
Polyureas derived from CO2 and diamines: highly efficient catalysts for C–H arylation of benzene†
Zhenghui
Liu‡
ab,
Zhenzhen
Yang‡
a,
Leiduan
Hao
a,
Xinwei
Liu
ab,
Hongye
Zhang
a,
Bo
Yu
a and
Zhimin
Liu
*ab
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Colloid, Interface and Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: liuzm@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 22nd November 2016
Abstract
Polyureas derived from CO2 and diamines were effective for C–H arylation of benzene in the presence of t-BuOK, producing biaryl products with various substituents in high yields up to 97%. Dual-activation of K+ and t-BuO−, and π,π-stacking interaction between the substrate and the polymer backbone may account for the superior activity.
Direct C–H arylation of benzene is a promising reaction to construct a C–C bond within biaryl moieties,1,2 which can be achieved via catalysis over Pd,3 Rh,4 Ru,5 Ir,6 and Fe7,8 complexes and organocatalysts.7–16 Recently, a variety of N-containing molecules (e.g., 1,10-phenanthroline9–11 or its derivatives,12N,N-dimethylethane-1,2-diamine,13MOF-253 involving bipyridine moieties,14 amino-linked nitrogen heterocyclic carbenes15) and O-containing compounds (e.g., alcohols16) have been proved to effectively promote the C–H arylation of benzene in the presence of a strong base (e.g. t-BuOK). Notably, molecules incorporated with both N- and O-functionalities, such as proline,17 macrocyclic aromatic pyridone pentamer,18 vasicine19 and 2-pyridyl carbinol,20,21 exhibited synergistic effects from N- and O-functionalities. Although these homogeneous catalysts showed efficient catalytic activity, heterogeneous catalysts are more desirable for industrial applications owing to the convenience of recovering and recycling of the catalysts.22 More recently, Ma et al. reported a heterogeneous graphene oxide catalytic system for the C–H bond arylation of benzene, in which both the oxygen functional groups and the graphene π system were found to facilitate the overall reaction.23 Inspired by the above progress, we envisaged that heterogeneous polymers functionalized by both nitrogen- and oxygen-containing groups may be favourable to catalyze the direct C–H arylation of benzene to biaryl compounds in the presence of a strong base.
Polyurea (PU) is a kind of polymer containing urea functional group, which shows high resistance to solvents, acids or bases, and superior thermal stability, and is thus widely being applied in coatings and corrosion protection areas. Recently, PU has been applied in catalysis due to its strong coordinating ability with metal complex and nanoparticles, showing promising application potentials.24 For example, PU-encapsulated Pd and Pd–Cu bimetallic catalysts displayed excellent performances in the hydrogenation of styrene oxide and reduction of aryl ketones.25,26 In our previous work, PU particles were synthesized via the reaction of CO2 with 1,4-butanediamine, and the PU supported Pd nanoparticles were found to have a mean size of <3.0 nm and show high efficiency for selective hydrogenation of o-chloronitrobenzene to o-chloroaniline.27
During our continued work on PU, we discovered that PU derived from CO2 and diamines could efficiently catalyze the direct C–H arylation of benzene to biaryl compounds in the presence of t-BuOK. In particular, PU with an aromatic ring in the skeleton displayed high efficiency for benzene arylation with aryl iodides containing both electron-donating and electron-withdrawing groups, producing a series of biaryl products with various substituents in high yields up to 97%. It was indicated that dual-activation of K+ and t-BuO− by the urea unit in PU together with π,π-stacking interaction between the substrate and the polymer backbone accounted for the superior activity of the PU catalysts. In addition, the PU catalysts had high stability and easy recyclability.
PUs were synthesized via the copolymerization of CO2 with 1,4-butanediamine, 1,4-cyclohexanediamine and p-xylylenediamine, respectively, catalyzed by [n-Bu4N]2WO4 using N-methyl pyrrolidone (NMP) as the solvent (for experimental details, see the ESI†), and their chemical structures are shown in the footnote of Table 1. The formation of PUs was revealed by Fourier transform infrared (FTIR) spectroscopy, elemental analysis (Table S1, ESI†) and cross-polarization magic-angle spinning (CP/MAS) 13C NMR. In the FTIR spectra of PU (Fig. S1, ESI†), the urea unit was confirmed by the characteristic bands at 1566 cm−1 (CO–NH), 1625 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 3324 cm−1 (N–H), and the respective bands for the methylene group around 2950 cm−1.27 The presence of chemical shifts around 159 ppm in CP/MAS 13C NMR spectra of PU belonged to the carbonyl carbon of the backbone (Fig. S2, ESI†), and those at 28–48 ppm belonged to methylene carbon, together with 131–138 ppm attributed to the aromatic carbons in PU-Benz. Thermogravimetric analysis (TGA) indicated that the resulting PUs were stable up to 300 °C in an air environment (Fig. S3, ESI†), which met the demands for potential applications in heterogeneous catalysis at high temperatures. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of PU showed morphologies of a lamellar structure (Fig. S4, ESI†), which may be due to the intramolecular and intermolecular hydrogen bond formation between urea groups.28
O), 3324 cm−1 (N–H), and the respective bands for the methylene group around 2950 cm−1.27 The presence of chemical shifts around 159 ppm in CP/MAS 13C NMR spectra of PU belonged to the carbonyl carbon of the backbone (Fig. S2, ESI†), and those at 28–48 ppm belonged to methylene carbon, together with 131–138 ppm attributed to the aromatic carbons in PU-Benz. Thermogravimetric analysis (TGA) indicated that the resulting PUs were stable up to 300 °C in an air environment (Fig. S3, ESI†), which met the demands for potential applications in heterogeneous catalysis at high temperatures. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of PU showed morphologies of a lamellar structure (Fig. S4, ESI†), which may be due to the intramolecular and intermolecular hydrogen bond formation between urea groups.28
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Yieldb/% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
a Reaction conditions: PhI 0.4 mmol, benzene 4 mL, t-BuOK 1.2 mmol, PU-Benz 12.5 mg/PU-cHex 10.7 mg/PU-nBu 9.0 mg (containing 0.07 mmol urea units based on the elemental analysis results in Table S1, ESI), 130 °C, 24 h.
b Determined by GC using dodecane as an internal standard.
c 1.2 mmol t-BuONa (entry 5), t-BuOLi (entry 6), KOH (entry 7) and K3PO4 (entry 8), respectively, were added instead of t-BuOK.
d 4-Phenylbutan-2-one 0.07 mmol, amine, 0.14 mmol for entries 10–13.
e 0.4 mmol TEMPO was added to the reaction mixture.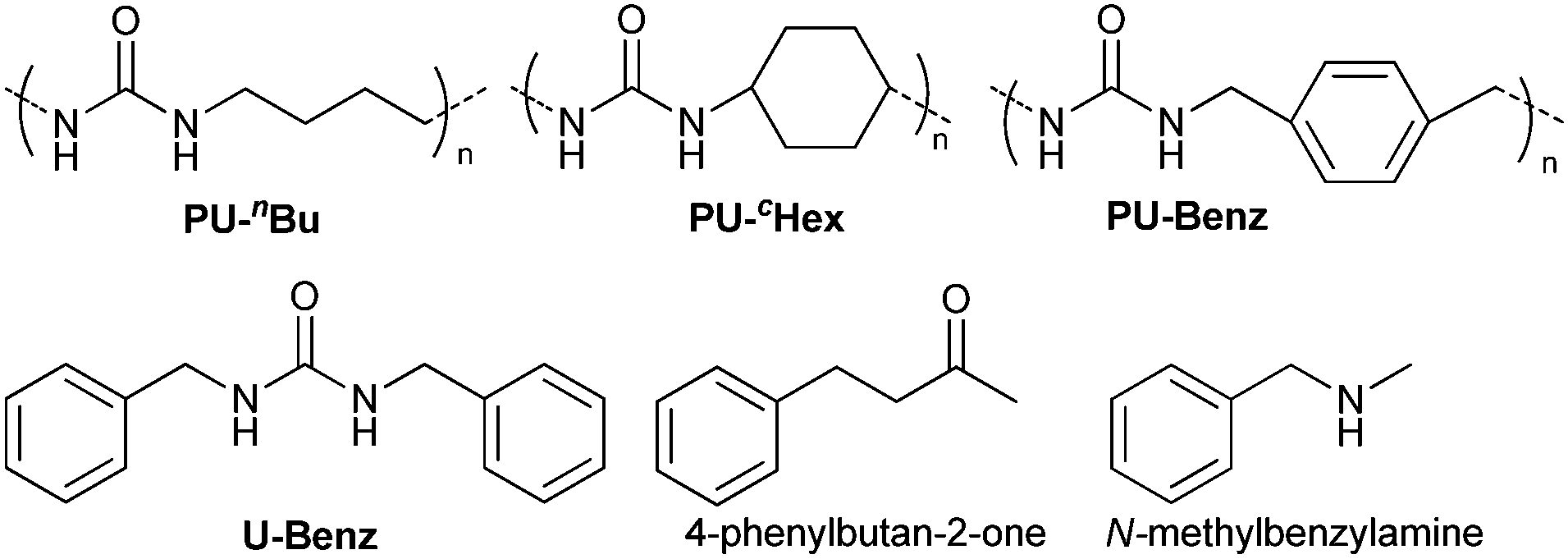
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | — | <1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | PU-Benz | 87 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | PU-cHex | 55 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | PU-nBu | 46 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5c | PU-Benz | <1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6c | PU-Benz | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7c | PU-Benz | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8c | PU-Benz | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9d | U-Benz | 65 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10d | N-Methylbenzylamine | 25 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11d | Benzylamine | 55 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12d | 4-Phenylbutan-2-one | 61 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13d | 4-Phenylbutan-2-one + N-methylbenzylamine | 88 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14e | PU-Benz | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The reaction of iodobenzene (1a) with benzene was taken as a model system to identify and optimize the key reaction parameters in the presence of PU and t-BuOK at 130 °C. As shown in Table 1, almost no biphenyl product (2a) was formed when the reaction was conducted in the absence of the PU catalysts (entry 1). Excitingly, all the resultant PUs were effective for the reaction, and especially, 87% yield of biphenyl was obtained in the presence of PU-Benz (entry 2), indicating that PU was indeed a promising catalyst for the arylation of benzene. This result was comparable to the reported heterogeneous graphene oxide (82% yield of 2a at 120 °C).23 In contrast, PU derived from aliphatic amines showed lower catalytic activities, with 55% and 46% yields of 2a being obtained in the presence of PU-cHex and PU-nBu, respectively (entries 2 vs. 3, 4), indicating that the chemical structures of PUs influenced the activities of the catalysts considerably. The π,π-stacking interaction29 between the aromatic ring of PU-Benz and the arene substrate together with the ion–π interaction of PU with t-BuOK may be favourable to facilitating the reaction.9,30 In this coupling system, a potassium cation was essential, while other bases with a tert-butoxide anion such as t-BuONa, t-BuOLi, as well as weaker potassium bases (KOH and K3PO4) (entries 5–8) were ineffective. For comparison, U-Benz as the corresponding urea monomer of PU-Benz was examined, and it exhibited inferior catalytic activity (2a yield: 65%) compared with the polymer PU-Benz (entries 9 vs. 2). This implies that the cross-linked hydrogen bonding and π,π-stacking interaction of the aromatic ring within the backbone of PU-Benz might facilitate the t-BuO− activation and further catalyse the reaction. Both the carbonyl and amino groups within the PU skeleton may show catalytic activity for C–H arylation of benzene in the presence of t-BuOK. To confirm this, several molecules with separate carbonyl and amino groups, respectively, were examined for the reaction. It was demonstrated that 25% yield of 2a was attained catalyzed by equimolar N-methylbenzylamine (Table 1, entry 10), while much higher catalytic activity was observed taking benzylamine as the catalyst (2a yield: 55%) (Table 1, entry 11). 4-Phenylbutan-2-one catalyzed C–H arylation to give 2a in 61% yield, demonstrating the superior catalytic activity of oxygen-containing groups.23 A physical mixture of homogeneous 4-phenylbutan-2-one and N-methylbenzylamine showed identical activity (2a yield: 88%) towards PU-Benz (entries 2 vs. 13). However, 4-phenylbutan-2-one was very unstable in the presence of strong base, e.g. t-BuOK, which reacted through nucleophilic attack on the carbonyl carbon and hydrogen transfer of the methylene group, as detected by 1H and 13 C NMR spectra (Fig. S5, ESI†), implying impossibility of its recyclability.21 Nonetheless, the carbonyl group of the urea unit in PUs was stable, confirmed by the fact that addition of t-BuOK to the suspension of U-Benz or PU-Benz did not result in any products detectable by NMR and FTIR (Fig. S6 and S7, ESI†). Subsequently, influences of various factors (e.g., catalyst loading, temperature, volume of benzene, amount of base and reaction time) on the reaction outcome were investigated, and the optimal reaction conditions involved the use of 12.5 mg of PU-Benz and 3 equivalents of t-BuOK at 130 °C for 24 h (Fig. S8, ESI†).
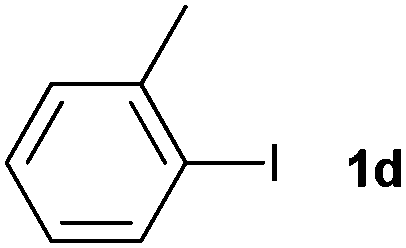
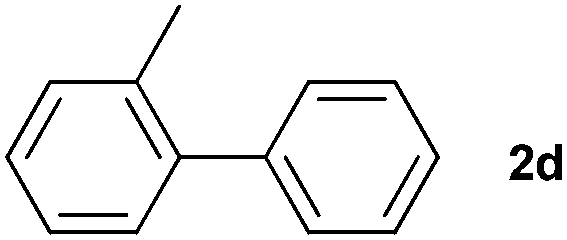
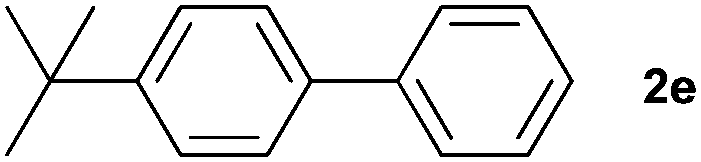
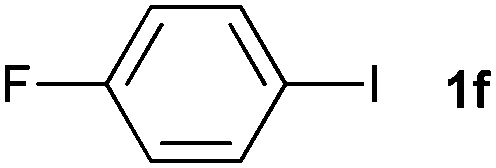
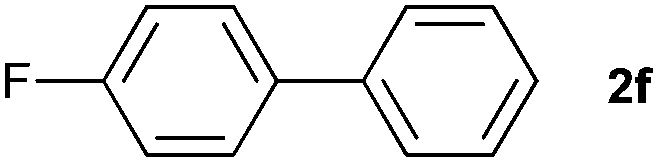
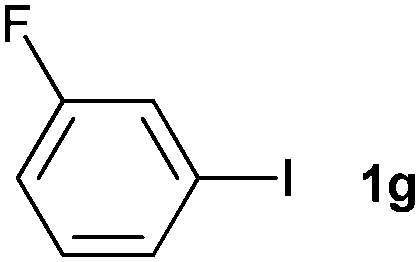
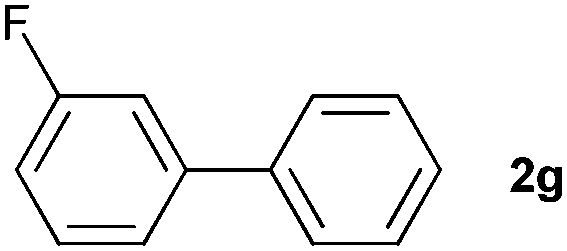
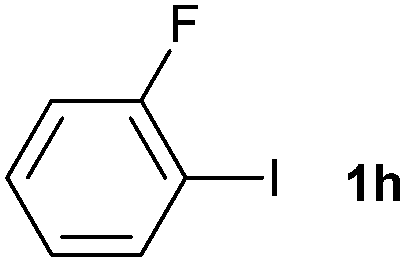
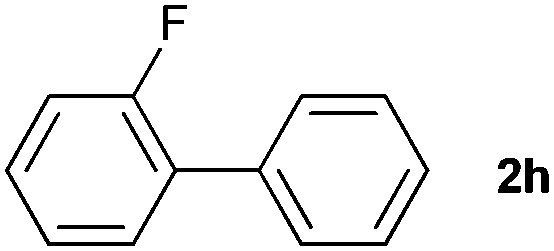
The generality of this protocol for other benzene arylation reactions was then examined and the results are summarized in Table 2. Generally, aryl iodides with electron-donating groups were more reactive than those with electron-neutral ones and those with electron-withdrawing groups. Aryl iodides with para-substituents were more reactive than those with meta- or ortho-ones owing to steric hindrance. For example, 97% yield of 2b was obtained taking 4-iodotulene (1b) as an arylation reagent, while the reactivities of iodobenzene (1a) and 1-fluoro-4-iodobenzene (1f) were much lower, giving 87% yield of 2a and 63% yield of 2f, respectively (entries 1, 2 vs. 6). In addition, the yield of 2f increased to 76% by prolonging the reaction time to 48 h. The coupling of benzene with 1-tert-butyl-4-iodobenzene (1e) was less efficient owing to the steric effect of the tert-butyl substituent, affording 58% yield of 2e, which increased to 72% within 48 h. Comparatively, only 28% yield of product (2i) was observed due to a combination of electronic and steric effects of the trifluoromethyl group of 1i (entries 5 vs. 9). The position of the substituents had a great effect on the reactivity. For methyl or fluoro-substituted iodobenzenes, the activity was decreased in the order of para- > meta- > ortho-substitution, that is, the yields decreased as 2b (97%) > 2c (85%) > 2d (74%), and 2f (63%) > 2g (15%) > 2h (10%), owing to the steric effect (entries 2–4 and 6–8). 83% yield of 2g and 81% yield of 2h were achieved by prolonging the reaction time to 72 h. Notably, less reactive 2-iodothiophene (1j) was also employed to arylate with benzene, producing 2j in 37% yield (entry 10). Aryl bromide was tested as well, but only 7% yield of biphenyl was obtained (entry 11).
| Entry | Substrate | Product | Yield/%b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1 0.4 mmol, benzene 4 mL, t-BuOK 1.2 mmol, PU-Benz 12.5 mg, 130 °C, 24 h. b Determined by GC using dodecane as an internal standard. c Yield in the bracket was obtained within 48 h. d Yield in the bracket was obtained within 72 h. e 4 mL toluene was added instead of benzene. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |
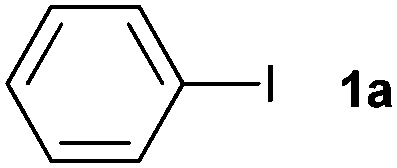
|
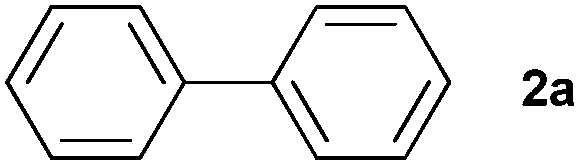
|
87 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |
|
|
97 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
|
|
85 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 |
|
|
74 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |

|
|
58(72)c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 |
|
|
63(76)c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 |
|
|
15(83)d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 |
|
|
10(81)d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 |
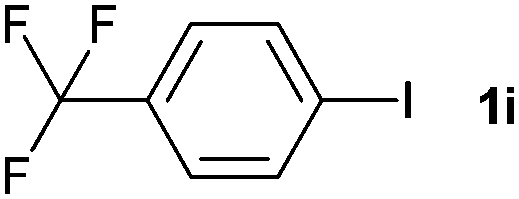
|
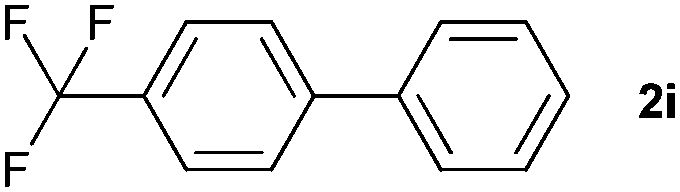
|
28 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 |

|
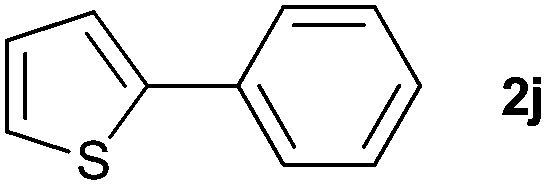
|
37 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 |

|
|
7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12e |
|
|
22/21/42 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The reusability of the catalyst PU-Benz was examined, and it was indicated that 86% yield of biphenyl 2a was still obtained after the catalyst was reused five times (Fig. 1) (for experimental details, see the ESI†), suggesting good reusability and stability. The structure of the recycled PU-Benz was well retained as detected by FTIR and CP/MAS 13C NMR (Fig. S9, ESI†).
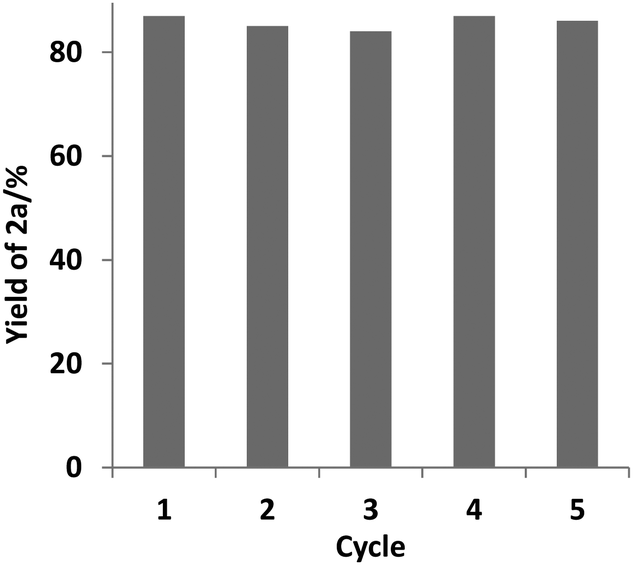 | ||
| Fig. 1 Recyclability test of PU-Benz. Reaction conditions: PhI 0.4 mmol, t-BuOK 1.2 mmol, PU-Benz 12.5 mg, benzene 4 mL, 130 °C, 24 h. Yield of 2a was determined by GC using dodecane as an internal standard. | ||
Further investigation was performed to gain a deep insight into the reaction mechanism. Firstly, consistent with the involvement of aryl radicals in the arylation reactions reported before,10,21 addition of one equivalent of free radical scavengers of TEMPO ceased the reaction and no product was obtained (Table 1, entry 14) in the presence of PU-Benz. The ESI-MS characterization of the CH3OH solution of U-Benz and t-BuOK with a 1:1 molar ratio showed a peak at 279, which was ascribed to [U-Benz + K]+ species from the coordination of K+ with the urea group. This implies that K+ may coordinate with the urea unit within the backbone of PU-Benz during the reaction process. Moreover, the interaction of the anion t-BuO− with the urea unit was also revealed by 1H NMR spectra of U-Benz with t-BuOK (Fig. S6, ESI†). The signal of the methylene group in U-Benz was located and split as dual peaks at 4.35 and 4.37 ppm by the adjacent N–H, which was merged as a single peak at 4.36 ppm after addition of equimolar t-BuOK, indicating the hydrogen bond formation between N–H and t-BuO−. This interaction was further evidenced by the signal of N–H in U-Benz located at 5.69 ppm, which became a broad one in the presence of t-BuOK. In the FTIR spectrum of PU-Benz, the sharp peak centred around 3324 cm−1 for N–H vibration obviously receded in the spectrum of the physical mixture of PU-Benz and t-BuOK (Fig. S7, ESI†).
Based on the above findings and related results reported by others,10,11,21,23 we speculated that interactions of the potassium cation and the tert-butoxide anion with the urea units and the aromatic ring in the backbone of PU-Benz acted synergistically to catalyze the C–C bond formation reaction. A radical-mediated catalytic mechanism was proposed (Scheme 1). The potassium cation and the tert-butoxide anion were activated by the urea unit in the skeleton of PU-Benz, which subsequently transferred a single electron to aryl iodide 1, affording the intermediate aryl radical anion I. And then, aryl radical II was formed by departure of an iodide anion, which then reacted with benzene to generate a biaryl radical III. Oxidation of III by the previously formed radical cation of t-BuOK produced the biaryl cation IV. Finally, deprotonation of IV by a tert-butoxide anion furnished the biaryl product 2. Notably, π,π-stacking interaction between the aryl ring within the polymer chains with the arene substrate played an important role in attracting the substrate to the vicinity of the catalytic center, and stabilizing the resultant intermediates.
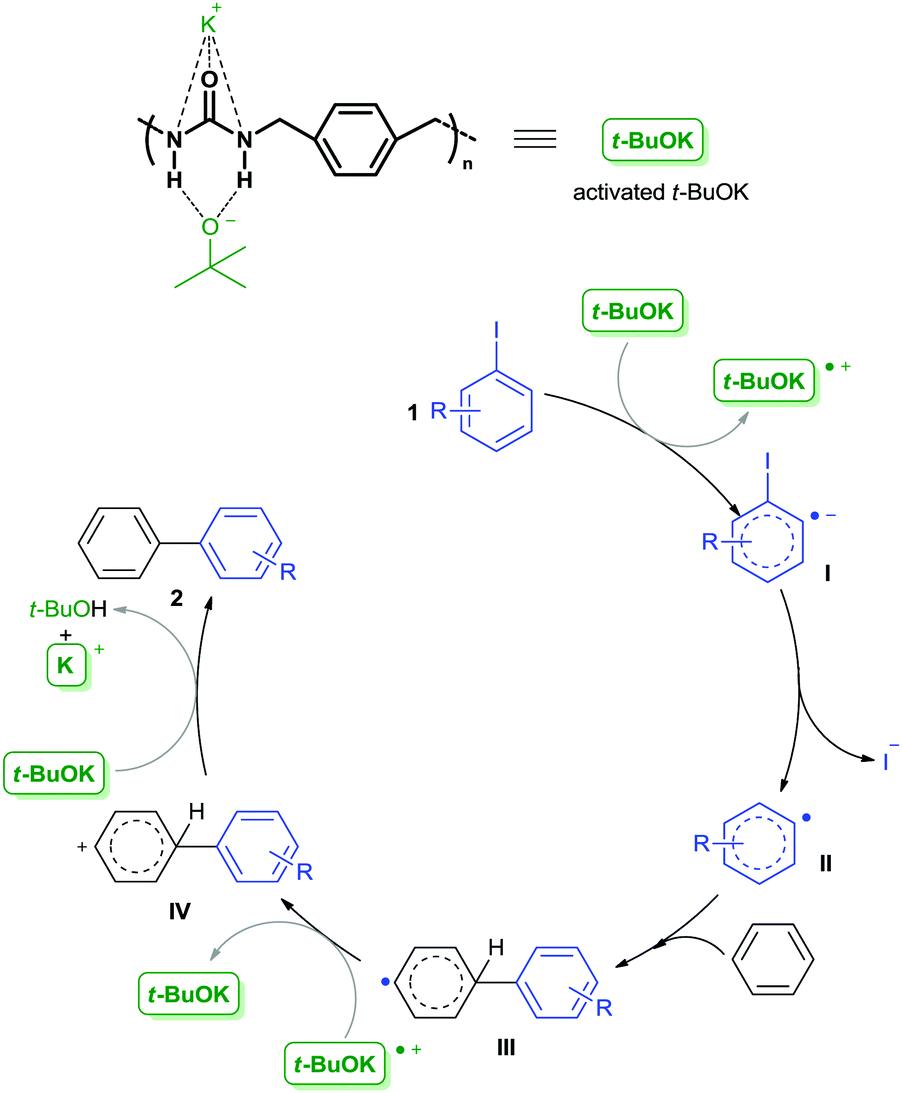 | ||
| Scheme 1 Proposed mechanism for the PU-Benz/t-BuOK catalyzed C–H arylation. | ||
In conclusion, PUs derived from CO2 and diamines, especially PU-Benz with an aromatic ring in the skeleton, was proved to be an efficient heterogeneous catalytic material for C–H arylation of benzene in the presence of t-BuOK, together with a broad substrate scope, high stability and easy recyclability. Both urea and benzene functionalities in the polymer backbone were important for the catalytic process, realizing dual-activation of K+ and t-BuO−, together with π,π-stacking interaction between the substrate and the polymer backbone, thus resulting in good catalytic activity. Based on the efficient coordination of urea units with alkali metal cations, applications of polyurea materials in hydrogen-transfer reactions are under investigation.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant no. 21402208, 21533011, and 21125314).Notes and references
- T. L. Chan, Y. Wu, P. Y. Choy and F. Y. Kwong, Chem. – Eur. J., 2013, 19, 15802–15814 CrossRef CAS PubMed.
- C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- S. Proch and R. Kempe, Angew. Chem., Int. Ed., 2007, 46, 3135–3138 CrossRef CAS PubMed.
- L. Ackermann, J. Pospech and H. K. Potukuchi, Org. Lett., 2012, 14, 2146–2149 CrossRef CAS PubMed.
- K.-i. Fujita, M. Nonogawa and R. Yamaguchi, Chem. Commun., 2004, 1926–1927 RSC.
- W. Liu, H. Cao and A. Lei, Angew. Chem., Int. Ed., 2010, 49, 2004–2008 CrossRef CAS PubMed.
- F. Vallée, J. J. Mousseau and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 1514–1516 CrossRef PubMed.
- C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed.
- H. Yi, A. Jutand and A. Lei, Chem. Commun., 2015, 51, 545–548 RSC.
- J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed.
- E. Shirakawa, K.-I. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537–15539 CrossRef CAS PubMed.
- W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS PubMed.
- H. Liu, B. Yin, Z. Gao, Y. Li and H. Jiang, Chem. Commun., 2012, 48, 2033–2035 RSC.
- W.-C. Chen, Y.-C. Hsu, W.-C. Shih, C.-Y. Lee, W.-H. Chuang, Y.-F. Tsai, P. P.-Y. Chen and T.-G. Ong, Chem. Commun., 2012, 48, 6702–6704 RSC.
- W. Liu, F. Tian, X. Wang, H. Yu and Y. Bi, Chem. Commun., 2013, 49, 2983–2985 RSC.
- K. Tanimoro, M. Ueno, K. Takeda, M. Kirihata and S. Tanimori, J. Org. Chem., 2012, 77, 7844–7849 CrossRef CAS PubMed.
- H. Zhao, J. Shen, J. Guo, R. Ye and H. Zeng, Chem. Commun., 2013, 49, 2323–2325 RSC.
- S. Sharma, M. Kumar, V. Kumar and N. Kumar, Tetrahedron Lett., 2013, 54, 4868–4871 CrossRef CAS.
- Y. Wu, P. Y. Choy and F. Y. Kwong, Org. Biomol. Chem., 2014, 12, 6820–6823 CAS.
- J. P. Barham, G. Coulthard, R. G. Kane, N. Delgado, M. P. John and J. A. Murphy, Angew. Chem., Int. Ed., 2016, 55, 4492–4496 CrossRef CAS PubMed.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- Y. Gao, P. Tang, H. Zhou, W. Zhang, H. Yang, N. Yan, G. Hu, D. Mei, J. Wang and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 3124–3128 CrossRef CAS PubMed.
- S. K. Dey, N. de Sousa Amadeu and C. Janiak, Chem. Commun., 2016, 52, 7834–7837 RSC.
- J.-Q. Yu, H.-C. Wu, C. Ramarao, J. B. Spencer and S. V. Ley, Chem. Commun., 2003, 678–679 RSC.
- G. D. Yadav and Y. S. Lawate, J. Supercrit. Fluids, 2011, 59, 78–86 CrossRef CAS.
- L. Hao, Y. Zhao, B. Yu, H. Zhang, H. Xu, J. Xu and Z. Liu, J. Colloid Interface Sci., 2014, 424, 44–48 CrossRef CAS PubMed.
- H. Zhang, Z. Yang, W. Gan, Y. Zhao, B. Yu, H. Xu, Z. Ma, L. Hao, D. Chen, S. Miao and Z. Liu, Chem. – Eur. J., 2015, 21, 14608–14613 CrossRef CAS PubMed.
- P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS PubMed.
- R. Kumpf and D. Dougherty, Science, 1993, 261, 1708–1710 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and additional data and figures. See DOI: 10.1039/c6nj02785g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |