
Gadolinium oxysulfide-coated gold nanorods with improved stability and dual-modal magnetic resonance/photoacoustic imaging contrast enhancement for cancer theranostics†
Tao
Guo
a,
Yan
Lin
a,
Zhi
Li
a,
Shan
Chen
a,
Guoming
Huang
*b,
Huirong
Lin
c,
Jun
Wang
d,
Gang
Liu
c and
Huang-Hao
Yang
*a
aThe Key Lab of Analysis and Detection Technology for Food Safety of the MOE, State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou 350116, P. R. China. E-mail: hhyang@fzu.edu.cn
bCollege of Biological Science and Engineering, Fuzhou University, Fuzhou 350116, P. R. China. E-mail: gmhuang@fzu.edu.cn
cState Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, Center for Molecular Imaging and Translational Medicine, School of Public Health, Xiamen University, Xiamen 361005, P. R. China
dSchool of Science, Xi'an Polytechnic University, Xi'an 710048, P. R. China
First published on 22nd November 2016
Abstract
Gold nanorods (GNRs) are emerging as a promising nanoplatform for cancer theranostics because of their unique optical properties. However, they still suffer from many limitations, such as high cytotoxicity, low thermodynamic and biological stability, and a tedious process for integrating other imaging modalities, for further practical biomedical applications. In this work, a strategy by one-step coating of Gd2O2S around GNRs is reported to address these limitations of GNRs. After the coating of the Gd2O2S shell, the as-fabricated Gd2O2S coated GNRs (GNRs@Gd2O2S) show enhanced biocompatibility and photostability, and tunable localized surface plasmon resonance. The strong absorption in the near-infrared region renders GNRs@Gd2O2S outstanding photoacoustic imaging and photothermal therapy capabilities. Moreover, owing to the T1 shortening ability of Gd2O2S, the GNRs@Gd2O2S also show an excellent T1 MRI contrast performance. The GNRs@Gd2O2S thus can serve as a versatile nanoplatform for cancer theranostics combining dual-modal imaging and photothermal therapy.
The ability to create agents incorporating multiple functionalities, such as targeting, imaging and therapy, is one of the main advantages of nanomedicine.1–3 In particular, the combination of diagnostic and therapeutic moieties into a single entity confers the theranostic nanoparticles’ unique capabilities.4–6 Due to the versatility of these nanoparticles, as well as their potential applications, interest in their synthesis and utility is rapidly increasing in recent years.7–11 Among the nanostructures being actively explored, gold nanorods (GNRs) are emerging as a promising nanoplatform for cancer theranostics because of their unique optical properties.12–14 In particular, the high absorption and tunable localized surface plasmon resonance (LSPR) across the near-infrared (NIR) region where tissues are nearly transparent, make GNRs an excellent candidate for photoacoustic imaging (PAI) and as photothermal therapy (PTT) agents.15–18 Despite these promising prospects of GNRs, many challenges remain to be addressed for further practical bioapplications, especially cancer theranostics. First, the typically synthesized GNRs are usually capped with stabilizing agents, such as cetyltrimethylammonium bromide (CTAB). The presence of these surfactant molecules would bring in toxicities to biological species at different levels.19,20 The removal of CTAB to reduce cytotoxicity by repeated centrifugation or ligand exchange reactions, however, may cause the aggregation and LSPR blue shift of GNRs. Second, the GNRs often suffer from poor photothermal stability due to the morphology melting and changing under the photothermal heating or femtosecond/nanosecond laser irradiation.21–23 The laser or high temperature induced melting effects lead to the shift of the longitudinal LSPR peak of GNRs from the desirable near-infrared (NIR) region to the visible region. Besides, when GNRs enter a complex environment, the occurrence of clustering and aggregating could also lead to the blue shift of the LSPR peak of GNRs.24–26 The loss of NIR deep-tissue penetration capability greatly hinders the application of GNRs for PTT. Third, the integration of other clinically used imaging modalities, such as magnetic resonance imaging (MRI), into the GNR nanoplatform in a facile and efficient way remains challenging. For example, current approaches for the fabrication of GNR nanoplatform with MRI function are typically through covalent chemistry, involving the covalent binding of GNRs and pre-prepared MRI contrast agents (e.g. Fe3O4 and Gd-based compounds).27–30 These covalent attachment strategies are complex and tedious and need a prior surface modification of the nanoparticles. Currently, to improve the biocompatibility and photostability of GNRs, the most common and effective method is coating biocompatible shells, such as silica, polymers, metal oxides and metal chalcogenides, around GNRs.26,31–38 The addition of protective shells imparts some steric constraint against reshaping and aggregating, thus provides better optical stability of GNRs. However, current coating materials, either silica or polymers, are usually monofunctional that merely serve as protective shells without an intrinsic imaging function. For multimodal cancer theranostics, the exploitation of novel coating materials with imaging abilities is highly desired to combine the LSPR properties of GNRs.
To address these challenges, we herein demonstrate our efforts in fabricating the gadolinium oxysulfide-coated GNRs (GNRs@Gd2O2S) as a novel cancer theranostic nanoplatform (Scheme 1). The GNRs@Gd2O2S can be synthesized by a one-step coating of the Gd2O2S shell without complicated modification. By controlling the thickness of the Gd2O2S shell, the LSPR peak of GNRs@Gd2O2S can be tuned to the NIR spectral region, which is highly beneficial for PTT and PAI. In addition, the coating of the Gd2O2S shell enhances the biocompatibility and photostability during photothermal heating or in complex biological environments of GNRs. More importantly, due to the ability of the Gd2O2S shell to shorten T1 relaxation times, the GNRs@Gd2O2S also exhibit excellent T1 MRI contrast enhancement. Combined with the PAI and PTT capabilities of the GNR core, the as-obtained GNRs@Gd2O2S thus can serve as a versatile and effective nanoplatform for in vivo cancer theranostics.
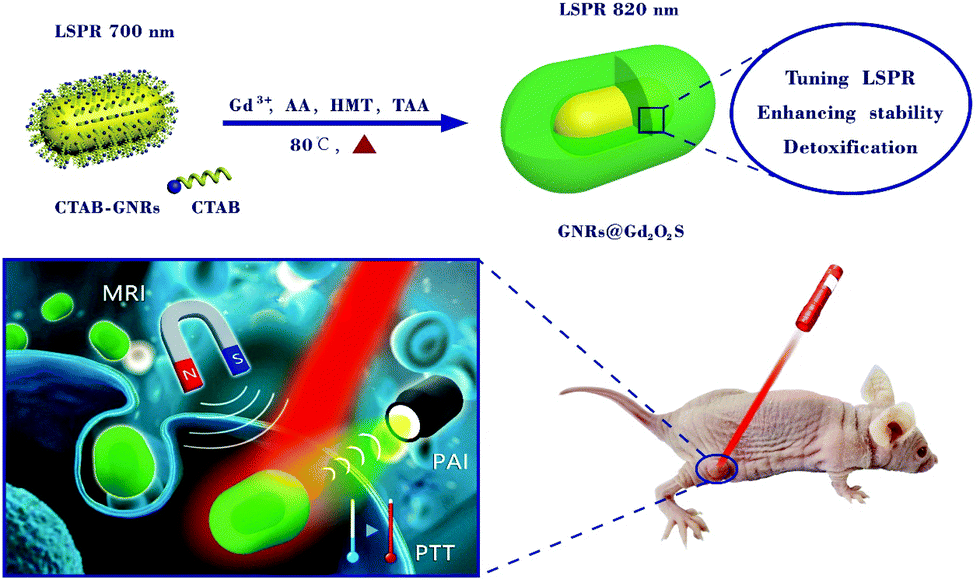 | ||
| Scheme 1 Schematic illustration of the synthesis of GNRs@Gd2O2S by incubating GNRs with Gd3+, ascorbic acid (AA), hexamethylenetetramine (HMT) and thioacetamide (TAA) at 80 °C and their potential applications in MRI, PAI and PTT. | ||
Results and discussion
We first synthesized CTAB-capped GNRs according to the previous method with a slight modification.39 The transmission electron microscopy (TEM) image showed that the as-synthesized GNRs were 55.7 ± 7.0 nm in length and 25.4 ± 3.2 nm in width, respectively (Fig. 1a). We then coated the GNRs with a Gd2O2S shell via a facile one-step process using Gd(NO3)3 as a metal precursor in the presence of ascorbic acid (AA) and thioacetamide (TAA) under mild reaction conditions. Previous studies have demonstrated that AA could induce the formation of AA–metal hydroxide complexes, and the complexes interact with the CTAB capping on the surface of the nanoparticles and subsequently form metal oxide shells via a dehydration process.40 TAA serving as a S source could also combine with metal ions and eventually lead to the formation of a metal oxysulfide shell deposited onto GNRs.41 To obtain better water dispersibility, the as-prepared GNRs@Gd2O2S were further modified with polyacrylic acid (PAA) through a simple ultrasonic treatment. The thickness of the Gd2O2S shell can be easily tuned by controlling the added concentration of the Gd(NO3)3 precursor. For example, we synthesized GNRs@Gd2O2S with different Gd2O2S shell thicknesses of 4 nm, 8 nm, and 20 nm, respectively (Fig. 1b–d). The high-resolution TEM (HRTEM) image revealed that the lattice spacing in the GNR core was about 0.235 nm, corresponding to the (111) plane of the face-centered cubic (fcc) Au, while the Gd2O2S shell was amorphous (Fig. 1e). The X-ray powder diffraction (XRD) pattern of GNRs@Gd2O2S showed no diffraction peaks of Gd2O2S, confirming that the Gd2O2S shell was amorphous (Fig. S1†). The survey scan X-ray photoelectron spectroscopy (XPS) spectrum of GNRs@Gd2O2S showed photoelectron lines at a binding energy of about 9 eV, 22 eV, 142 eV, 171 eV, 272 eV, 284 eV, 531 eV, 976 eV, 1187 eV and 1220 eV attributed to Gd 4f, Gd 5p, Gd 4d, S 2p, Gd 4p, C 1s, O 1s, O KLL, Gd 3d5/2, and Gd 3d3/2,42 respectively (Fig. S2†). All these peaks are very similar to that of previously reported Gd2O2S:Eu3+ nanostructures.43 TEM-associated energy-dispersive X-ray spectroscopy (EDS) analysis showed typical peaks of Gd, O, and S, and the atomic ratio of Gd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O:S was about 25.92:27.54:12.03, indicating the successful coating of Gd2O2S (Fig. S3†). The energy-dispersive X-ray (EDX) elemental mapping analysis of GNRs@Gd2O2S further revealed that the coating of Gd2O2S on the nanorods was homogeneous (Fig. 1f). The mass ratio of Au and Gd in GNRs@Gd2O2S with a 20 nm shell thickness determined by inductively coupled plasma mass spectrometry (ICP-MS) is about 1.82 ± 0.13.
:O:S was about 25.92:27.54:12.03, indicating the successful coating of Gd2O2S (Fig. S3†). The energy-dispersive X-ray (EDX) elemental mapping analysis of GNRs@Gd2O2S further revealed that the coating of Gd2O2S on the nanorods was homogeneous (Fig. 1f). The mass ratio of Au and Gd in GNRs@Gd2O2S with a 20 nm shell thickness determined by inductively coupled plasma mass spectrometry (ICP-MS) is about 1.82 ± 0.13.
 | ||
| Fig. 1 TEM images of (a) GNRs, and GNRs@Gd2O2S with different shell thicknesses of (b) 4 nm, (c) 8 nm, and (d) 20 nm, respectively. Inset: Corresponding photographs of GNRs and GNRs@Gd2O2S with different shell thicknesses. (e) High-resolution TEM image and (f) EDX mapping and HAADF-STEM (inset) images of GNRs@Gd2O2S. | ||
In general, a strong resonance peak in the NIR region is desirable for biomedical applications due to the high transparency of soft tissues in this region.18,44 By controlling the thickness of the Gd2O2S shell, we can tune the longitudinal LSPR peak of GNRs@Gd2O2S to the NIR region. The as-prepared GNRs had a longitudinal LSPR peak at 700 nm. After coating the Gd2O2S shell of 4 nm, 8 nm and 20 nm, the longitudinal LSPR peak was red-shifted to 754 nm, 788 nm and 818 nm, respectively (Fig. 2a). Increasing the Gd2O2S shell thickness could increase the effective dielectric constant surrounding the GNR cores, therefore causing an increased red shift of the LSPR modes.13 The strong absorbance in the NIR region indicates that GNRs@Gd2O2S are highly appropriate for biomedical applications. We therefore only investigated the performance of GNRs@Gd2O2S with a 20 nm Gd2O2S shell thickness in the subsequent experiments because of their absorbance at a longer wavelength of 818 nm. To evaluate the photothermal conversion capability of GNRs@Gd2O2S, the GNR@Gd2O2S aqueous solution was exposed to an 808 nm laser and the heating curves and images were collected. Irradiated by the 808 nm laser, the GNR@Gd2O2S aqueous solutions showed a rapid increase in temperature within 5 min, and the temperature rising rate and the final temperature were proportional to the concentration and laser power density (Fig. 2b and c). The photothermal conversion efficiency of GNRs@Gd2O2S was calculated as about 22.6%, which was similar to that of GNRs (23.2%), indicating that the coating of the Gd2O2S shell does not influence the photothermal conversion efficiency of GNRs@Gd2O2S (Fig. S4†). However, the GNRs@Gd2O2S exhibited enhanced photothermal stability compared to the GNRs. Over four laser irradiation cycles, the photothermal conversion efficiency of GNRs@Gd2O2S remained stable without significant changes, while a downtrend could be observed in that of GNRs (Fig. 2d). The laser illumination induced instability of GNRs not only influence the photothermal effect of GNRs, but also may hinder GNRs as PAI agents, since the photon fluencies in PAI always induce a shape deformation of GNRs.45 To further confirm the photostability of GNRs@Gd2O2S, we investigated the spectrum and morphology changes of GNRs@Gd2O2S under the irradiation of high-fluence nanosecond laser pulses (7 ns pulse duration, 10 Hz). We also synthesized GNRs with a longitudinal LSPR peak at about 815 nm (60.9 ± 4.9 nm in length and 13.4 ± 2.5 nm in width) as control samples. Under the nanosecond laser irradiation even up to 50 mJ cm−2 for 20 min, the longitudinal LSPR absorbance of GNRs@Gd2O2S remained highly stable with only a slight shift (Fig. 2e). In contrast, an obvious blue-shift could be observed for GNRs (Fig. S5†). The TEM images further confirmed that the morphology of GNRs@Gd2O2S remained the same after the irradiation, while the shape of GNRs changed obviously due to laser melting effects (Fig. S6†). These results clearly demonstrated that GNRs@Gd2O2S with the protection of the Gd2O2S shell have enhanced photothermal stability. The unpredictable aggregation of GNRs in complicated environments, such as serum, within cells, also limits their further biomedical applications due to the resulted loss of desirable NIR absorbance. To investigate the optical stability of GNRs@Gd2O2S in complex biological environments, we measured the absorbance spectrum of GNRs@Gd2O2S in different aqueous media including 100 mM NaCl, phosphate buffered saline (PBS, pH 7.4), and cell culture media OptiMEM (containing 10% fetal bovine serum). No matter what solution was dispersed in, the LSPR absorbance at 818 nm of GNRs@Gd2O2S remained stable with relatively small changes over 2 days (Fig. 2f). In contrast, after only 1 h incubation, the LSPR peaks of GNRs became broadened, red-shifted or even disappeared, and the absorption intensity greatly decreased (Fig. S7†). Besides, no leakage of Gd ions was detected by ICP-MS in the supernatant 7 days after GNRs@Gd2O2S was dispersed in these solutions, indicating that GNRs@Gd2O2S is also structurally stable. To better demonstrate the protection of the Gd2O2S shell on LSPR of GNRs@Gd2O2S, we further performed the finite-difference time-domain (FDTD) simulation. Since the GNRs aggregated mainly in a side-by-side (SS) manner, we simulated the coupling of longitudinal LSPR absorption in the SS dimer.46 The simulated longitudinal LSPR peak of the GNR@Gd2O2S SS dimer was at 804 nm, exhibiting little blue-shift compared to 815 nm of individual GNRs@Gd2O2S, while the longitudinal LSPR peak of GNRs could largely shift from 815 nm to 756 nm upon SS dimer formation (Fig. S8†). Additionally, the electric field distribution of the GNR@Gd2O2S SS dimer showed only a slight change (Fig. 2g). In contrast, the electric field intensity of the GNR SS dimer significantly reduced or almost disappeared (Fig. S9†). These results confirmed that the Gd2O2S shell can produce steric constraints against plasmon coupling, and therefore provides better optical stability of Gd2O2S in complex environments. The stabilizing agent CTAB is essential and widely used for the synthesis of high-quality GNRs. However, the non-covalent adsorption and desorption between the CTAB and GNRs is a dynamic process, and the detached CTAB is highly harmful to cells.19,20 To evaluate the cytotoxicity of GNRs@Gd2O2S, we conducted the tetrazolium-based colorimetric assay (MTT assay). After being incubated with HeLa, Hep G2, and L02 cells for 24 h, the GNRs@Gd2O2S had no significant cytotoxicity even at the highest concentration, while CTAB capped GNRs showed obvious cytotoxicity that less than 45% of cells were viable even at the concentration as low as 12.5 μg mL−1 (Fig. 2h). These results indicated that the coating of Gd2O2S could reduce the toxicity caused by CTAB, rendering GNRs@Gd2O2S good biocompatibility.
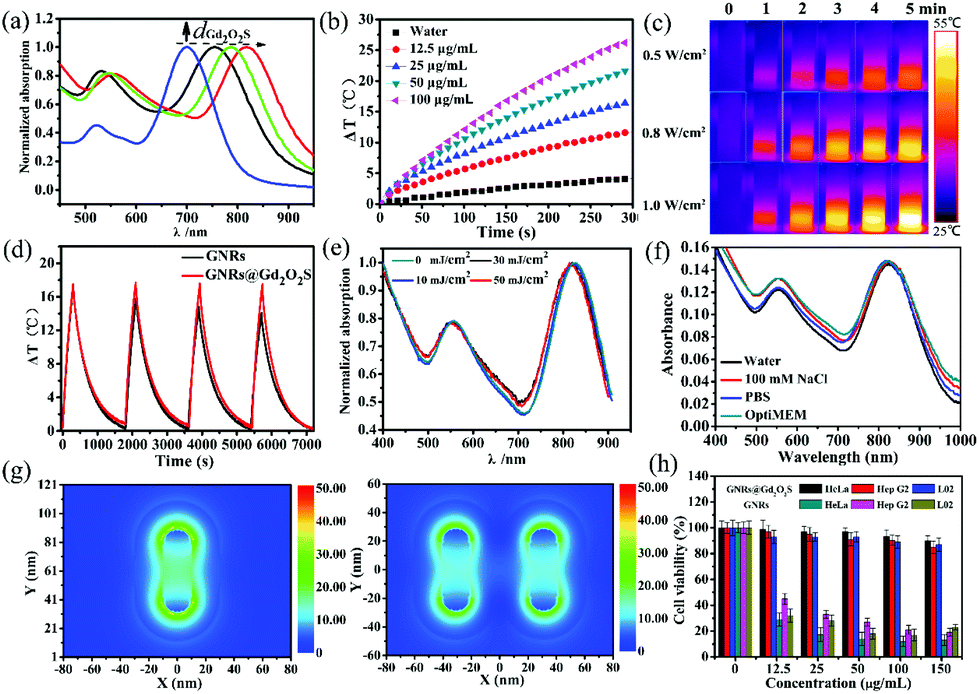 | ||
| Fig. 2 (a) Normalized absorption spectra of GNRs@Gd2O2S with different thicknesses of the Gd2O2S shell (dGd2O2S). (b) Temperature elevation curves of GNR@Gd2O2S aqueous solutions of different Au ion concentrations as a function of irradiation time under the 808 nm laser irradiation (1 W cm−2). (c) Infrared (IR) thermograph of GNR@Gd2O2S aqueous solutions (100 μg mL−1) under the 808 nm laser irradiation with different power densities. (d) Temperature elevation curves over four cycles of GNRs and GNR@Gd2O2S aqueous solutions (25 μg mL−1), respectively. (e) Normalized absorption spectra of GNRs@Gd2O2S under nanosecond laser (7 ns pulse duration, 10 Hz) irradiation with different laser fluences. (f) Absorption spectra of GNRs@Gd2O2S dispersed in water, 100 mM NaCl, PBS, and OptiMEM, respectively. (g) FDTD simulated localized electric field distributions of GNRs@Gd2O2S and GNR@Gd2O2S SS dimer, respectively. (h) Cell viability of HeLa, Hep G2 and L02 cells after incubated with GNRs and GNRs@Gd2O2S with different Au ion concentrations at 37 °C for 24 h, respectively. | ||
PAI has emerged as a versatile imaging modality for various kinds of biomedical applications.47 Gold nanomaterials have been widely used as contrast agents for PAI because of their tunable LSPR peak positions and large absorption cross sections.12 We then measured the photoacoustic signals from GNR@Gd2O2S aqueous solutions with different concentrations. The photoacoustic signal increased with the increase of the concentration, demonstrating that GNRs@Gd2O2S could effectively convert the energy harvested from light into heat and thus serve as a novel contrast agent for PAI (Fig. 3a and b). Gadolinium-based complexes or nanostructures have been widely exploited as T1 MRI contrast agents owing to the high magnetic moment and long electron spin relaxation time of Gd3+ ions.48,49 To investigate the MRI performance of the GNRs@Gd2O2S, we conducted phantom studies and relaxivity measurements on a 0.5 T MRI system. T1-Weighted phantom images showed that GNRs@Gd2O2S exhibit a stronger positive contrast effect (brighten signal) than Gd-DTPA for a given Gd concentration, suggesting the excellent T1 contrast ability of GNRs@Gd2O2S (Fig. 3c). We then measured the longitudinal relaxivity (r1) value according to the linear relationship of longitudinal relaxation rate (1/T1) versus Gd ion concentrations (Fig. 3d). The r1 value of GNRs@Gd2O2S was determined as 9.13 ± 0.3 mM−1 s−1, which was more than twice as high as that of commercial T1 contrast agents Gd-DTPA (4.44 ± 0.5 mM−1 s−1), further confirming that GNRs@Gd2O2S could afford more effective chemical exchange relaxations to the water protons around the nanoparticles and therefore resulted in the greater T1 relaxivity. Inspired by the excellent PAI and MRI contrast performance of GNRs@Gd2O2S, we further investigated the potential of GNRs@Gd2O2S for in vivo PAI/MRI dual-modal tumor imaging using Hep G2 tumor bearing BALB/c nude mice as models. We used intratumoral injection for animal studies because it enables dispensing extremely high doses of drug throughout the tumor with minimal systemic toxicity. After injection of GNRs@Gd2O2S (100 μL, 50 μg mL−1), PAI images of the tumor site were collected. The tumor region presented a significant increase in the photoacoustic signal after the injection, confirming the feasibility of GNRs@Gd2O2S for PAI contrast enhancement (Fig. 3e and f). We then examined the in vivo MRI contrast effects of GNRs@Gd2O2S on a 7 T MRI scanner. After the injection, a brighter signal in the tumor region could be observed on T1 weighted MRI images, suggesting that GNRs@Gd2O2S can produce strong T1 contrast enhancements in the tumor (Fig. 3g and h). The presented results therefore demonstrated that GNRs@Gd2O2S hold great promise in serving as a dual-modal contrast agent combining PAI and MRI for cancer diagnosis.
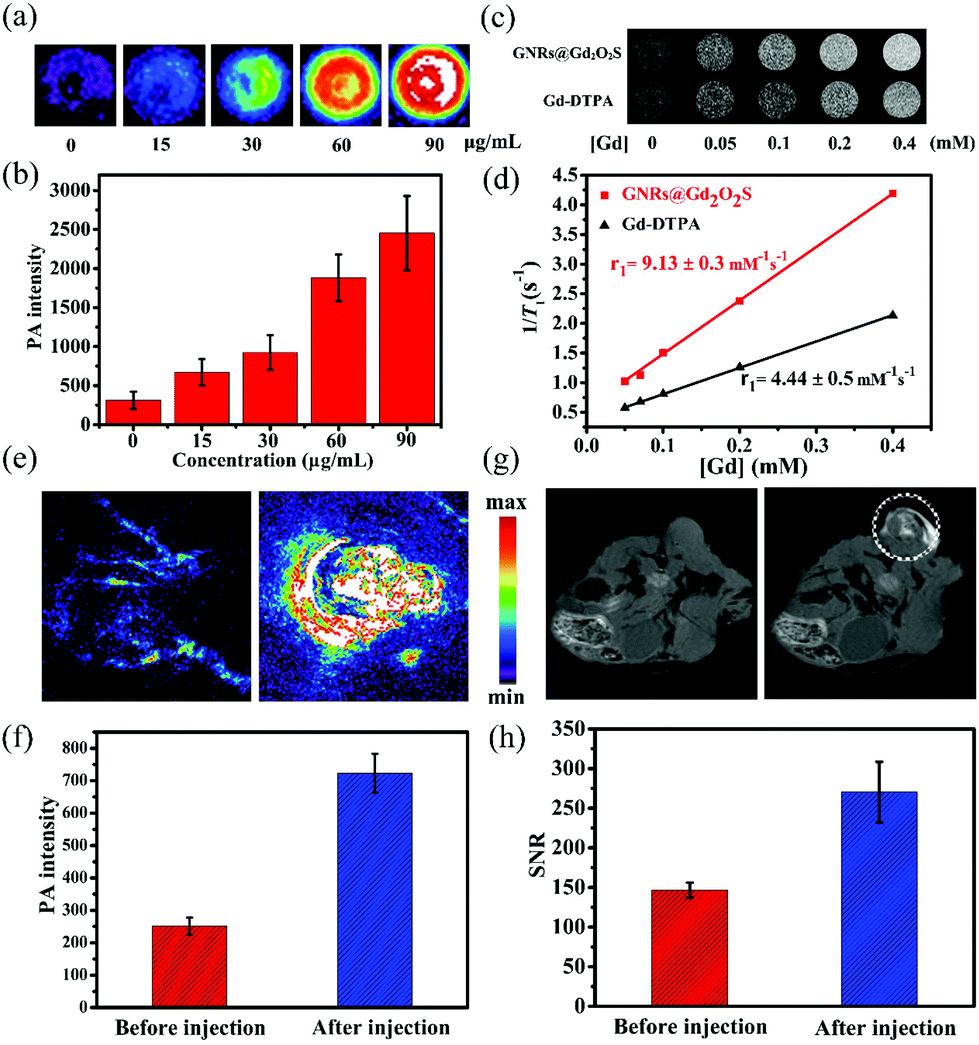 | ||
| Fig. 3 (a) PAI images and (b) PAI intensity of GNR@Gd2O2S aqueous solutions with different Au concentrations. (c) T1-Weighted phantom images of GNR@Gd2O2S and Gd-DTPA aqueous solutions with different Gd concentrations at 0.5 T, respectively. (d) The linear fitting of longitudinal relaxation rates at 0.5 T versus Gd concentrations for GNRs@Gd2O2S and Gd-DTPA, respectively. The r1 values were determined by three independent experiments. (e, f) In vivo PAI and corresponding quantification of intensity, and (g, h) T1-weighted MRI images and corresponding quantification of the signal-to-noise ratio (SNR) of mice tumor sites before and after the injection of GNRs@Gd2O2S (100 μL, 50 μg mL−1). | ||
PTT is emerging as one of the most powerful techniques for cancer therapy, because of its high selectivity and minimal invasiveness.50–53 Nanomaterials with NIR absorption have been widely developed as PTT agents for cancer treatment.18 We have showed that GNRs@Gd2O2S exhibit strong NIR absorption and high photothermal conversion efficiency under 808 nm laser irradiation, indicating that GNRs@Gd2O2S are highly appropriate for use as PTT agents. We first investigated the in vitro PTT performance of GNRs@Gd2O2S. Hep G2 cells were incubated with GNRs@Gd2O2S of different concentrations for 12 h, and then treated with 808 nm laser irradiation (1 W cm−2) for 5 min. The MTT assay showed that the cell survival rate decreases sharply with the increase of GNR@Gd2O2S concentration. When the concentration of GNRs@Gd2O2S reached 50 μg mL−1, the cell survival rate was only less than 20% (Fig. S10†). The merged fluorescence images also confirmed that almost all the cells were dead after they were incubated with GNRs@Gd2O2S of 50 μg mL−1 for 4 h and treated with 1 W cm−2 laser irradiation for 5 min (Fig. S11†). Motivated by the in vitro results that GNRs@Gd2O2S can efficiently kill the cancer cells under NIR laser irradiation, we further investigated the in vivo PTT performance of GNRs@Gd2O2S. We first examined the in vivo photothermal conversion effect of GNRs@Gd2O2S by using an infrared thermograph to record the temperature changes in the tumor site of the mice. The IR thermographic images showed that the temperature at the tumor area could rapidly reach ∼52.1 °C in the presence of GNRs@Gd2O2S (100 μL of 50 μg mL−1) under 808 nm laser irradiation (1 W cm−2) within 8 min (Fig. 4a). In comparison with the injection of PBS, the maximum temperature at the tumor area reached only about 34.9 °C under the same irradiation conditions, confirming that GNRs@Gd2O2S have an excellent photothermal conversion capability even in solid tumors. The tumor bearing mice were then randomly divided into four groups, mice were injected with GNRs@Gd2O2S (100 μL, 50 μg mL−1) and then irradiated with a 808 nm laser (1 W cm−2) for 8 min (GNRs@Gd2O2S + laser group), mice without any treatment (blank group), mice were only injected with GNRs@Gd2O2S (GNRs@Gd2O2S only group), and mice injected with PBS and then irradiated with a laser (PBS + laser group). The co-treatment with both GNRs@Gd2O2S and 808 nm laser irradiation could induce serious empyrosis at the tumor sites of mice, leading to the complete elimination of tumor in several days. In marked contrast, the mice in the other control groups exhibited rapid tumor growth within 16 days (Fig. 4b and c). The body weights of the mice for all groups were measured during the treatments, and no significant weight drop was observed, indicating the low toxicity of all treatments (Fig. S12†). The hematoxylin and eosin (H&E) staining of tumor sections revealed the pycnosis and karyolysis of cancer cells in the GNRs@Gd2O2S + laser group, confirming that the tumor was severely burned after PTT treatment (Fig. 4d). Meanwhile, no noticeable organ damage or inflammatory lesion was observed in major organs, such as heart, liver, spleen, lung, kidney, of mice 16 days after the therapy, demonstrating the safety of PTT treatments (Fig. S13†).
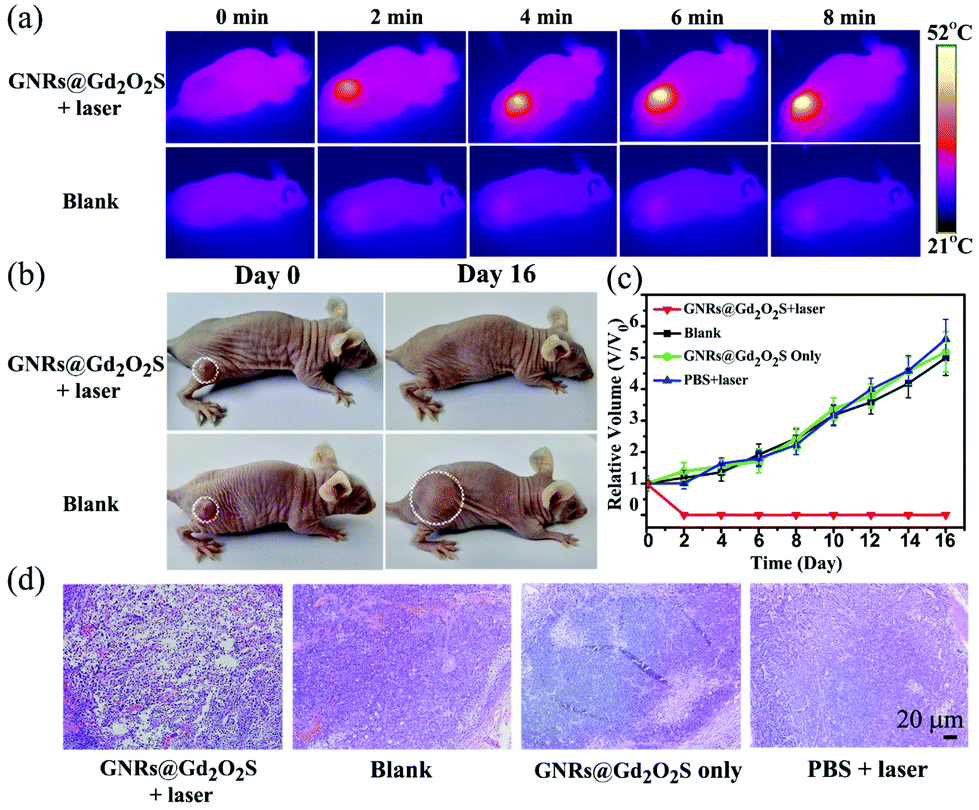 | ||
| Fig. 4 (a) IR thermograph of mice under laser irradiation (808 nm, 1 W cm−2) at different time points with (upper row) and without (lower row) the injection of GNRs@Gd2O2S. (b) Representative photographs of Hep G2 tumor-bearing BALB/c mice with (upper row) and without (lower row) PTT treatments. Photos were collected at day 0 and day 16, respectively. (c) Tumor growth curves of the Hep G2 tumor-bearing mice after different treatments. (d) Representative H&E stained histological images of tumor sections after different treatments. All images share the same scale bar. | ||
Conclusions
In summary, we have successfully synthesized GNRs@Gd2O2S and investigated their potential to serve as a multifunctional cancer theranostic nanoplatform. Owing to the protection of the Gd2O2S shell, the GNRs@Gd2O2S exhibit high stability either under high-fluence nanosecond laser irradiation or in complex biological environments. Additionally, the coating of Gd2O2S also reduces the toxicity caused by CTAB, rendering GNRs@Gd2O2S good biocompatibility. Both in vitro and in vivo studies demonstrate that the GNRs@Gd2O2S have excellent PAI and MRI contrast enhancement, showing great capability for the use of dual-modal contrast agents. Furthermore, the GNRs@Gd2O2S with strong NIR absorption and high photothermal conversion efficiency can effectively inhibit tumor growth through the photothermal effect. We thus believe that the stable and powerful GNR@Gd2O2S nanoplatform holds a great promise for biomedical applications, particularly cancer imaging and therapy.Acknowledgements
This research was supported by the National Natural Science Foundation of China (no. 21475026, U1505221, 21505021, 21622502, 21635002, 81501461 and 11547172), Natural Science Foundation of Fujian Province of China (no. 2015H6011, 2016J05035), the Program for Changjiang Scholars and Innovative Research Team in the University (no. IRT15R11), and the Independent Research Project of State Key Laboratory of Photocatalysis on Energy and Environment (no. 2014B02).References
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef CAS PubMed.
- T. Lammers, S. Aime, W. E. Hennink, G. Storm and F. Kiessling, Acc. Chem. Res., 2011, 44, 1029–1038 CrossRef CAS PubMed.
- Y. Gao, J. Lim, S.-H. Teoh and C. Xu, Chem. Soc. Rev., 2015, 44, 6306–6329 RSC.
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS PubMed.
- E. K. Lim, T. Kim, S. Paik, S. Haam, Y. M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS PubMed.
- R. Wang, H. Hu, Q. Cai, N. Zhao, Y. Zhu and F. Xu, Sci. China: Chem., 2015, 58, 1461–1470 CrossRef CAS.
- L. S. Lin, Z. X. Cong, J. B. Cao, K. M. Ke, Q. L. Peng, J. Gao, H. H. Yang, G. Liu and X. Chen, ACS Nano, 2014, 8, 3876–3883 CrossRef CAS PubMed.
- L. S. Lin, Z. X. Cong, J. Li, K. M. Ke, S. S. Guo, H. H. Yang and G. N. Chen, J. Mater. Chem. B, 2014, 2, 1031–1037 RSC.
- X. R. Song, X. Wang, S. X. Yu, J. Cao, S. H. Li, J. Li, G. Liu, H. H. Yang and X. Chen, Adv. Mater., 2015, 27, 3285–3291 CrossRef CAS PubMed.
- G. Huang, X. Zhu, H. Li, L. Wang, X. Chi, J. Chen, X. Wang, Z. Chen and J. Gao, Nanoscale, 2015, 7, 2667–2675 RSC.
- L. Cheng, A. Kamkaew, H. Sun, D. Jiang, H. F. Valdovinos, H. Gong, C. G. England, S. Goel, T. E. Barnhart and W. Cai, ACS Nano, 2016, 10, 7721–7730 CrossRef CAS PubMed.
- X. Yang, M. Yang, B. Pang, M. Vara and Y. Xia, Chem. Rev., 2015, 115, 10410–10488 CrossRef CAS PubMed.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- X. Huang, S. Neretina and M. A. El-Sayed, Adv. Mater., 2009, 21, 4880–4910 CrossRef CAS PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- S. Manohar, C. Ungureanu and T. G. Van Leeuwen, Contrast Media Mol. Imaging, 2011, 6, 389–400 CrossRef CAS PubMed.
- J. V. Jokerst, A. J. Cole, D. Van de Sompel and S. S. Gambhir, ACS Nano, 2012, 6, 10366–10377 CrossRef CAS PubMed.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS PubMed.
- A. M. Alkilany, P. K. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701–708 CrossRef CAS PubMed.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647–1671 RSC.
- S. Link, C. Burda, M. B. Mohamed, B. Nikoobakht and M. A. El-Sayed, J. Phys. Chem. A, 1999, 103, 1165–1170 CrossRef CAS.
- S. Link, C. Burda, B. Nikoobakht and M. A. El-Sayed, J. Phys. Chem. B, 2000, 104, 6152–6163 CrossRef CAS.
- M. B. Mohamed, K. Z. Ismail, S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1998, 102, 9370–9374 CrossRef CAS.
- H. C. Huang, S. Barua, D. B. Kay and K. Rege, ACS Nano, 2009, 3, 2941–2952 CrossRef CAS PubMed.
- X. Hu and X. Gao, Phys. Chem. Chem. Phys., 2011, 13, 10028–10035 RSC.
- Z. Zhang, L. Wang, J. Wang, X. Jiang, X. Li, Z. Hu, Y. Ji, X. Wu and C. Chen, Adv. Mater., 2012, 24, 1418–1423 CrossRef CAS PubMed.
- C. Wang, J. Chen, T. Talavage and J. Irudayaraj, Angew. Chem., Int. Ed., 2009, 48, 2759–2763 CrossRef CAS PubMed.
- H. W. Yang, H. L. Liu, M. L. Li, I. W. Hsi, C. T. Fan, C. Y. Huang, Y. J. Lu, M. Y. Hua, H. Y. Chou, J. W. Liaw, C. C. Ma and K. C. Wei, Biomaterials, 2013, 34, 5651–5660 CrossRef CAS PubMed.
- H. Sun, Q. Yuan, B. Zhang, K. Ai, P. Zhang and L. Lu, Nanoscale, 2011, 3, 1990–1996 RSC.
- J. Wang, J. Liu, Y. Liu, L. Wang, M. Cao, Y. Ji, X. Wu, Y. Xu, B. Bai, Q. Miao, C. Chen and Y. Zhao, Adv. Mater., 2016, 28, 8950–8958 CrossRef CAS PubMed.
- C.-S. Chang and L. J. Rothberg, Chem. Mater., 2015, 27, 3211–3215 CrossRef CAS.
- Z. Zhang, J. Wang, X. Nie, T. Wen, Y. Ji, X. Wu, Y. Zhao and C. Chen, J. Am. Chem. Soc., 2014, 136, 7317–7326 CrossRef CAS PubMed.
- Y. Zhang, J. Qian, D. Wang, Y. Wang and S. He, Angew. Chem., Int. Ed., 2013, 52, 1148–1151 CrossRef CAS PubMed.
- H. Hou, L. Chen, H. He, L. Chen, Z. Zhao and Y. Jin, J. Mater. Chem. B, 2015, 3, 5189–5196 RSC.
- M. Ji, M. Xu, W. Zhang, Z. Yang, L. Huang, J. Liu, Y. Zhang, L. Gu, Y. Yu, W. Hao, P. An, L. Zheng, H. Zhu and J. Zhang, Adv. Mater., 2016, 28, 3094–3101 CrossRef CAS PubMed.
- M. A. Muhammed, M. Doblinger and J. Rodriguez-Fernandez, J. Am. Chem. Soc., 2015, 137, 11666–11677 CrossRef CAS PubMed.
- H. Wang, Z. Sun, Q. Lu, F. Zeng and D. Su, Small, 2012, 8, 1167–1172 CrossRef CAS PubMed.
- J. Zhao, P. Xu, Y. Li, J. Wu, J. Xue, Q. Zhu, X. Lu and W. Ni, Nanoscale, 2016, 8, 5417–5421 RSC.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- Y. Yang, S. Han, G. Zhou, L. Zhang, X. Li, C. Zou and S. Huang, Nanoscale, 2013, 5, 11808–11819 RSC.
- L. Ma, S. Liang, X.-L. Liu, D.-J. Yang, L. Zhou and Q.-Q. Wang, Adv. Funct. Mater., 2015, 25, 898–904 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer Corporation, Eden Prairie, 1992 Search PubMed.
- J. Thirumalai, R. Chandramohan, S. Valanarasu, T. A. Vijayan, R. M. Somasundaram, T. Mahalingam and S. R. Srikumar, J. Mater. Sci., 2009, 44, 3889–3899 CrossRef CAS.
- E. Hemmer, A. Benayas, F. Légaré and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC.
- L. C. Chen, C. W. Wei, J. S. Souris, S. H. Cheng, C. T. Chen, C. S. Yang, P. C. Li and L. W. Lo, J. Biomed. Opt., 2010, 15, 016010 CrossRef PubMed.
- P. K. Jain and M. A. El-Sayed, Chem. Phys. Lett., 2010, 487, 153–164 CrossRef CAS.
- M. Xu and L. V. Wang, Rev. Sci. Instrum., 2006, 77, 041101 CrossRef.
- E. J. Werner, A. Datta, C. J. Jocher and K. N. Raymond, Angew. Chem., Int. Ed., 2008, 47, 8568–8580 CrossRef CAS PubMed.
- H. Chen, G. D. Wang, W. Tang, T. Todd, Z. Zhen, C. Tsang, K. Hekmatyar, T. Cowger, R. B. Hubbard, W. Zhang, J. Stickney, B. Shen and J. Xie, Adv. Mater., 2014, 26, 6761–6766 CrossRef CAS PubMed.
- K. Dong, Z. Liu, Z. Li, J. Ren and X. Qu, Adv. Mater., 2013, 25, 4452–4458 CrossRef CAS PubMed.
- Z. Zha, X. Yue, Q. Ren and Z. Dai, Adv. Mater., 2013, 25, 777–782 CrossRef CAS PubMed.
- X. R. Song, S. X. Yu, G. X. Jin, X. Wang, J. Chen, J. Li, G. Liu and H. H. Yang, Small, 2016, 12, 1506–1513 CrossRef CAS PubMed.
- L. S. Lin, X. Yang, G. Niu, J. Song, H. H. Yang and X. Chen, Nanoscale, 2016, 8, 2116–2122 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details on experimental information and supplementary figures. See DOI: 10.1039/c6nr08281e |
| This journal is © The Royal Society of Chemistry 2017 |