
α-Fe2O3/TiO2 3D hierarchical nanostructures for enhanced photoelectrochemical water splitting†
Hyungkyu
Han
a,
Francesca
Riboni
b,
Frantisek
Karlicky
ac,
Stepan
Kment
a,
Anandarup
Goswami
a,
Pitchaimuthu
Sudhagar
d,
Jeongeun
Yoo
b,
Lei
Wang
b,
Ondrej
Tomanec
a,
Martin
Petr
a,
Ondrej
Haderka
a,
Chiaki
Terashima
d,
Akira
Fujishima
d,
Patrik
Schmuki
*be and
Radek
Zboril
*a
aRegional Centre of Advanced Technologies and Materials, Department of Physical Chemistry, Faculty of Science, Palacky University, Slechtitelu 11, 783 71 Olomouc, Czech Republic. E-mail: radek.zboril@upol.cz
bDepartment of Materials Science and Engineering, University of Erlangen-Nuremberg, Martensstrasse 7, D-91058 Erlangen, Germany
cDepartment of Physics, Faculty of Science, University of Ostrava, 30. dubna 22, 701 03 Ostrava, Czech Republic
dPhotocatalysis International Research Center, Research Institute for Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan
eDepartment of Chemistry, Faculty of Science, King Abdulaziz University, P.O. Box 80203, Jeddah 21569, Saudi Arabia. E-mail: schmuki@ww.uni-erlangen.de
First published on 2nd November 2016
Abstract
We report the fabrication of 3D hierarchical hetero-nanostructures composed of thin α-Fe2O3 nanoflakes branched on TiO2 nanotubes. The novel α-Fe2O3/TiO2 hierarchical nanostructures, synthesized on FTO through a multi-step hydrothermal process, exhibit enhanced performances in photo-electrochemical water splitting and in the photocatalytic degradation of an organic dye, with respect to pure TiO2 nanotubes. An enhanced separation of photogenerated charge carriers is here proposed as the main factor for the observed photo-activities: electrons photogenerated in TiO2 are efficiently collected at FTO, while holes are transferred to the α-Fe2O3 nanobranches that serve as charge mediators to the electrolyte. The morphology of α-Fe2O3 that varies from ultrathin nanoflakes to nanorod/nanofiber structures depending on the Fe precursor concentration was shown to have a significant impact on the photo-induced activity of the α-Fe2O3/TiO2 composites. In particular, it is shown that for an optimized photo-electrochemical structure, a combination of critical factors should be achieved such as (i) TiO2 light absorption and photo-activation vs. α-Fe2O3-induced shadowing effect and (ii) the availability of free TiO2 surface vs. α-Fe2O3-coated surface. Finally, theoretical analysis, based on DFT calculations, confirmed the optical properties experimentally determined for the α-Fe2O3/TiO2 hierarchical nanostructures. We anticipate that this new multi-step hydrothermal process can be a blueprint for the design and development of other hierarchical heterogeneous metal oxide electrodes suitable for photo-electrochemical applications.
Introduction
Titanium dioxide (TiO2) represents the most widely investigated semiconductor photocatalyst for e.g., photo-oxidation reactions, photo-electrochemical (PEC) water splitting, and anodes in solar cells.1–3 The intrinsic key advantages of TiO2 are well established (i.e., it is cheap, abundant, environmentally friendly, and corrosion-resistant); however, (i) its relatively large optical band-gap (3.0–3.2 eV) that hampers the use of sunlight to promote photoreactions, (ii) the high rate of recombination of photogenerated electron/hole (e−/h+) pairs and (iii) the low rate of charge transfer to reactants still represent serious challenges to be overcome. In particular, the generation of long-living and spatially separated charge carriers appears of primary importance. For this, one-dimensional arrangements of TiO2 (e.g., in the form of nanotubes, nanorods and nanofibers grown on a conductive substrate) are beneficial, since the orthogonal carrier separation in these structures is facilitated by the preferential percolation of e− to the back contact and the parallel accumulation of h+ at the semiconductor/electrolyte interface.3,4 Also, combining TiO2 with another semiconductor with a suitable band gap and band offsets has been often proposed as a valuable approach to achieve larger e−/h+ separation.5 In a composite photocatalytic system, the intimate contact between two or more semiconductors promotes the mobility of charge carriers between the different components, enhancing their separation and, hence, the photoactivity of the composite system compared to that of the single counterparts. Owing to the availability of a large number of semiconducting materials, an adequate selection in terms of band alignment (specific for different reactions) is clearly required. For instance, coupling TiO2 and α-Fe2O3 nanostructures is, in principle, a promising approach for increasing the efficiency of titania for PEC applications, as long as TiO2 is the light absorbing material and the major contributor to the photogeneration of charge carriers. With this arrangement, from the mechanistic point of view, electrons photopromoted in TiO2 are transferred to the back contact (also, taking advantage of the long electron diffusion length typical of TiO2 nanotubes, i.e., ∼20 μm),6 while holes can be trapped in the valence band of α-Fe2O3, which that lies at a more negative potential than that of TiO2, and easily conveyed to the electrolyte. This spatial separation reduces e−/h+ recombination probability and leads to an enhanced performance of the α-Fe2O3/TiO2 composite materials. Clearly, an essential prerequisite, often overlooked in the literature,7 is that the hematite layer does not shade the TiO2 nanostructures and allows its photoactivation. Indeed, if e− are photo-generated in α-Fe2O3, they cannot be transferred to TiO2 due to an unfavorable conduction band alignment of the two oxides. Hence, in this regard, also a fine tuning of the composite nanoarchitecture has to be achieved to avoid the undesired detrimental effect of classic α-Fe2O3/TiO2 heterostructures.7We report the fabrication of α-Fe2O3/TiO2 3D hierarchical nanostructures, with a specific composition and tailored architecture that meet all the aforementioned requirements. Aligned TiO2 nanotubes of a pure anatase composition (highly desired for PEC applications) were grown on FTO glass via a sacrificial template-based hydrothermal approach; their surface was modified with α-Fe2O3 nanostructures, whose morphologies varied from thin nanoflakes to nanorods, to surface agglomerates, depending on the amount of Fe precursor. This approach was shown to have a significant impact on the photo-induced activity of the α-Fe2O3/TiO2 composites. In particular, under specific preparation conditions, α-Fe2O3 nanobranches were grown perpendicular to the TiO2 NTs, leaving the TiO2 surface partially exposed to both the electrolyte and light; this configuration provided a twofold increase in the PEC and photocatalytic (PC) activities of bare TiO2 nanotubes, since it enabled an optimized light absorption and photoactivation of TiO2, coupled with an efficient collection of photo-generated charges (i.e., e− to FTO and h+ to α-Fe2O3). We believe that the experimental control over the morphology and related light-promoted activity of α-Fe2O3/TiO2 hierarchical materials can be extended to other semiconductor oxide combinations and be adopted as a defined strategy to improve their performance in photo-electrochemical reactions.
Experimental
Fabrication of TiO2 nanotube arrays on FTO glass
The procedure for the fabrication of α-Fe2O3/TiO2 3D hierarchical nanostructures is reported in Scheme 1. First, a thin ZnO seed layer was deposited on FTO glass by spin coating a 20 mM zinc acetate dihydrate [Zn(O2CCH3)2·2H2O] aqueous solution at 4000 rpm for 35 s. ZnO nanorods (NRs), which served as a sacrificial template for the growth of TiO2 nanotubes (see below), were grown hydrothermally from the seed layer at 85 °C for 10 h starting from an aqueous solution of 25 mM zinc nitrate hexahydrate [Zn(NO3)2·6H2O] and 25 mM hexamethylenetetramine (C6H12N4). The as-synthesized ZnO NRs on FTO were immersed for 30 min in an aqueous solution containing 75 mM ammonium hexafluorotitanate [(NH4)2TiF6] and 0.2 M boric acid (H3BO3). (NH4)2TiF6 hydrolyzes to TiOx on the individual ZnO nanorods, and the ZnO template is simultaneously dissolved in the acidic environment (where acids are produced by (NH4)2TiF6 hydrolysis and H3BO3). To achieve a complete removal of residual ZnO inside the TiO2 tubes, the NT arrays were immersed in a 0.5 M boric acid solution for 1 h. Finally, the samples were rinsed with DI water and annealed in Ar at 500 °C for 30 min.8,9 | ||
| Scheme 1 Schematic representation of the α-Fe2O3/TiO2 3D hierarchical nanostructure formation process: (a) first a ZnO seed layer is deposited on FTO and (b) ZnO nanorods are grown; (c) afterwards, TiO2 nanotubes are grown via a hydrothermal process and the ZnO sacrificial template is simultaneously dissolved; (d) finally, α-Fe2O3 nanobranches are formed on the surface of the TiO2 NTs. | ||
Preparation of Fe2O3/TiO2 hierarchical nanotube arrays on FTO glass
TiO2 nanotubes on FTO glass were immersed for 2 days at room temperature in an aqueous solution of iron(III) nitrate nonahydrate [Fe(NO3)3·9H2O], containing different amounts of Fe precursor (0.01–0.05 M). The samples, immersed in the iron(III) solution, were then transferred to an oven and kept at 90 °C for 2 h. Finally, they were rinsed with DI water and annealed in air at 450 °C for 2 h to convert amorphous FeOOH into crystalline α-Fe2O3.Following the same procedure, pure α-Fe2O3 layers were also grown directly on FTO glass from 0.01–0.05 M precursor solutions.
Characterization
For the morphological characterization of the α-Fe2O3/TiO2 hierarchical composites, a field-emission scanning electron microscope (FE-SEM, Hitachi S4800, Japan), a field-emission transmission electron microscope (FE-TEM, JEOL 2010F) and a high-resolution transmission electron microscope (HRTEM, FEI TITAN G2 60-300) were employed. The crystallographic properties of the materials were analyzed by X-ray diffraction (XRD) performed with an X'pert Philips MPD (equipped with a Panalytical X'celerator detector), using iron-filtered Co-Kα radiation (λ = 1.78901 Å, 40 kV, 30 mA). XRD data were then converted to Cu-Kα radiation, according to the relationships in (θCu) = (λCu/λCo) × sin(θCo), where λCu is 1.54056 Å. X-ray photoelectron spectroscopy (XPS, PHI 5600, US) was used to characterize the chemical composition of the materials.Photo-electrochemical characterization and photocatalytic activity
The photo-electrochemical experiments were carried out in a three-electrode configuration, under simulated AM 1.5G (100 mW cm−2) illumination provided by a solar simulator (150 W Xe with optical filter). TiO2 and α-Fe2O3/TiO2 NT electrodes served as photoanodes, an Ag/AgCl (3 M KCl) as the reference electrode, and a platinum foil as the counter electrode. 1 M NaOH aqueous solution was used as the electrolyte, and the solution was bubbled with N2 for 30 min prior to measurements. Photocurrent versus voltage (I–V) characteristics were recorded by scanning the potential from −0.8 to 1.3 V (vs. Ag/AgCl (3 M KCl)) with a scan rate of 10 mV s−1 using a Series 300 Potentiostat/Galvanostat/ZRA potentiostat. Photocurrent transients for the TiO2 and Fe2O3/TiO2 NT electrodes were measured at +0.26 V (vs. Ag/AgCl (3 M KCl)) in 1 M NaOH.Electrochemical impedance was measured in a Princeton Applied Research potentiostat (PARSTAT 2273). The frequency was varied in the 1 MHz–1 mHz range, with a potential amplitude of 10 mV. The measurements were carried out in a three-electrode configuration, with Pt serving as the counter electrode and Ag/AgCl as the reference electrode, at +0.26 V (vs. Ag/AgCl (3 M KCl)) and under simulated AM 1.5G (100 mW cm−2) illumination provided by a solar simulator (150 W Xe with an optical filter). 1 M NaOH was used as the electrolyte.
The photocatalytic degradation of Rhodamine B (RhB) was carried out in a 5 mL quartz cuvette filled with a 1 mM dye solution. A 300 W Xe lamp, equipped with an AM 1.5G filter, was used as an irradiation source and the degradation of RhB was monitored with a Lambda XLS – Perkin Elmer spectrophotometer measuring the decrease in the intensity of the dye main absorption band (λ = 554 nm) from samples withdrawn from the photoreactor every 30 min. Overall, each photodegradation experiment lasted for 2 h.
Theoretical analysis
We performed electronic structure calculations using a density functional theory (DFT) corrected by an additional Hubbard-like term (DFT+U). For modeling bulk anatase TiO2 and bulk α-Fe2O3 hematite, the experimental lattice constants a1 = 3.785 Å and c1 = 9.514 Å (tetragonal unit cell)10 and a2 = 5.0355 Å and c2 = 13.7471 Å (rhombohedral unit cell)11 were used, respectively. We used the Perdew–Burke–Ernzerhof (PBE)12 generalized gradient approximation (GGA) to DFT and we selected the effective on-site Coulomb interaction parameters U = 7.5 eV for Ti atoms and U = 4.3 eV for Fe atoms. This choice provides good agreement with the band gaps of bulk TiO2 and bulk α-Fe2O3 (Fig. S1†). We also created the corresponding layers. The predicted dominant growth face of the α-Fe2O3 surface is the (0001) surface. Therefore, following ref. 13, we used a single-iron terminated (0001) layer. For simplicity, we used an oxygen terminated (001) TiO2 anatase layer, following ref. 14. Because of the lattice mismatch between α-Fe2O3 (0001) and TiO2 (001) surfaces, it was necessary to introduce a larger computational supercell for simulating the α-Fe2O3 (0001)/TiO2 (001) interface. Within all those computed by using the Cell Match utility,15 the 15.14 Å × 38.04 Å supercell with 142 atoms (36 Fe, 86 O, 20 Ti) was selected – the thickness of the layer was in some cases reduced to 81 atoms (18 Fe, 51 O, 12 Ti).All the DFT+U calculations were performed using the VASP code16 with the PAW formalism.17 For the initial bulk calculations, the first Brillouin zones were sampled by 10 × 10 × 4 k-points. For surface calculations, 10 × 10 × 2 and 7 × 7×1 k-points were used for TiO2 (001) and α-Fe2O3 (0001), respectively, and the atomic positions were relaxed until the change in forces was less than 10−2 eV A−1. Finally, for the large supercell of TiO2/Fe2O3 interfaces, 2 × 1 × 2 k-points were used and atomic positions were relaxed until the change in energy was less than 10−3 eV. A cut-off energy of 300 eV was applied together with normal accuracy of VASP. The optical absorption spectrum corresponded to the imaginary part of the dielectric function ε(ω). A Gaussian broadening of 50 meV and 128 sampling points were used for the dielectric function.
Results and discussion
Materials characterization
Scheme 1 represents the procedure for growing α-Fe2O3-branched TiO2 nanotubes (Fe2O3/TiO2 NTs) on FTO glass. In detail, upon the deposition of a ZnO seed layer (a), ZnO NRs are hydrothermally grown on the seeded substrate (b) and subsequently converted to TiO2 NTs via a liquid-phase deposition, with the simultaneous dissolution of the ZnO backbone, i.e., the sacrificial template (c). Due to the lattice mismatch between FTO and ZnO, the seeding procedure is crucial as it overcomes the high-energy nucleation step typical of hydrothermal methods based on a crystal nucleation-growth sequence.18 According to previous studies, the selective etching of the ZnO NR template may be ascribed to fluoroboric acid (HBF4) formed in solution from the Ti-precursor hydrolysis in combination with H3BO3.8 SEM images (Fig. 1a, S2 and S3(a)†) show that vertically aligned TiO2 nanotubes (∼1.5 μm in thickness and with a ∼80 nm diameter) are formed well anchored on the FTO substrate. TEM and selected-area electron diffraction (Fig. 2(a)) confirm that, upon annealing, amorphous TiO2 is converted into crystalline anatase. The NTs exhibit ∼20 nm thick walls and a lattice fringe d = 3.5 Å (inset of Fig. 2(a)) that is assigned to the interplanar distance of the (101) plane of anatase. This is also consistent with the XRD results showing a full anatase phase composition for pure TiO2 NTs (Fig. 3(a)). Finally, evidence for the formation of pure TiO2 NTs on FTO glass is also provided by energy-dispersive X-ray (EDX) analysis (Fig. S4†), where only the peaks corresponding to Ti, O and Sn (from the FTO support) can be observed. | ||
| Fig. 1 Top view SEM images of (a) TiO2 NTs and (b–f) α-Fe2O3/TiO2 NT composites prepared from Fe(III) solutions with different hematite precursor concentrations (i.e., 0.01–0.05 M). | ||
 | ||
| Fig. 2 (a) TEM image of TiO2 NTs; insets: HRTEM image (lower corner) and the corresponding SAED pattern (upper corner). (b) TEM image of 0.02 Fe2O3/TiO2 NTs; insets: HRTEM image (lower corner) and the corresponding SAED pattern (upper corner). (c) HAADF image of 0.02 Fe2O3/TiO2 NTs. (d–f) Ti, O and Fe elemental mapping of the 0.02 Fe2O3/TiO2 NT sample. | ||
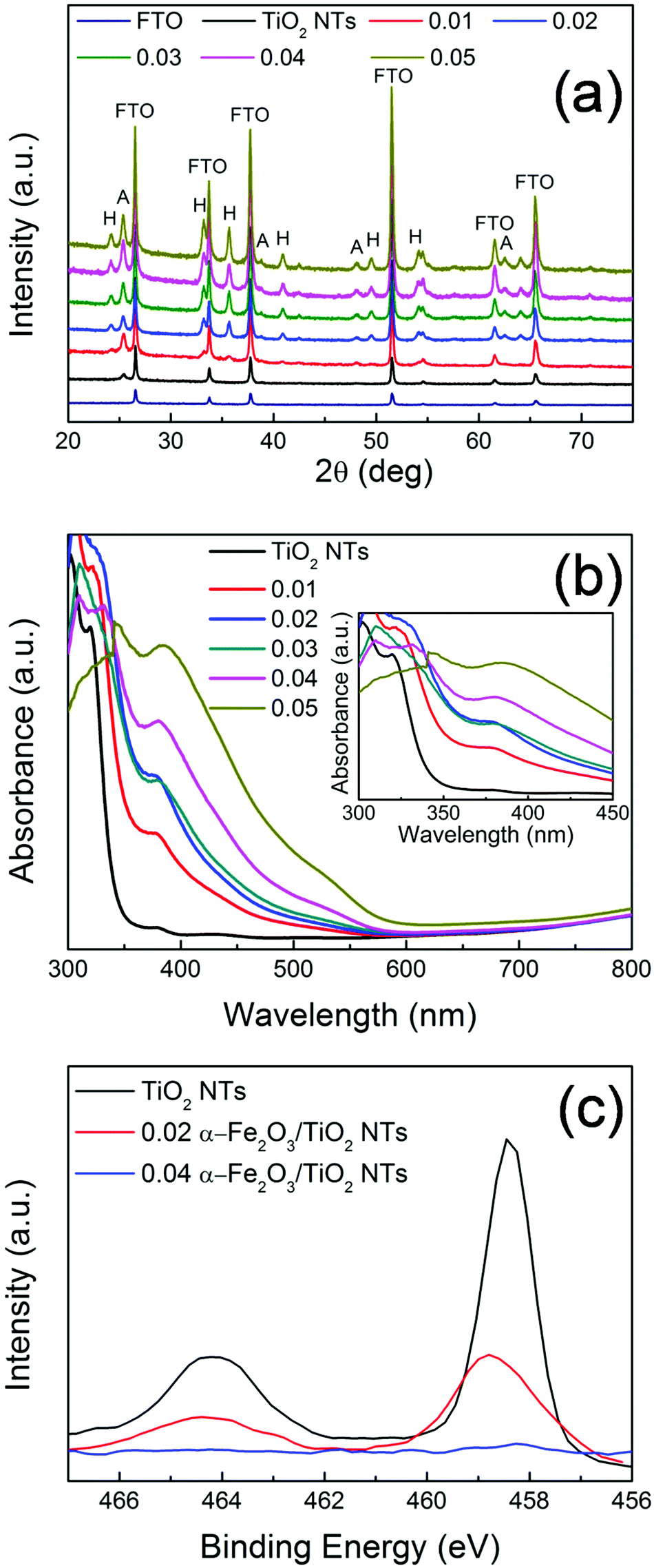 | ||
| Fig. 3 (a) XRD of TiO2 NTs and α-Fe2O3/TiO2 NT composites; A = anatase reflections, H = hematite reflections. (b) UV-vis absorption spectra of TiO2 NTs and α-Fe2O3/TiO2 NT composites; the inset shows the absorption characteristics of the samples in the 300–450 nm wavelength region. (c) XPS spectra of TiO2 NTs, 0.02 and 0.04 Fe2O3/TiO2 NT composites. In (a, b) 0.01–0.05 labels indicate the different concentrations of the starting Fe(III) precursor solutions. | ||
Immersion of TiO2 NTs in the Fe(NO3)3 solution at 90 °C results in the seeding and growth of FeOOH-branches on TiO2 NTs, that is, under optimized conditions the surface of TiO2 nanotubes is active for FeOOH nucleation (Scheme 1(d)). Different concentrations of Fe-precursor were investigated (0.01–0.05 M) that correspondingly led to different nanoarchitectures (i.e., from a homogeneous thin layer to dense and packed nanobranches/agglomerates); annealing at 450 °C in air for 2 h converted the initially amorphous FeOOH into crystalline α-Fe2O3. Table 1 summarizes the diameter and length of the α-Fe2O3 nanostructures, as a function of the Fe(NO3)3 solution concentration. Except for the 0.01 Fe2O3/TiO2 sample that exhibits a limited thickening of the TiO2 tube walls consistent with the formation of a conformal hematite shell layer (Fig. 1(b)), in all the other cases, a 3D-hierarchical α-Fe2O3/TiO2 composite is formed (Fig. 1, S2, S3† and Scheme 1(d)). In detail, for the 0.02 Fe2O3/TiO2 sample, short α-Fe2O3 nanobranches (d ∼ 10 nm, l ∼ 30–140 nm) grow on the TiO2 NT surface; a further increase in the precursor concentration results instead in longer and more densely packed nanobranches. The observed trend suggests that the thin FeOOH conformal layer that forms on TiO2 tubes at very low precursor concentrations acts as a seed layer for the growth of hierarchical nanostructures promoted by a larger amount of Fe3+ ions in solution.
| [Fe(NO3)3] (M) | d (nm) | Length (nm) |
|---|---|---|
| 0.01 | n/a | ∼10 (shell) |
| 0.02 | 8–10 | 30–140 |
| 0.03 | 20–30 | 60–140 |
| 0.04 | 40–50 | 80–250 |
| 0.05 | 30–50 | 80–280 |
However, in contrast to the formation of energy-minimized structures (i.e., the nanobranches) for 0.02–0.04 M Fe(NO3)3 solutions, when the iron precursor concentration exceeds the optimum (i.e., for [Fe(NO3)3] > 0.04 M), the Fe3+ species adsorbed on the surface of TiO2 NTs tend to aggregate and promote the formation of iron oxide-based clusters (see for instance the ∼300 nm long aggregates for the 0.05 M Fe(NO3)3 solution in Fig. 1(f), S2 and S3(f)†).
The formation of surface aggregates is certainly to be avoided in heterocomposite materials for light-induced applications where the core structure (in this case, the TiO2 nanotubes) is the major photoactive semiconductor (see below). HRTEM, exemplarily shown for the 0.02 Fe2O3/TiO2 sample (Fig. 2(b)), confirms (i) the formation of a 3D-Fe2O3/TiO2 nanobranched architecture and (ii) the conversion of amorphous FeOOH into crystalline α-Fe2O3 hematite, upon adequate annealing. In line with this, the distance between adjacent lattice fringes (d = 2.5 Å and d = 3.7 Å) in the α-Fe2O3 nanobranches can be assigned to the interplanar distance of the (110) and (012) planes of hematite, respectively (inset of Fig. 2(b)).19–21
HAADF image and EDX elemental mapping (Fig. 2(c–f)) clearly show a TiO2 nanotube-backbone with α-Fe2O3 dendritic nanostructures and also confirm the formation of a heterocomposite material where Ti, O and Fe co-exist. In this regard, experimental evidence is also provided by means of Auger Electron Spectroscopy (AES) horizontal mapping across the α-Fe2O3/TiO2 layers, performed for the different Fe-precursor solutions. As shown by the concentration scans of Ti, O and Fe superimposed on the original SEM image of the 0.02 Fe2O3/TiO2 sample (Fig. S5†), iron concentration peaks in the outer-shell and extends up to ∼60 nm from the TiO2 NT walls; correspondingly, titanium concentration is higher in the core, while oxygen is homogeneously distributed within the inner and outer layers, in line with the formation of two distinct oxides that is, Fe2O3 and TiO2. These observations on the morphology and chemical composition of 0.02 Fe2O3/TiO2 also hold for the other Fe-precursor concentrations; see, for instance, HAADF images and AES horizontal mapping of 0.01 and 0.03 Fe2O3/TiO2 in Fig. S6 and S7,† respectively. In particular, the horizontal mapping of 0.01 Fe2O3/TiO2 shows a lower amount of α-Fe2O3 and, most importantly, a ∼10 nm thick hematite layer that covers the TiO2 inner backbone (Fig. S6(g)†).
XRD patterns of the different heterocomposites are reported in Fig. 3. Aside from the reflections assigned to FTO (i.e., SnO2 cassiterite – see also Fig. S8†), all the other peaks belong to TiO2 (A) and α-Fe2O3 (H). Typically, TiO2 nanostructures grown via the hydrothermal method on FTO are mainly composed of rutile phase22 due to a favorable matching between rutile and cassiterite crystal lattices.23 Here, contrary to this expectation and in line with TEM (Fig. 2a), pure anatase TiO2 nanotubes were obtained (main reflections at 2θ ∼ 25.4°), most likely due to the presence of the ZnO sacrificial template that is dissolved during the tubes formation and that seemingly prevents TiO2 preferential seeding in the form of rutile.9
Besides the low-intensity peaks (e.g., 2θ ∼ 25°) corresponding to the (012) plane, the most intense diffraction peaks of α-Fe2O3 (i.e., 2θ ∼ 34° and 36°) correspond to the (104) and (110) planes, respectively. This, combined with TEM analysis (Fig. 2b), confirms the polycrystalline nature of the film.24,25 It is noteworthy that the (110) orientation of hematite, with its surface termination dominated by Fe(III) ions features a high conductivity.24–26 In addition, the prevalence of the (110) reflection indicates a strong preferential orientation growth of hematite with the [110] axis vertical to the substrate. Both aspects are beneficial for facilitating charge transfer from the substrate (TiO2) to the electrolyte, through α-Fe2O3, in view of photo-electrochemical applications (see discussion below).27
The optical properties of the investigated samples were determined by UV-vis spectroscopy (Fig. 3(b)). All the samples show (or partially maintain) the typical absorption edge at ca. 380–400 nm owing to TiO2 optical band-gap excitation (∼3.2 eV). In addition, the α-Fe2O3/TiO2 heterocomposites also exhibit absorption features that extend to the visible region. In particular, the light absorption onset around 600 nm corresponds to the ∼2.1 eV optical band-gap of hematite.24,28 This reflects the bi-composite nature of the 0.01–0.05 Fe2O3/TiO2 samples that exhibit the distinct optical properties of both oxide counterparts and indicates a tandem contribution of the two components to the photo-electrochemical activity of the investigated materials.
The effect of the bi-composite nature on the optical properties of α-Fe2O3/TiO2 is also confirmed by theoretical calculations performed on a TiO2 layer and an α-Fe2O3/TiO2 heterocomposite layer, modeled as in Fig. 4a and S9.† DFT calculations revealed an absorption edge for pure anatase (001) TiO2 in the 380–400 nm range (black curve in Fig. 4b), while the presence of α-Fe2O3 on TiO2 significantly extends the simulated optical absorption to the visible range (red curve in Fig. 4b). This trend is qualitatively in line with the experimental absorption spectra reported in Fig. 3b.
 | ||
| Fig. 4 (a) Schematic representation of α-Fe2O3 nanobranches on TiO2 nanotubes. (b) Optical absorption spectra of pure TiO2 and α-Fe2O3/TiO2 composite, determined through a DFT+U method. | ||
Moreover, a more careful analysis of the experimental UV-vis absorption characteristics (inset of Fig. 3b) points out that the structure decorated with the largest amount of α-Fe2O3 (i.e., 0.05 M Fe(NO3)3) shows the lowest absorbance in the 300–350 nm range, where typically TiO2 absorbs photons. This is attributed to a “shading effect”, i.e., the increase in the density and thickness of hematite-based nanobranches optically shades the underneath structure, so that TiO2 is actually exposed to a lower specific photon flux and light absorption consequently drops. The shading effect on TiO2, that may also physically prevent the percolation of the electrolyte to the tube surface with a detrimental consequence on the PEC activity, is also confirmed by XPS (Fig. 3(c)): for pure TiO2 and α-Fe2O3/TiO2 heterocomposites with a small amount of hematite (e.g., 0.02 Fe2O3/TiO2), the typical Ti 2p3/2 and 2p1/2 peaks were observed at ca. 458 eV and 464 eV, respectively; however, for larger nominal amounts of α-Fe2O3, the intensity of Ti 2p features drastically drops (it nearly vanishes for 0.04 Fe2O3/TiO2). This further supports the observation that, with increasing iron precursor concentration, longer and denser hematite nanobranches grow on the supporting TiO2 tubes, eventually leading to clusters formation that act as a barrier for both light penetration and electrolyte percolation.
Therefore, for an optimized photo-electrochemical structure, a combination of critical factors should be achieved such as (i) TiO2 light absorption and photoactivation vs. α-Fe2O3-induced shadowing effect and (ii) the availability of free TiO2 surface vs. α-Fe2O3-coated surface.
Photo-electrochemical and photocatalytic activities
Fig. 5(a) provides the current–potential curves of the electrodes under AM 1.5G illumination in 1 M NaOH electrolyte. All the electrodes exhibit a photocurrent onset at ca. −0.8 V vs. Ag/AgCl that is typical of TiO2. This demonstrates that, in α-Fe2O3/TiO2 composites, TiO2 absorbs UV photons and generates charge carriers. Due to the natural upwards band bending (enhanced by the applied bias) in TiO2 tube walls, electrons photopromoted in the conduction band are collected in the center of material and, through a preferential orthogonal pathway, are transported along the tubes to the back-contact (FTO). By contrast, positive holes migrate from the valence band of TiO2 to that of α-Fe2O3 that mediates their transfer to the environment, i.e., the electrolyte, and decreases the probability of charge recombination.29,30 A sketch of the proposed mechanism is reported in Fig. 5(e). An increase in photocurrent density is indeed observed for the composite materials, with respect to plain TiO2 NTs.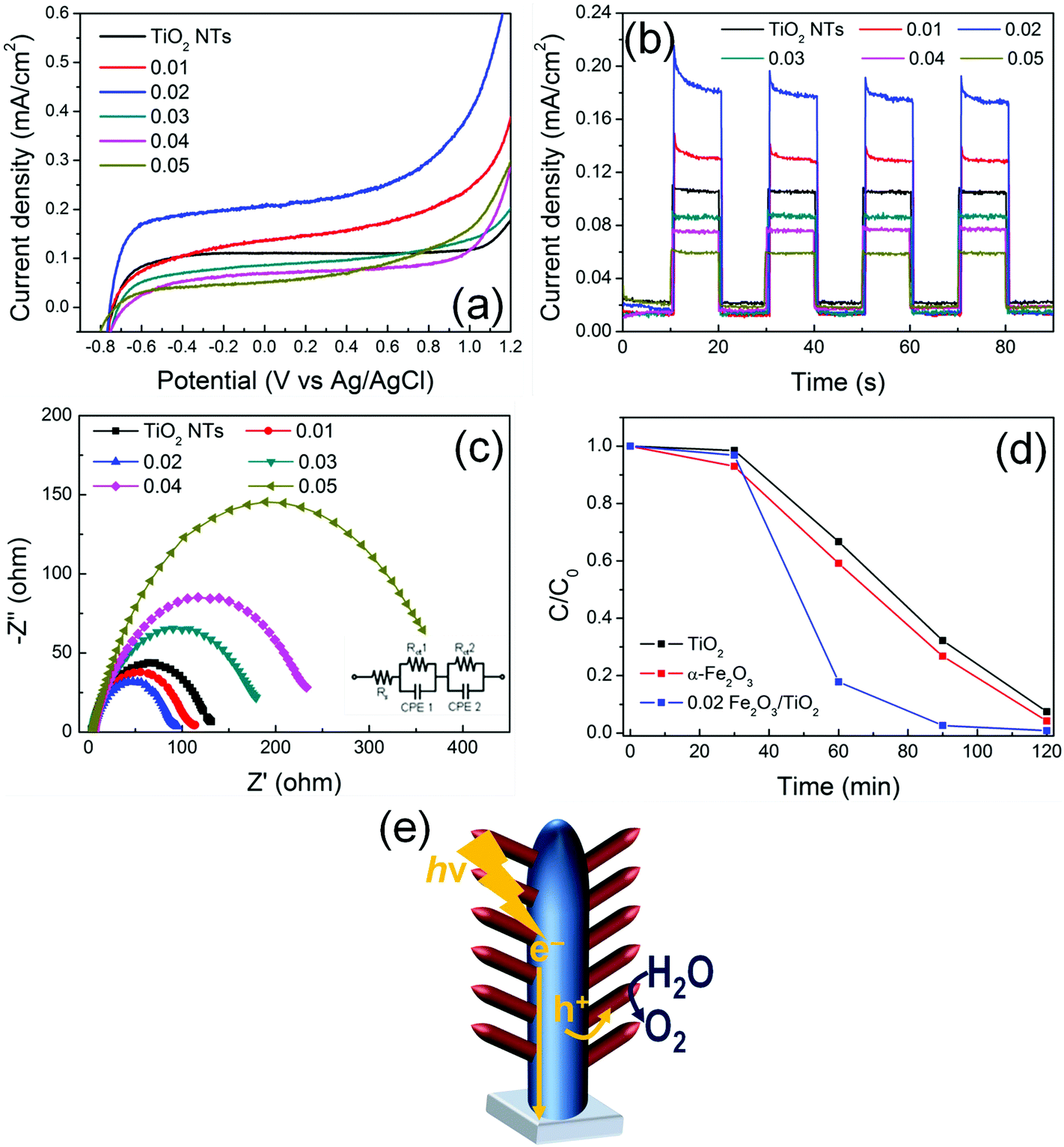 | ||
| Fig. 5 (a) Current–potential characteristics of TiO2 and α-Fe2O3/TiO2 NT layers on FTO, measured in 1 M NaOH at 10 mV s−1 scan rate, under AM 1.5G illumination. (b) Photo-transient characteristics of TiO2 and α-Fe2O3/TiO2 NT layers on FTO, measured in 1 M NaOH at +0.26 V vs. Ag/AgCl and under AM 1.5G chopped light illumination. (c) Nyquist plots of electrochemical impedance measurements of TiO2 and α-Fe2O3/TiO2 NT layers; inset: equivalent circuit for the nanotube structures. (d) Photocatalytic degradation of RhB (C0 = 1 mM) performed under AM 1.5G illumination. (e) Sketch of the proposed charge photogeneration and transfer mechanism in the optimized 0.02 α-Fe2O3/TiO2 NT layer. | ||
The beneficial effect of combining the two oxides is observed with the composite heterostructures decorated with a relatively low amount of hematite (i.e., with a ∼10–30 nm thick Fe2O3 layer). In particular, the 0.02 Fe2O3/TiO2 sample provides an optimized arrangement that leads to the highest photocurrent; moreover, Fig. S10† shows that the photo-electrode features an adequate stability, for continuous illumination under AM 1.5G simulated sunlight at 0.8 V vs. Ag/AgCl for extended times.
The same relative trend is also observed in transient photoresponse measurements (Fig. 5(b)). All the transients have a similar shape and exhibit an initial decay of photocurrent, particularly evident for the Fe2O3-modified TiO2 electrodes. This indicates that beneficial charge carrier separation is enabled in the system, with hematite that works as a hole-transfer mediator (from TiO2) to the electrolyte. Nevertheless, also a minor contribution of α-Fe2O3 as an active photocatalyst should be considered.
In addition, I–V and photo-transient characteristics of pure α-Fe2O3 layers (Fig. S11†) confirmed that hematite suffers from a low PEC ability compared to both pure TiO2 and Fe2O3/TiO2 arrays. The photocurrent enhancement (i.e., ca. 4×) can be attributed to the Fe2O3/TiO2 heterojunction, which reduces electron–hole recombination by enhancing photogenerated charge carrier separation.
Typically, the formation of titania/hematite core/shell heterocomposites is reported to limit the performance of TiO2 in photo-electrochemical applications. This stems from the fact that, in most common configurations, a hematite layer is grown/deposited over TiO2 and usually shades the core, hence preventing TiO2 surface reactive sites from absorbing light.7 Instead, the observed enhanced PEC performance of the heterocomposite materials is here attributed to the dendritic nanostructure which simultaneously exposes the TiO2 nanotubes to the AM 1.5G-filtered irradiation, enabling the photogeneration of charges also in the supporting tubes, and efficiently separates e− and h+ while minimizing the distance that photogenerated holes have to diffuse through to reach the electrolyte.24 Moreover, the α-Fe2O3 1D nanobranches grow directly connected to the TiO2 NTs (that is, no polymers or binders were used in the reported procedure). This configuration not only provides a preferential direction for hole diffusion from TiO2 with no extra-layers to be tunneled, but is also compatible with the short diffusion length of holes that typically affects both TiO2 (i.e., ca. 10 nm) and α-Fe2O3 (i.e., 2–4 nm).31 Finally, the directional electron flow along the axial direction of the 1D TiO2 NTs is further beneficial in view of reducing the probability of electron–hole recombination since it promotes the collection of negative charges at FTO.
The Electrochemical Impedance Spectroscopy (EIS) results show that the charge transfer resistance (Rct) in the low-frequency zone correlates with the photocurrent responses (Fig. 5c). Indeed, the 0.02 α-Fe2O3/TiO2 sample exhibits the lowest Rct value not only compared to pure TiO2 nanotubes and pure α-Fe2O3 thin layers (Fig. S12†), but also compared to all the other investigated α-Fe2O3/TiO2 hierarchical heterostructures, thus confirming that thin α-Fe2O3 nanoflakes branched on TiO2 nanotubes and the formation of a Fe2O3/TiO2 heterojunction facilitate charge transfer and, hence, are beneficial in terms of the PEC performance.32
The proposed mechanism is also supported by the results of Rhodamine B (RhB) photodegradation (Fig. 5(d)). The experiment was performed with a pure TiO2 NT sample, a pure α-Fe2O3 layer and a 0.02 Fe2O3/TiO2 sample. RhB is typically decomposed by the ˙OH radicals generated in solution through the reaction of photogenerated charges with water.33 In line with an enhanced e−/h+ separation and, hence, a more favorable holes injection into the solution, ∼90% of the initial RhB was degraded within the first 60 min of irradiation with 0.02 Fe2O3/TiO2, in contrast with ∼30–40% RhB photodegraded with the pure single oxides.
Conclusion
α-Fe2O3/TiO2 3D-hierarchical nanostructured materials that consist of vertically aligned TiO2 nanotubes modified with different amounts of α-Fe2O3 nanoflakes were successfully fabricated on FTO glass via a multi-step hydrothermal approach. In contrast with previous studies that reported on the detrimental effect of α-Fe2O3 on the TiO2 PEC activity, the here proposed α-Fe2O3/TiO2 dendritic nanostructures provide an ideal architecture for (i) the optimized light absorption and activation of TiO2 and (ii) a straightforward transfer of photo-generated charges, enabled by the direct connection of TiO2 NTs with both the e− collector (i.e., FTO) and the h+ collector (i.e., α-Fe2O3). The morphology of the α-Fe2O3 nanostructures that varies from ultra thin nanoflakes (for dilute Fe precursor solution) to nanorod/nanofiber structures (for higher Fe precursor concentration) was shown to have a significant impact on the photo-induced activity of the α-Fe2O3/TiO2 composites. In particular, a two-times higher photocurrent density and faster photocatalytic degradation of RhB in aqueous solution were measured with the 0.02 α-Fe2O3/TiO2 hierarchical nanocomposite. For larger hematite precursor concentrations, the decrease in the α-Fe2O3/TiO2 PEC activity was attributed either to (i) the formation of hematite clusters – in this case, the short diffusion length of holes in α-Fe2O3 could no longer be overcome and charge carriers are more likely prone to recombine in TiO2, or (ii) an α-Fe2O3 dense layer that blocks the active surface sites of TiO2 and reduces the amount of photons available to promote e−/h+.Follow-up studies may be beneficial in view of improving the light-promoted activity of the reported α-Fe2O3/TiO2 hierarchical materials; however, this new multi-step hydrothermal process holds large potential as it can be finely tuned and adjusted also for the design and development of other hierarchical heterogeneous metal oxide electrodes with suitable photo-electrochemical characteristics.
Acknowledgements
The authors gratefully acknowledge the support by Project No. LO1305 and Project No. 8E15B009 of the Ministry of Education, Youth and Sports of the Czech Republic, Project No. 15-19705S of the Grant Agency of the Czech Republic, and the Research Infrastructure NanoEnviCz, supported by the Ministry of Education, Youth and Sports of the Czech Republic under Project No. LM2015073. The DFG, and the DFG cluster of excellence “Engineering of Advanced Materials”, as well as DFG “funCOS” are also gratefully acknowledged for financial support.References
- A. L. Linsebigler, G. Lu and J. T. Yates Jr., Chem. Rev., 1995, 95(3), 735 CrossRef CAS.
- A. Fujishima, X. Zhang and D. a. Tryk, Surf. Sci. Rep., 2008, 63, 515 CrossRef CAS.
- K. Lee, A. Mazare and P. Schmuki, Chem. Rev., 2014, 114, 9385 CrossRef CAS PubMed.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904 CrossRef CAS PubMed.
- M. Dahl, Y. Liu and Y. Yin, Chem. Rev., 2014, 114, 9853 CrossRef CAS PubMed.
- R. P. Lynch, A. Ghicov and P. Schmuki, J. Electrochem. Soc., 2010, 157(3), G76 CrossRef CAS.
- T. H. Jeon, W. Choi and H. Park, J. Phys. Chem. C, 2011, 115(14), 7134 CAS.
- C. Xu, P. H. Shin, L. Cao, J. Wu and D. Gao, Chem. Mater., 2010, 22(1), 143 CrossRef CAS.
- H. Han, T. Song, E. K. Lee, A. Devadoss, Y. Jeon, J. Ha, Y. C. Chung, Y. M. Choi, Y. G. Jung and U. Paik, ACS Nano, 2012, 6(9), 8308 CrossRef CAS PubMed.
- J.-Y. Liao, B.-X. Lei, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, Energy Environ. Sci., 2012, 5(2), 5750 CAS.
- E. N. Maslen, V. a. Streltsov, N. R. Streltsova and N. Ishizawa, Acta Crystallogr., Sect. B: Struct. Sci., 1994, 50(4), 435 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- P. Liao and E. a. Carter, Phys. Chem. Chem. Phys., 2011, 13, 15189 RSC.
- H. Han, T. Song, J.-Y. Bae, L. F. Nazar, H. Kim and U. Paik, Energy Environ. Sci., 2011, 4(11), 4532 CAS.
- P. Lazic, Comput. Phys. Commun., 2015, 197, 324 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54(16), 11169 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter, 1999, 59(3), 1758 CrossRef CAS.
- E. J. W. Crossland, N. Noel, V. Sivaram, T. Leijtens, J. a. Alexander-Webber and H. J. Snaith, Nature, 2013, 495(7440), 215 CrossRef CAS PubMed.
- D. E. Janney, J. M. Cowley and P. R. Buseck, Clays Clay Miner., 2000, 48(1), 111 CAS.
- S. Li, G. Qin, X. Meng, Y. Ren and L. Zuo, J. Mater. Sci., 2013, 48(17), 5744 CrossRef CAS.
- Y. Li, X. Wei, B. Zhu, H. Wang, Y. Tang, T. C. Sum and X. Chen, Nanoscale, 2016, 8, 11284 RSC.
- J. Xi, O. Wiranwetchayan, Q. Zhang, Z. Liang, Y. Sun and G. Cao, J. Mater. Sci.: Mater. Electron., 2012, 23(9), 1657 CrossRef CAS.
- W. H. Baur, Acta Crystallogr., 1956, 9(6), 515 CrossRef CAS.
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128(49), 15714 CrossRef CAS PubMed.
- S. Kment, P. Schmuki, Z. Hubicka, L. Machala, R. Kirchgeorg, N. Liu, L. Wang, K. Lee, J. Olejnicek, M. Cada, I. Gregora and R. Zboril, ACS Nano, 2015, 9(7), 7113 CrossRef CAS PubMed.
- X. Zhang, P. Klaver, R. van Santen, M. C. M. van de Sanden and A. Bieberle-Hütter, J. Phys. Chem. C, 2016, 120(32), 18201 CAS.
- Y. Fu, J. Chen and H. Zhang, Chem. Phys. Lett., 2001, 350(5–6), 491 CrossRef CAS.
- A. Duret and M. Grätzel, J. Phys. Chem. B, 2005, 109(36), 17184 CrossRef CAS PubMed.
- S. J. a. Moniz, S. a. Shevlin, X. An, Z. X. Guo and J. Tang, Chem. – Eur. J., 2014, 20(47), 15571 CrossRef CAS PubMed.
- Z. Li, S. Feng, S. Liu, X. Li, L. Wang and W. Lu, Nanoscale, 2015, 7(45), 19178 RSC.
- L. Wang, C. Y. Lee and P. Schmuki, Electrochem. Commun., 2014, 44, 49 CrossRef CAS.
- L. Wang, N. T. Nguyen and P. Schmuki, ChemSusChem, 2016, 1 Search PubMed.
- L. You-ji and C. Wei, Catal. Sci. Technol., 2011, 1(5), 802 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr06908h |
| This journal is © The Royal Society of Chemistry 2017 |