procedure is quite promising for the counting of CTCs, and would be more successful on a pre-enriched sample.
Fluorescence based methods have the advantage of well-understood biomarkers and allow scientists to analysis cell contents. These methods can include computer-based thresholding and flow cytometry procedures. One disadvantage of the technique is that if the cells are fixed or permeabilized it renders the cells unusable for culture and further analysis.
8.2 Nanomaterial-based detection
Nanomaterials such as gold nanoparticles are exploited for detection of CTCs bound to them. The local surface plasmon resonance (LSPR) of gold nanoparticles allows visual observation, colorimetry, UV-Vis spectroscopy, ELISA,126 and ultrasensitive surface-enhanced Raman scattering (SERS).127–129 SERS measures the scattering at a noble metal surface and is the most significant detection method involving gold nanorods, but the requirement for a large-scale SERS imaging technologies is a disadvantage.130 Nima et al. conjugated four sets of Raman-active molecules and antibody probes specific to breast cancer onto multicolored, silver-coated nanoparticles, and they enhanced 129-fold the SERS signal, shortened the detection time, and very specifically detected a single MCF-7 breast cancer cell in whole blood (1 CTC per 90000 fibroblast cells).129 Furthermore, they were able to discriminate between the four targets selected.129
Additionally, gold nanoparticles, which are biocompatible, can be conjugated with probes to capture CTCs in vivo and photoacoustic imaging detects them.131–135 Simply, photoacoustics swiftly heats nanoparticles within tissues with light; these heated nanoparticles emit photoacoustic waves in response which can be detected with an ultrasonic transducer.131,136 Multilayer biocompatible nanoparticles consisting of a gold core, a silica coated middle, an outer layer of iron oxide nanocrystals, and capture probes to synchronously capture and detect CTCs in vivo with photoacoustic/photothermal flow cytometry.131,135 This method efficiently captured 1 CTC per mL and detected 90% of CTCs captured.131,135 Additionally, it could potentially ablate CTCs with lasers, which would bridge a gap between surgical biopsies, minimal biopsies, and liquid biopsies if it can overcome the shear stress of blood circulation, prevent accumulation in organs, and reduce non-specific binding and background noise.131,137 These disadvantages could be overcome by integrating this method into microchip technologies.
Another nanomaterial, graphene and graphene oxides, detects CTCs via fluorescence quenching, electrochemical impedance spectroscopy, and cyclic voltammetry.131,138–140 Graphene/graphene oxides bind to dye-labeled molecules through pi–pi stacking. Essentially, fluorescent molecules are quenched when bound to graphene/graphene oxides, and when fluorescently tagged probes have captured their target, they recover their fluorescence. Nellore et al. demonstrated using this strategy and aptamers as probes, a 95% capture efficiency and 97% cell viability of SKBR3 breast cancer cells, LNCaP prostate cancer cells and SW-948 colon cancer cells spiked into 105 cells per mL of peripheral blood with mononuclear cells and HaCaT skin cells.131,138 Electrochemical impedance spectroscopy and cyclic voltammetry measure the change in electrical current through a graphene/graphene oxide electrode when a CTC is captured by a probe conjugated to the electrodes surface.131,139 Graphene/graphene oxide detection methods must prevail over issues of scaling-up and quality synthesis of graphene in order to viable method of clinically detecting CTCs.131
8.3 Label-free detection
Label-free detection of CTCs is beneficial due to the detection of a more heterogeneous population of CTCs. Chiu et al. engineered a microfluidic-based optical sensing device for label-free detection of CTCs through their lactic acid metabolism.141 CTCs were captured in water-in-oil cell-encapsulating micro-droplets on a microfluidic chip, and fluorescence-based detection of lactic acid produced within the micro-droplets ensued. A known number of OEC-M1 liver cancer cells were spiked into a suspension of leukocytes and loaded onto the device. Chiu et al. showed that there was no significant discrepancy between the number of OEC-M1 liver cancer cells spiked into the solution and the number of OEC-M1 liver cancer cells detected by the device, and were able to maintain 96% cell viability. Fluorescence detection of lactic acid in a micro-droplet was linearly proportional to the number of CTCs in the droplet. Leukocyte contamination did not interfere with the detection of the CTCs.141 For this method to translate into the clinical setting, Chiu et al. must demonstrate that they can isolate by centrifugation CTCs and leukocytes from whole blood, which contains lactic acid, suspend the CTCs and leukocytes in cell medium, and detect CTCs with their method. They must also run experiments with their device in parallel with the clinical gold standard.The detection methodologies reviewed in this section include fluorescence-based cell detection, nanotechnology, and label-free detection of captured cells. While the fluorescence-based detection allows for quick enumeration and validation, it limits the scope of analysis that can be performed on captured CTCs. The nanoparticle and label-free approaches allow for a wider range of cell culture and molecular analysis.
9. Analysis technologies
Once the CTCs are captured and verified, a number of analyses can be performed based on the goal of the liquid biopsy. For predictive screening, diagnosis, and monitoring, the clinician may just want to enumerate the CTCs and measure surface expression and receptor availability. For determination of prognosis and targeting therapy, a more in-depth analysis requiring cell culture can be used for measurement of drug response. The two major classes of analysis are molecular analysis, which typically uses a molecular biology technique such as PCR to determine the presence of biological molecules, followed by bioinformatics analysis to glean usable knowledge from the experiments. This section will detail some molecular and computational analysis approaches.9.1 Molecular analysis
One molecular analysis approach concerns the capture and analysis of DNA and RNA present in circulating tumor cells. These studies use methods of PCR and RT-PCR to characterize the DNA and RNA respectively that are present in captured cells.142 Knowledge of the genetic composition of tumor cells may provide insight into the causes of their behavior as well as their possible responses to treatment forms. In 94% of cases, genetic mutations found in captured CTCs matched mutations in the solid tumor mass.143 The non-matching percentage may be due to the heterogeneity of the cancer population, and therefore may suggest that CTC sampling provides a better and more diverse genetic profile than a solid biopsy.144 This sort of study shows the comparison to a gold standard tissue biopsy and presents the possibility that liquid biopsy can fill gaps in existing technologies. The presence or absence of certain genetic traits and biomarkers, such as HER-2, ERA, and a trisomy of chromosome 8, are known to influence patient responsiveness to chemical agents.144,145 Prior knowledge of a cell's genetic profile could therefore influence the choice of treatment. There are limitations to this approach: such limitations include the lack of singular, identifiable tumor specific genes, the ambiguity of positive and negative results, and the knowledge that the presence of a gene does not necessarily indicate the presence of its post-translational protein products.142CTC characterization through immunostaining is another approach for analysis. CTCs, including subpopulations of CTCS, can be qualified and differentiated through the targeting of specific biomarkers. Huang et al. used FISH to visually differentiate between HER-2 positive (SK-BR3) and HER-2 negative (MDA-MB-231) cancer cell lines in a population of immunomagnetic captured CTCs. Since a large copy number of HER-2 is associated with shorter survival rate and a higher risk of cancer re-occurrence,146 the ability to identify this trait in patient samples may influence options and prognosis.147 A similar differentiation of HER-2 screening was performed with Liquid Permanent Red targeted toward HER-2 positive SK-BR3 cells while HER-2 negative MCF7 cells remained unstained.144 This approach comes with its challenges. It requires pre-emptive targeting of specific biomarkers. Common stains meant to identify across most CTC subpopulations are imperfect as well. Cytokeratin stain (CK) is most common for mass CTC identification. Yet CTCs downregulate their CK expression during EMT, which corresponds to increased invasiveness and metastatic potential.148–150 This phenomenon means that staining may not be the best tool for wide CTC quantification. It is suggested that immunostaining and FISH approaches by themselves are not enough to fully characterize a population of CTCs.148,151,152
One of the standard methods of cancer analysis and categorization has been based on the histology, analysis of the stained pattern of fixed cancer cells.153 With recent advances in molecular biology, there is a lot more information that can be gleaned from DNA arrays to understand the specific expression of an individual tumor. This allows for dealing with the tumor heterogeneity, because it provides an understanding of the full tumor rather than each individual cell. Transcriptional profiling is a method of assigning cancers to specific classes for treatment and prognosis that uses gene expressions to correlate with outcome.153 DNA microarrays have been used to understand and characterize a number of molecular subtypes of cancer at once, and then relate the distinct types to different progressions of disease and treatments.154 The kinds of bioinformatics that can be performed on this data will be described in the next section.
The lack of purity of CTC samples has posed a major challenge to single cell proteomic analysis approaches. Recently, Deng et al. have demonstrated single-cell proteomic profiling of CTCs from whole blood using microfluidics and photocleavable antibody conjugates (Fig. 12).155 The technique involves a number of CTC separation approaches including ones mentioned previously such as a herringbone structure and spiral channel structure.155 The crux of the technology is the placement of high-purity CTCs to a single-cell barcode chip that employs a poly-L-lysine barcode pattern to detect secreted proteins.155 These barcode patterns enable single CTCs to be efficiently captured into microchambers for proteomic analysis. Using a combination of the barcode pattern and photocleavable ssDNA-antibody conjugates, the CTCs can efficiently be captured and released, purified, and subjected to single-cell proteomic characterization.155 This device limitedly requires a relatively high concentration of cells as input, and results in single cells in only about 1/4 of the 2720 microchambers.155 The combined device has a relatively low capture efficiency (55%) which limits its use for retaining target CTCs on chip for analysis.155 The group was able to demonstrate selections of CTCs based on cell surface marker phenotypes, efficient cell capture, and release of these selected cells. This combined microfluidic device shows great promise towards phenotypic analysis of single-cell samples and towards applications such as determination of phosphorylation, signaling proteins, and secretions.155
 | ||
| Fig. 12 Integrated system for single cell secretion profiling. Overall workflow including photocleavable capture probes, blood cell depletion, and single-cell proteomic profiling using barcoding patterns. Reprinted from Scientific Reports, 4, Deng, An Integrated Microfluidic Chip System for Single-Cell Secretion Profiling of Rare Circulating Tumor Cells, 7499, Copyright (2014), with permission from NPG. | ||
The downstream use of CTCs has more abstract purposes. Outside their predictive use for individual patients, they can also be used in research settings. Cell lines can be formed from captured CTCs, which can then be used for behavior modeling.143 One such approach resulted in the identification of a novel gene associated with the metastasis of brain cancer.156 Other such cell lines of breast and colon cancers have been isolated from CTCs.157,158 Stable lines have resulted in the successful xenografting of murine models for further downstream study.159
9.2 Computational analysis-bioinformatics
Once CTCs are molecularly characterized, there are a number of high-throughput bioinformatics approaches to score patients and improve diagnosis, prognosis, and therapeutics.160 These bioinformatics approaches can focus on signaling mechanisms, metabolites, and metastasis data.161 More broadly, bioinformatics is the use of mathematics, particularly statistics, and computer science to try to understand large sets of biological data.162 Beyond the applications to understanding existing cases, this sort of analysis helps to determine new drug targets because it sheds lights on which genes are present or mutated in a given patient.162Statistical analyses relate the presence of type of CTCs found in patient studies to the patients' prognosis. Muller et al. found that among CTC positive patients, median survival time was 18.1 months (95% CI 15.1–22.1) while among CTC negative patients, the median survival time was 27 months (23.5–30.7 months p < 0.001). The correlation did not extend to progression free survival.163 The source of the CTCs also seems to impact the patient prognosis. In a study comparing CTCs gathered from the hepatic vein to those gathered from the peripheral veins, the presence of HV CTCs showed strong relation to shorter disease-free survival and overall survival in patients. The presence of PV CTCs did not correlate with either. For the hepatic-sourced CTCs, a presence of three or more per 7.5 mL of blood corresponded to a hazard ratio of 3.97 for overall survival, and 2.55 for disease free survival. The peripheral-sourced CTCs showed no such relation.164
The DNA microarray approach described in the molecular analysis section can be used to collect meta-data to be integrated into computational analysis to fully understand gene profiling.154 Rhodes et al. has presented a technology called ONCOMINE, a microarray database for cancer that allows for data-mining.165 This database allows for data queries to select for specific gene expression across cancer subtypes, experiments, and pathology-based analyses.165 One of the particularly useful features is a color-map for visualization of standard gene expression, known as the STUDY module. This portal is a useful synthesis of microarray, clinical, and pathological studies for data mining cancer genomic analysis.
One of the most commonly used cancer bioinformatics tools predicts protein structure and function.166 For protein structure prediction, predicted secondary and tertiary structure can be used for designing further experiments. Protein function tools use gene function prediction methods to try to determine the information about the activities of each of the gene clusters that may be unknown. They typically use gene sequence homology because the sequence is well known, and these methods use similarities is sequence to determine similarities in function. Similarly, there are database and analysis methods to try to understand protein–protein interactions in model organisms of interest.166
Cheng et al. used models combined with a large set of compiled data to determine the relationship between transcription factor binding and gene expression.167 In the K562 cells used for this study, five transcription factors were found to be most important: YY1, E2F4, MYC, MAX, and ELF1.167 These were the transcription factors that most impacted gene expression based on binding data at transcription start sites. This indicates some of the important factors for prediction of expression levels.167 The E2F4 score is a prognostic signature that has been predictive of breast cancer patient outcome regardless of the treatment and other frequently considered variables.168 The signature used is E2F4, a ubiquitous tissue factor, was defined based on target genes in various tissues, and these genes were found to be 21-fold more likely to be correlated to patient survival. The group used chromatin-immunoprecipitation sequencing to identify the target genes of interest, and then used computational methods to calculate a regulatory activity score of E2F4. The correlation to survival was confirmed using almost two thousand samples and controlled for a number of additional factors including age, tumor size and grading, and therapeutic condition.168 The group found that the E2F4 target gene is related to cell cycle regulators, and the related genes correlate with the survival of patients. In humans, the gene cluster related to cell cycle regulation, mitosis, DNA repair and replication, and nucleotide binding.168 The regulatory activity scores were compared to two commonly used survival prediction variables, lymph node and estrogen receptor status. Multiple poolings and data sets were used to show that this regulatory activity score methodology held up against other analyses. A number of breast cancer subtypes, such as ER-positive, PR-positive, MYC-negative, and intrinsic subtypes were shown to be predicted by the regulatory activity scoring methodology.168
Among the options for analysis of captured CTCs are molecular methods and bioinformatics approaches. Many of the molecular methods, such as PCR and surface marker expression can be benchmarked to the gold standard of tissue biopsy. Some early studies suggest that CTC liquid biopsy actually provides greater heterogeneity than a tissue biopsy, meaning that the results of molecular analysis could be more meaningful. The results of the molecular analysis can be inputted into bioinformatics analyses that could provide greater understanding of important genes and biomarkers in the future.
10. Discussion and conclusion
The resounding evidence that earlier detection of cancer correlates with survival rates has spurred a recent push to enhance diagnosis and monitoring. While current gold standard diagnostics require excisional tissues, cutting-edge scientific research spotlights liquid biopsy on chip for capture of CTCs from a mere blood sample. The preliminary studies and FDA approval of CellSearch validate the utility of incorporating immunomagnetic capture of CTCs. However, none of these devices fully demonstrate high throughput, high capture efficiency, low contamination, and high cell viability for analysis of the full snapshot of genomic fluctuations of a tumor. There remains a need for an automated, portable, and a simple – yet sophisticated and robust chip for the capture, detection, and single cell analysis of CTCs.The boost in the clinical insight into the mechanisms of cancer and EMT illustrates not only the basis of CTC heterogeneity but the complexity of surmounting to these challenges. Gaps in our understanding of a cell's metastatic journey and the lack of stratification of CTC phenotypes raise a challenge against integrating biology into liquid biopsy chips. Opportunities for refined and well-designed studies to determine the ideal biomarkers for a particular clinical need are still out there.
Capture of CTCs has progressed from macroscale commercial technologies to miniaturized microfluidic devices, requiring only scant reagent volumes and permitting greater sample control. Most technologies have only optimized a few of the key parameters; so there remain opportunities to integrate existing strategies in parallel. Furthermore, most of the developed technologies have not yet been validated at biological concentrations, which are most often far lower than spiking concentrations. Of the prevailing strategies, immunomagnetic separation typically has very high throughput and capture efficiency and has been widely studied. The technologies for capture and release and single cell analysis have not yet been optimized to this degree. The clinical applicability of on-demand capture and analysis due to the heterogeneity and ever-changing nature of cancer necessitates the improvement of these technologies.
Similarly, immune affinity strategies for CTC detection are well corroborated, but they do not lend themselves well to further analysis for targeted therapies. With recent improvements in nanotechnology, many groups have considered alternative CTC detection methods that show promising sensitivity and viability, including nanoparticle label-free methods. These grant an opportunity for lowering the limit of detection because they work at the nanoscale, allowing a move towards biological concentrations while maintaining the low cost and portability of future devices.
Most current CTC analyses are corollaries to existing tissue or molecular biology assays. The challenge is that current molecular assays require a larger input sample concentration than liquid biopsies on chip output, especially at the single cell level. In the past few years, improved computing power has allowed for the crunching of larger datasets from multiple studies, improved bioinformatics capabilities, data pooling, and correlation-based analysis. One opportunity in the analysis field is a targeted, metadata analysis of a variety of factors to determine the optimal design conditions for a given application.
Liquid biopsy can potentially offer a minimal invasive tool for dynamic monitoring cancer metastasis, as specific design criteria allow for analysis of cancer heterogeneity. The procedure outlined in the paper describes the capture and detection methods that are compatible with different biological analyses to further our understanding of metastatic potential. A series of design and validation experiments should follow to be consistent with current clinical methods and the FDAs clinical evaluation criteria. Collaborations between clinicians, biomedical researchers and technologists are necessary for development of elegant platforms that proceed through the FDA pipeline into mainstream use. Numerous agencies and initiatives have poised researchers well to build upon existing work, optimize designs, and validate their clinical efficacy in order to continue on the path towards better technologies and improved understanding of the complicated biology at play.
References
- R. Siegel, J. Ma, Z. Zou and A. Jemal, Cancer statistics, 2014, Ca-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- H. I. Vargas, R. V. Agbunag and I. Khaikhali, State of the art of minimally invasive breast biopsy: principles and practice, Breast Cancer, 2000, 7, 370–379 CrossRef CAS PubMed.
- C. DeSantis, R. Siegel, P. Bandi and A. Jemal, Breast cancer statistics 2011, Ca-Cancer J. Clin., 2011, 61, 408–418 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, 2015, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- B. Hong and Y. Zu, Detecting circulating tumor cells: Current challenges and new trends, Theranostics, 2013, 3, 377–394 CrossRef CAS PubMed.
- A. Liga, A. D. Vliengenthart, W. Oosthuyzen, J. Dear and M. Kersaudy-Kerhoas, Exosome isolation: A microfluidic road-map, Lab Chip, 2015, 15, 2388–2394 RSC.
- M. Colombo, G. Raposo and C. Théry, Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles, Annu. Rev. Cell Dev. Biol., 2014, 30, 255–292 CrossRef CAS PubMed.
- S. S. Kanwar, C. J. Dunlay, D. M. Simeone and S. Nagrath, Microfluidic device (ExoChip) for on-chip isolation, quantification and characterization of circulating exosomes, Lab Chip, 2014, 14, 1891–1900 RSC.
- H. Im, et al., Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor, Nat. Biotechnol., 2014, 32, 490–495 CrossRef CAS PubMed.
- A. Schramm, et al., Therapeutic intervention based on circulating tumor cell phenotype in metastatic breast cancer: concept of the DETECT study program, Arch. Gynecol. Obstet., 2016, 293, 271–281 CrossRef CAS PubMed.
- N. Chen, et al., Proof of concept for inhibiting metastasis: circulating tumor cell-triggered localized release of anticancer agent via a structure-switching aptamer, Chem. Commun., 2016, 52, 6789–6792 RSC.
- X. Sun, J. Bao and Y. Shao, Mathematical Modeling of Therapy-induced Cancer Drug Resistance: Connecting Cancer Mechanisms to Population Survival Rates, Sci. Rep., 2016, 6, 22498 CrossRef CAS PubMed.
- A. F. Sarioglu, et al., A microfluidic device for label-free, physical capture of circulating tumor cell clusters, Nat. Methods, 2015, 12, 1–10 CrossRef PubMed.
- L. Hajba and A. Guttman, Circulating tumor-cell detection and capture using microfluidic devices, TrAC, Trends Anal. Chem., 2014, 59, 9–16 CrossRef CAS.
- K. Hoshino, et al., Microchip-based immunomagnetic detection of circulating tumor cells, Lab Chip, 2011, 11, 3449 RSC.
- A. Brouwer, et al., Evaluation and consequences of heterogeneity in the circulating tumor cell compartment, Oncotarget, 2016, 7, 48625–48643 Search PubMed.
- S. J. Werden, et al., Phosphorylation of serine 367 of FOXC2 by p38 regulates ZEB1 and breast cancer metastasis, without impacting primary tumor growth, Oncogene, 2016, 1–12, DOI:10.1038/onc.2016.203.
- K. J. Cheung, et al., Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 201508541 Search PubMed.
- Q. Shi, et al., Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 419–424 CrossRef CAS PubMed.
- C. A. Klein, et al., Combined transcriptome and genome analysis of single micrometastatic cells, Nat. Biotechnol., 2002, 20, 387–392 CrossRef CAS PubMed.
- F. Fabbri, et al., Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs, Cancer Lett., 2013, 335, 225–231 CrossRef CAS PubMed.
- W. Wei, et al., Microchip platforms for multiplex single-cell functional proteomics with applications to immunology and cancer research, Genome Med., 2013, 5, 75 CrossRef PubMed.
- X. Ni, et al., Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 21083–21088 CrossRef CAS PubMed.
- A. Diamantis, E. Magiorkinis and H. Koutselini, Fine-needle aspiration (FNA) biopsy: historical aspects, Folia Histochem. Cytobiol., 2009, 47, 191–197 Search PubMed.
- U. Veronesi, et al., A randomized comparison of sentinel-node biopsy with routine axillary dissection in breast cancer, N. Engl. J. Med., 2003, 349, 546–553 CrossRef PubMed.
- B. E.-B. Practice, Core Needle and Open Surgical Biopsy for Diagnosis of Breast Lesions, An Update to the 2009 Report, 2009 Search PubMed.
- H. Wada, et al., Expression of Somatostatin Receptor Type 2A and PTEN in Neuroendocrine Neoplasms Is Associated with Tumor Grade but Not with Site of Origin, Endocr. Pathol., 2016, 27, 179–187 CrossRef CAS PubMed.
- U. Kettritz, Minimally Invasive Biopsy Methods – Diagnostics or Therapy? Personal Opinion and Review of the Literature, Breast Cancer, 2011, 6, 94–97 Search PubMed.
- L. Liberman, et al., Impact of core biopsy on the surgical management of impalpable breast cancer, Am. J. Roentgenol., 1997, 168, 495–499 CrossRef CAS PubMed.
- K. R. Gundry and W. A. Berg, Treatment issues and core needle breast biopsy: clinical context, Am. J. Roentgenol., 1998, 171, 41–49 CrossRef CAS PubMed.
- H. M. Verkooijen, et al., Impact of stereotactic large-core needle biopsy on diagnosis and surgical treatment of non-palpable breast cancer, Eur. J. Surg. Oncol., 2001, 27, 244–249 CrossRef CAS PubMed.
- M. R. Newman, et al., Diagnosis of breast microcalcifications: a comparison of stereotactic FNA and core imprint cytology as adjuncts to core biopsy, Pathology, 2001, 33, 449–453 CrossRef CAS PubMed.
- S. H. Parker, et al., US-guided automated large-core breast biopsy, Radiology, 1993, 187, 507–511 CrossRef CAS PubMed.
- G. Kocjan, Fine needle aspiration cytology, Cytopathology, 2003, 14, 307–308 CrossRef CAS PubMed.
- E. A. M. O'Flynn, A. R. M. Wilson and M. J. Michell, Image-guided breast biopsy: state-of-the-art, Clin. Radiol., 2010, 65, 259–270 CrossRef PubMed.
- G. Schueller, C. Schueller-Weidekamm and T. H. Helbich, Accuracy of ultrasound-guided, large-core needle breast biopsy, Eur. J. Radiol., 2008, 18, 1761–1773 CrossRef CAS PubMed.
- H.-L. Park and L. S. Kim, The Current Role of Vacuum Assisted Breast Biopsy System in Breast Disease, Breast Cancer, 2011, 14, 1–7 CrossRef PubMed.
- J. Dong, A. Ly, R. Arpin, Q. Ahmed and E. Brachtel, Breast fine needle aspiration continues to be relevant in a large academic medical center: experience from Massachusetts General Hospital, Breast Cancer Res. Treat., 2016, 158, 297–305 CrossRef PubMed.
- M. G. Krebs, et al., Molecular analysis of circulating tumour cells-biology and biomarkers, Nat. Rev. Clin. Oncol., 2014, 11, 129–144 CrossRef CAS PubMed.
- W.-C. Chie, Y.-H. Chang and H.-H. Chen, A novel method for evaluation of improved survival trend for common cancer: early detection or improvement of medical care, J. Eval. Clin. Pract., 2007, 13, 79–85 CrossRef PubMed.
- C. Bettegowda, et al., Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies, Sci. Transl. Med., 2014, 6, 224ra24 CrossRef PubMed.
- M. Stroun, J. Lyautey, C. Lederrey, A. Olson-Sand and P. Anker, About the possible origin and mechanism of circulating DNA, Clin. Chim. Acta, 2001, 313, 139–142 CrossRef CAS.
- E. Racila, et al., Detection and characterization of carcinoma cells in the blood, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4589–4594 CrossRef CAS.
- N. Karachaliou, C. Mayo-de-las-Casas, M. A. Molina-Vila and R. Rosell, Real-time liquid biopsies become a reality in cancer treatment, Ann. Transl. Med., 2015, 3, 36 Search PubMed.
- I. Baccelli, et al., Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay, Nat. Biotechnol., 2013, 31, 539–544 CrossRef CAS PubMed.
- P. Vicki, D. Charlotte and Z. W. Koopman, Circulating Tumor Cells, Science, 2013, 341, 1186–1188 CrossRef PubMed.
- D. R. Parkinson, et al., Considerations in the development of circulating tumor cell technology for clinical use, J. Transl. Med., 2012, 10, 138 CrossRef PubMed.
- D. Hanahan and R. A. Weinberg, The hallmarks of cancer, Cell, 2000, 100, 57–70 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Hallmarks of cancer: The next generation, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- P. Fedi, S. R. Tronick and S. A. Aaronson, in Cancer Medicine, ed. J. F. Hollandet al.,, Williams and Wilkins, 1997, pp. 41–64 Search PubMed.
- R. A. Weinberg, The retinoblastoma protein and cell cycle control, Cell, 1995, 81, 323–330 CrossRef CAS PubMed.
- A. H. Wyllie, J. F. Kerr and A. R. Currie, Cell death: the significance of apoptosis, Int. Rev. Cytol., 1980, 68, 251–306 CAS.
- L. Hayflick, Mortality and immortality at the cellular level: a review, Biochemistry, 1997, 62, 1180–1190 CAS.
- D. Hanahan and J. Folkman, Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis, Cell, 1996, 86, 353–364 CrossRef CAS PubMed.
- N. Bouck, V. Stellmach and S. C. Hsu, How tumors become angiogenic, Adv. Cancer Res., 1996, 69, 135–174 CrossRef CAS PubMed.
- M. B. Sporn, The war on cancer, Lancet, 1996, 347, 1377–1381 CrossRef CAS.
- J. P. Thiery, Epithelial-mesenchymal transitions in development and pathologies S0955067403001339 [pii], Curr. Opin. Cell Biol., 2003, 15, 740–746 CrossRef CAS PubMed.
- R. Kalluri and E. G. Neilson, Epithelial-mesenchymal transition and its implications for fibrosis.pdf, J. Clin. Invest., 2003, 112, 1776–1784 CrossRef CAS PubMed.
- R. Kalluri and R. A. Weinberg, Review series The basics of epithelial-mesenchymal transition, J. Clin. Invest., 2009, 119, 1420–1428 CrossRef CAS PubMed.
- D. C. Radisky, Epithelial-mesenchymal transition, J. Cell Sci., 2005, 118, 4325–4326 CrossRef CAS PubMed.
- R. Tannishtha, S. J. Morrison, M. F. Clarke and I. L. Weissman, Stem cells, cancer, and cancer stem cells, Nature, 2001, 414, 105–111 CrossRef PubMed.
- S. Sun and X. S. Qiu, Cancer stem cells and tumor metastasis, J. Cancer Res. Ther., 2013, suppl 9, S150–S152 Search PubMed.
- D. S. Micalizzi, S. M. Farabaugh and H. L. Ford, Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression, J. Mammary Gland Biol. Neoplasia, 2010, 15, 117–134 CrossRef PubMed.
- M. M. Ferreira, V. C. Ramani and S. S. Jeffrey, Circulating tumor cell technologies, Mol. Oncol., 2016, 1–21, DOI:10.1016/j.molonc.2016.01.007.
- A. A. Powell, et al., Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines, PLoS One, 2012, 7, e33788 CAS.
- C. Gasch, et al., Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer, Clin. Chem., 2013, 59, 252–260 CAS.
- A. Ulmer, et al., Immunomagnetic Enrichment, Genomic Characterization, and Prognostic Impact of Circulating Melanoma Cells Immunomagnetic Enrichment, Genomic Characterization, and Prognostic Impact of Circulating Melanoma Cells, Clin. Cancer Res., 2004, 10, 531–537 CrossRef CAS PubMed.
- D. Ramsköld, et al., Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells, Nat. Biotechnol., 2012, 30, 777–782 CrossRef PubMed.
- K. Strimbu and J. A. Tavel, What are Biomarkers?, Curr. Opin. HIV AIDS, 2011, 5, 463–466 CrossRef PubMed.
- G. Barriere, et al., Circulating tumor cells and epithelial, mesenchymal and stemness markers: characterization of cell subpopulations, Ann. Transl. Med., 2014, 2, 109 Search PubMed.
- H. Iinuma, et al., Clinical significance of circulating tumor cells, including cancer stem-like cells, in peripheral blood for recurrence and prognosis in patients with dukes' stage B and C colorectal cancer, J. Clin. Oncol., 2011, 29, 1547–1555 CrossRef PubMed.
- M. Chimonidou, G. Kallergi, V. Georgoulias, D. R. Welch and E. S. Lianidou, Breast Cancer Metastasis Suppressor-1 promoter methylation in primary breast tumors and corresponding Circulating Tumor Cells, Mol. Cancer Res., 2013, 11, 1248–1257 CrossRef CAS PubMed.
- S. V. Litvinov, Ep-CAM: a human epithelial antigen is a homophilic cell-cell adhesion molecule, J. Cell Biol., 1994, 125, 437–446 CrossRef CAS PubMed.
- A. M. Sieuwerts, et al., Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells, J. Natl. Cancer Inst., 2009, 101, 61–66 CrossRef CAS PubMed.
- T. M. Gall and A. E. Frampton, Gene of the month: E-cadherin (CDH1), J. Clin. Pathol., 2013, 66, 928–932 CrossRef CAS PubMed.
- H. Peinado, F. Portillo and A. Cano, Transcriptional regulation of cadherins during development and carcinogenesis, Int. J. Dev. Biol., 2004, 48, 365–375 CrossRef CAS PubMed.
- J. P. Thiery and J. P. Sleeman, Complex networks orchestrate epithelial-mesenchymal transitions, Nat. Rev. Mol. Cell Biol., 2006, 7, 131–142 CrossRef CAS PubMed.
- S. A. Joosse, et al., Changes in keratin expression during metastatic progression of breast cancer: impact on the detection of circulating tumor cells, Clin. Cancer Res., 2012, 18, 993–1003 CrossRef CAS PubMed.
- R. B. Hazan, R. U. I. Qiao, R. Keren, I. Badano and K. Suyama, Cadherin Switch in Tumor Progression, Ann. N. Y. Acad. Sci., 2004, 1014, 155–163 CrossRef CAS PubMed.
- A. Satelli and S. Li, Vimentin in cancer and its potential as a molecular target for cancer therapy, Cell. Mol. Life Sci., 2011, 68, 3033–3046 CrossRef CAS PubMed.
- M. B. Lustberg, et al., Heterogeneous atypical cell populations are present in blood of metastatic breast cancer patients, Breast Cancer Res., 2014, 16, R23 CrossRef PubMed.
- T. Yokobori, et al., Plastin3 Is a Novel Marker for Circulating Tumor Cells Undergoing the Epithelial–Mesenchymal Transition and Is Associated with Colorectal Cancer Prognosis, Cancer Res., 2013, 73, 2059–2069 CrossRef CAS PubMed.
- K. Sugimachi, et al., Aberrant Expression of Plastin-3 Via Copy Number Gain Induces the Epithelial–Mesenchymal Transition in Circulating Colorectal Cancer Cells, Ann. Surg. Oncol., 2014, 21, 3680–3690 CrossRef PubMed.
- C. Ginestier, et al., ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome, Cell Stem Cell, 2007, 1, 555–567 CrossRef CAS PubMed.
- G. Barriere, A. Riouallon, J. Renaudie, M. Tartary and M. Rigaud, Mesenchymal characterization: alternative to simple CTC detection in two clinical trials, Anticancer Res., 2012, 32, 3363–3370 Search PubMed.
- M. Balic, et al., Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype, Clin. Cancer Res., 2006, 12, 5615–5621 CrossRef CAS PubMed.
- M. Al-Hajj, M. S. Wicha, A. Benito-Hernandez, S. J. Morrison and M. F. Clarke, Prospective identification of tumorigenic breast cancer cells, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3983–3988 CrossRef CAS PubMed.
- Y. Zhang, K. A. Toy and C. G. Kleer, Metaplastic breast carcinomas are enriched in markers of tumor-initiating cells and epithelial to mesenchymal transition, Mod. Pathol., 2011, 25, 178–184 Search PubMed.
- M. Todaro, et al., CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis, Cell Stem Cell, 2014, 14, 342–356 CrossRef CAS PubMed.
- W. Harb, et al., Mutational Analysis of Circulating Tumor Cells Using a Novel Microfluidic Collection Device and qPCR Assay, Transl. Oncol., 2013, 6, 528–538 CrossRef PubMed.
- G. Vona, et al., Isolation by size of epithelial tumor cells: a new method for the immunomorphological and molecular characterization of circulatingtumor cells, Am. J. Pathol., 2000, 156, 57–63 CrossRef CAS PubMed.
- F. Farace, et al., A direct comparison of CellSearch and ISET for circulating tumour-cell detection in patients with metastatic carcinomas, Br. J. Cancer, 2011, 105, 847–853 CrossRef CAS PubMed.
- P. Chen, Y.-Y. Huang, K. Hoshino and X. Zhang, Multiscale immunomagnetic enrichment of circulating tumor cells: from tubes to microchips, Lab Chip, 2014, 14, 446–458 RSC.
- N. Saucedo-Zeni, et al., A novel method for the in vivo isolation of circulating tumor cells from peripheral blood of cancer patients using a functionalized and structured medical wire, Int. J. Oncol., 2012, 41, 1241–1250 Search PubMed.
- K.-A. Hyun and H.-I. Jung, Advances and critical concerns with the microfluidic enrichments of circulating tumor cells, Lab Chip, 2014, 14, 45–56 RSC.
- G. M. Whitesides, The origins and the future of microfluidics, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- D. J. Beebe, G. A. Mensing and G. M. Walker, Physics and applications of microfluidics in biology, Annu. Rev. Biomed. Eng., 2002, 4, 261–286 CrossRef CAS PubMed.
- D. N. Breslauer, P. J. Lee and L. P. Lee, Microfluidics-based systems biology, Mol. Biosyst., 2006, 2, 97 RSC.
- C. Alix-Panabières and K. Pantel, Challenges in circulating tumour cell research, Nat. Rev. Cancer, 2014, 14, 623 CrossRef PubMed.
- D. R. Gossett, et al., Label-free cell separation and sorting in microfluidic systems, Anal. Bioanal. Chem., 2010, 397, 3249–3267 CrossRef CAS PubMed.
- Z. Liu, et al., Rapid isolation of cancer cells using microfluidic deterministic lateral displacement structure, Biomicrofluidics, 2013, 7, 11801 CrossRef PubMed.
- S. Shim, et al., Antibody-independent isolation of circulating tumor cells by continuous-flow dielectrophoresis, Biomicrofluidics, 2013, 7, 11807 CrossRef PubMed.
- I.-F. Cheng, et al., Antibody-free isolation of rare cancer cells from blood based on 3D lateral dielectrophoresis, Lab Chip, 2015, 15, 2950–2959 RSC.
- H.-S. Moon, et al., Continuous separation of breast cancer cells from blood samples using multi-orifice flow fractionation (MOFF) and dielectrophoresis (DEP), Lab Chip, 2011, 11, 1118–1125 RSC.
- G. Gutierrez-Juarez, et al., Detection of melanoma cells in vitro using an optical detector of photoacoustic waves, Lasers Surg. Med., 2010, 42, 274–281 CrossRef PubMed.
- M. Hosokawa, et al., Size-based isolation of circulating tumor cells in lung cancer patients using a microcavity array system, PLoS One, 2013, 8, e67466 CAS.
- S. Zheng, et al., 3D microfilter device for viable circulating tumor cell (CTC) enrichment from blood, Biomed. Microdevices, 2011, 13, 203–213 CrossRef PubMed.
- F. I. Thege, et al., Microfluidic immunocapture of circulating pancreatic cells using parallel EpCAM and MUC1 capture: characterization, optimization and downstream analysis, Lab Chip, 2014, 14, 1775–1784 RSC.
- E. Ozkumur, et al., Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells, Sci. Transl. Med., 2013, 5, 179ra47 Search PubMed.
- J. H. Kang, et al., A combined micromagnetic-microfluidic device for rapid capture and culture of rare circulating tumor cells, Lab Chip, 2012, 12, 2175–2181 RSC.
- C. M. Earhart, et al., Isolation and mutational analysis of circulating tumor cells from lung cancer patients with magnetic sifters and biochips, Lab Chip, 2014, 14, 78–88 RSC.
- P. Chen, Y.-Y. Huang, K. Hoshino and J. X. J. Zhang, Microscale Magnetic Field Modulation for Enhanced Capture and Distribution of Rare Circulating Tumor Cells, Sci. Rep., 2015, 5, 8745 CrossRef CAS PubMed.
- K. Hoshino, et al., An Immunofluorescence-assisted Microfluidic Single Cell Quantitative Reverse Transcription Polymerase Chain Reaction Analysis of Tumour Cells Separated from Blood, J. Circ. Biomarkers, 2015, 4, 1–5 CrossRef.
- N. M. Karabacak, et al., Microfluidic, marker-free isolation of circulating tumor cells from blood samples, Nat. Protoc., 2014, 9, 694–710 CrossRef CAS PubMed.
- H. Liu, et al., Hydrophobic interaction-mediated capture and release of cancer cells on thermoresponsive nanostructured surfaces, Adv. Mater., 2013, 25, 922–927 CrossRef CAS PubMed.
- H. Liu, et al., Dual-responsive surfaces modified with phenylboronic acid-containing polymer brush to reversibly capture and release cancer cells, J. Am. Chem. Soc., 2013, 135, 7603–7609 CrossRef CAS PubMed.
- W. Zhao, et al., Bioinspired multivalent DNA network for capture and release of cells, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19626–19631 CrossRef CAS PubMed.
- C.-H. Lin, et al., A Microfluidic Dual-well Device for High-throughput Single-Cell Capture and Culture, Lab Chip, 2015, 15, 2928–2938 RSC.
- A. R. Wheeler, et al., Microfluidic device for single-cell analysis, Anal. Chem., 2003, 75, 3581–3586 CrossRef CAS PubMed.
- B. L. Khoo, et al., Liquid biopsy and therapeutic response: Circulating tumor cell cultures for evaluation of anticancer treatment, Sci. Adv., 2016, 2, e1600274 Search PubMed.
- S. J. Tan, et al., Versatile label free biochip for the detection of circulating tumor cells from peripheral blood in cancer patients, Biosens. Bioelectron., 2010, 26, 1701–1705 CrossRef CAS PubMed.
- S. J. Tan, L. Yobas, G. Y. H. Lee, C. N. Ong and C. T. Lim, Microdevice for the isolation and enumeration of cancer cells from blood, Biomed. Microdevices, 2009, 11, 883–892 CrossRef PubMed.
- C. W. Shields, C. D. Reyes and G. P. López, Microfluidic cell sorting: a review of the advances in the separation of cells from debulking to rare cell isolation, Lab Chip, 2015, 15, 1230–1249 RSC.
- A. A. S. Bhagat, H. W. Hou, L. D. Li, C. T. Lim and J. Han, Pinched flow coupled shear-modulated inertial microfluidics for high-throughput rare blood cell separation, Lab Chip, 2011, 11, 1870–1878 RSC.
- T. Hristozova, R. Konschak, V. Budach and I. Tinhofer, A simple multicolor flow cytometry protocol for detection and molecular characterization of circulating tumor cells in epithelial cancers, Cytometry, Part A, 2012, 81, 489–495 CrossRef PubMed.
- T. S. Safaei, R. M. Mohamadi, E. H. Sargent and S. O. Kelley, In situ electrochemical ELISA for specific identification of captured cancer cells, ACS Appl. Mater. Interfaces, 2015, 7, 14165–14169 CAS.
- X. Wu, et al., Improved SERS Nanoparticles for Direct Detection of Circulating Tumor Cells in the Blood, ACS Appl. Mater. Interfaces, 2015, 7, 9965–9971 CAS.
- A. Pallaoro, M. R. Hoonejani, G. B. Braun, C. D. Meinhart and M. Moskovits, Rapid identification by surface-enhanced raman spectroscopy of cancer cells at low concentrations flowing in a microfluidic channel, ACS Nano, 2015, 9, 4328–4336 CrossRef CAS PubMed.
- Z. A. Nima, et al., Circulating tumor cell identification by functionalized silver-gold nanorods with multicolor, super-enhanced SERS and photothermal resonances, Sci. Rep., 2014, 4, 1–8 Search PubMed.
- P. Zhang, R. Zhang, M. Gao and X. Zhang, Novel Nitrocellulose Membrane Substrate for Efficient Analysis of Circulating Tumor Cells Coupled with Surface-Enhanced Raman Scattering Imaging, ACS Appl. Mater. Interfaces, 2014, 6, 370–376 CAS.
- J. H. Myung, K. A. Tam, S. J. Park, A. Cha and S. Hong, Recent advances in nanotechnology-based detection and separation of circulating tumor cells, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 223–239 CrossRef PubMed.
- E. I. Galanzha, J. W. Kim and V. P. Zharov, Nanotechnology-based molecular photoacoustic and photothermal flow cytometry platform for in-vivo detection and killing of circulating cancer stem cells, J. Biophotonics, 2009, 2, 725–735 CrossRef CAS PubMed.
- E. I. Galanzha, et al., In vivo magnetic enrichment and multiplex photoacoustic detection of circulating tumour cells, Nat. Nanotechnol., 2009, 4, 855–860 CrossRef CAS PubMed.
- D. A. Nedosekin, et al., Photoacoustic and photothermal detection of circulating tumor cells, bacteria and nanoparticles in cerebrospinal fluid in vivo and ex vivo, J. Biophotonics, 2013, 6, 523–533 CrossRef CAS PubMed.
- X. Hu, et al., Trapping and photoacoustic detection of CTCs at the single cell per milliliter level with magneto-optical coupled nanoparticles, Small, 2013, 9, 2046–2052 CrossRef CAS PubMed.
- V. P. Zharov, et al., In vivo photoacoustic flow cytometry for monitoring of circulating single cancer cells and contrast agents, Opt. Lett., 2006, 31, 3623–3625 CrossRef PubMed.
- W. He, H. Wang, L. C. Hartmann, J.-X. Cheng and P. S. Low, In vivo quantitation of rare circulating tumor cells by multiphoton intravital flow cytometry, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11760–11765 CrossRef CAS PubMed.
- B. P. Viraka Nellore, et al., Aptamer-conjugated graphene oxide membranes for highly efficient capture and accurate identification of multiple types of circulating tumor cells, Bioconjugate Chem., 2015, 26, 235–242 CrossRef CAS PubMed.
- L. Feng, Y. Chen, J. Ren and X. Qu, A graphene functionalized electrochemical aptasensor for selective label-free detection of cancer cells, Biomaterials, 2011, 32, 2930–2937 CrossRef CAS PubMed.
- H. J. Yoon, et al., Sensitive capture of circulating tumour cells by functionalized graphene oxide nanosheets, Nat. Nanotechnol., 2013, 8, 735–741 CrossRef CAS PubMed.
- T.-K. Chiu, et al., Development of a microfluidic-based optical sensing device for label-free detection of circulating tumor cells (CTCs) through their lactic acid metabolism, Sensors, 2015, 15, 6789–6806 CrossRef CAS PubMed.
- H. Lin, M. Balic, S. Zheng, R. Datar and R. J. Cote, Disseminated and circulating tumor cells: role in effective cancer management, Crit. Rev. Oncol. Hematol., 2011, 77, 1–11 CrossRef PubMed.
- Z. Zhang, N. Ramnath and S. Nagrath, Current Status of CTCs as Liquid Biopsy in Lung Cancer and Future Directions, Front. Oncol., 2015, 5, 1–11 Search PubMed.
- H. Frithiof, C. Welinder, A.-M. Larsson, L. Rydén and K. Aaltonen, A novel method for downstream characterization of breast cancer circulating tumor cells following CellSearch isolation, J. Transl. Med., 2015, 13, 1–10 CrossRef CAS PubMed.
- P. P. Lin, Integrated EpCAM-independent subtraction enrichment and iFISH strategies to detect and classify disseminated and circulating tumors cells, Clin. Transl. Med., 2015, 4, 38 CrossRef PubMed.
- P. P. Lee, et al., Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients, Nat. Med., 1999, 5, 677–685 CrossRef CAS PubMed.
- S. K. Huang and D. S. B. Hoon, ScienceDirect Liquid biopsy utility for the surveillance of cutaneous malignant melanoma patients, Mol. Oncol., 2015, 5267, 1–14 Search PubMed.
- S. D. Mikolajczyk, et al., Detection of EpCAM-negative and cytokeratin-negative circulating tumor cells in peripheral blood, J. Oncol., 2011, 2011, 252361 Search PubMed.
- P. K. Grover, A. G. Cummins, T. J. Price, I. C. Roberts-Thomson and J. E. Hardingham, Circulating tumour cells: The evolving concept and the inadequacy of their enrichment by EpCAM-based methodology for basic and clinical cancer research, Ann. Oncol., 2014, 25, 1506–1516 CrossRef CAS PubMed.
- B. Willipinski-Stapelfeldt, et al., Changes in cytoskeletal protein composition indicative of an epithelial-mesenchymal transition in human micrometastatic and primary breast carcinoma cells, Clin. Cancer Res., 2005, 11, 8006–8014 CrossRef CAS PubMed.
- A. Krtolica, S. Parrinello, S. Lockett, P. Y. Desprez and J. Campisi, Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12072–12077 CrossRef CAS PubMed.
- U. Woelfle, G. Sauter, S. Santjer, R. Brakenhoff and K. Pantel, Down-Regulated Expression of Cytokeratin 18 Promotes Progression of Human Breast Cancer Down-Regulated Expression of Cytokeratin 18 Promotes Progression of Human Breast Cancer, Clin. Cancer Res., 2004, 10, 2670–2674 CrossRef CAS PubMed.
- L. Liotta and E. Petricoin, Molecular Profiling of Human Cancer, Nat. Rev. Genet., 2001, 1, 48–56 CrossRef PubMed.
- D. R. Rhodes and A. M. Chinnaiyan, Integrative analysis of the cancer transcriptome, Nat. Genet., 2005,(suppl 37), 0–7 Search PubMed.
- Y. Deng, et al., An Integrated Microfluidic Chip System for Single-Cell Secretion Profiling of Rare Circulating Tumor Cells, Sci. Rep., 2014, 4, 7499 CrossRef CAS PubMed.
- L. Zhang, et al., The identification and characterization of breast cancer CTCs competent for brain metastasis, Sci. Transl. Med., 2013, 5, 180ra48 Search PubMed.
- M. Yu, et al., Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility, Science, 2014, 345, 216–220 CrossRef CAS PubMed.
- L. Cayrefourcq, et al., Establishment and characterization of a cell line from human Circulating colon cancer cells, Cancer Res., 2015, 75, 892–901 CrossRef CAS PubMed.
- M. Yu, S. Stott, M. Toner, S. Maheswaran and D. A. Haber, Circulating tumor cells: approaches to isolation and characterization, J. Cell Biol., 2011, 192, 373–382 CrossRef CAS PubMed.
- G. Gómez-López and A. Valencia, Bioinformatics and cancer research: Building bridges for translational research, Clin. Transl. Oncol., 2008, 10, 85–95 CrossRef.
- D. Wu, C. M. Rice and X. Wang, Cancer bioinformatics: A new approach to systems clinical medicine, BMC Bioinf., 2012, 13, 71 CrossRef PubMed.
- R. Simon, Bioinformatics in cancer therapeutics—hype or hope?, Nat. Clin. Pract. Oncol., 2005, 2, 223 CrossRef PubMed.
- V. Müller, et al., Prognostic impact of circulating tumor cells assessed with the CellSearch System™ and AdnaTest Breast™ in metastatic breast cancer patients: the DETECT study, Breast Cancer Res., 2012, 14, R118 CrossRef PubMed.
- A. A. Connor, et al., Central, But Not Peripheral, Circulating Tumor Cells are Prognostic in Patients Undergoing Resection of Colorectal Cancer Liver Metastases, Ann. Surg. Oncol., 2016, 23, 2168–2175 CrossRef PubMed.
- D. R. Rhodes, et al., ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform1, Neoplasia, 2004, 6, 1–6 CrossRef CAS PubMed.
- D. Kihara, Y. D. Yang and T. Hawkins, Bioinformatics resources for cancer research with an emphasis on gene function and structure prediction tools, Cancer Inf., 2006, 2, 25–35 CAS.
- C. Cheng, et al., Understanding transcriptional regulation by integrative analysis of transcription factor binding data, Genome Res., 2012, 1658–1667, DOI:10.1101/gr.136838.111.
- S. S. Khaleel, E. H. Andrews, M. Ung, J. DiRenzo and C. Cheng, E2F4 regulatory program predicts patient survival prognosis in breast cancer, Breast Cancer Res., 2014, 16, 486 CrossRef PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |