
Probing the hydrolytic reactivity of 2-difluoromethyl pyrroles†
Jennifer A.
Melanson
,
Carlotta
Figliola
,
Deborah A.
Smithen
,
Aleksandra K.
Kajetanowicz
and
Alison
Thompson
*
Department of Chemistry, Dalhousie University, P.O. Box 15000, Halifax, NS B3H 4R2, Canada. E-mail: Alison.Thompson@dal.ca
First published on 14th November 2016
Abstract
α-Difluoromethyl pyrroles were found to be stable while N-protected with an electron-withdrawing group. Due to the propensity of pyrroles to access azafulvenium-like intermediates, the C–F bonds of an α-difluoromethyl substituent are labile under hydrolytic conditions. The presence of certain electron-withdrawing substituents about the pyrrolic ring can accelerate this process, as determined through a kinetic comparison of the deprotection and subsequent hydrolysis reactions of N-protected β-aryl α-difluoromethyl pyrroles.
Introduction
Porphyrinogens have been intensely studied, particularly as regards to chemical and biological relevance of the corresponding porphyrins.1–4 Although spontaneous oxidation of porphyrinogens to porphyrins can render the isolation of these precursors rather challenging,5 calix[4]pyrroles6 bear eight alkyl or aryl groups at the meso positions of the tetrapyrrolic skeleton and thus resist oxidation.7–14 The presence of a meso sp3 carbon confers to the structure a significant conformational freedom,5 enabling potential binding of these macrocycles for a wide range of applications.7–14 As part of an ongoing effort to develop functionalised dipyrrolic structures, we began to investigate the synthesis of pyrroles featuring a –CF2– moiety at the α-position, contrasting with the –CR2– unit (R = alkyl or aryl) used as precursors to calix[4]pyrroles. The strength of the C–F bond,15 alongside its ability to sterically mimic the C–H unit, has driven growing interest in the use of fluorine as an isostere of hydrogen, a common strategy in agrochemical16 and pharmaceutical industries.17,18 In tandem, the development of more efficient and diverse methods through which to incorporate this functional moiety, either directly19–22 or via the fluorination of existing functionality,23 renders this group even more suitable for incorporation into a wide variety of frameworks. However, there are few reports describing pyrroles bearing a difluoromethyl substituent directly appended to the heterocyclic core. Baran and co-workers developed a fluorinating reagent for the installation of a difluoromethyl group on nitrogen-containing heteroaromatic compounds via a radical mechanism (Scheme 1, top):19N-unsubstituted 2- and 3-acyl pyrroles 1a–b were shown to react promptly with ZnDFMS in the presence of tBuOOH and gave the α-difluoromethyl-substituted analogues 2a–b in moderate yield. Note that 2a and 2b both feature conjugated electron-withdrawing acyl groups that serve to stabilise the heterocyclic core. | ||
| Scheme 1 Examples of α-difluoromethyl-substituted pyrroles. | ||
In 1995 Scott reported the fluorination of several 2-formyl pyrroles using DAST, with the goal of synthesising an inhibitor of the uroporphyrinogen III cosynthase.24 Pyrrole 3, which features a substitution pattern matching that of the enzyme substrate, uroporphomethene, reacted with DAST to render 4 in high yield (87%, Scheme 1, middle). Subsequent deprotection of 4 under standard hydrolysis conditions was reported to give the desired α-difluoromethyl pyrrole 5 apparently without loss of fluorine. However, experimental conditions or isolated yields for this step are not described, nor does the report include any evidence for the stated maintenance of fluoro substituents. As reported by Haufe (Scheme 1, bottom),25 upon exposure to acid the stability of the difluoromethyl group is strongly influenced by the electronic nature of ring substitution. Indeed, in the presence of the electron-donating methyl group the difluoromethyl group was hydrolysed to the corresponding 2-formyl pyrrole 6a, while, under the same reaction conditions, pyrrole 6b was reported to be stable (Scheme 1). With the backdrop of these limited reports, we report herein upon the reactivity of α-difluoromethyl pyrroles under hydrolytic conditions,24 as well as an approach for the preparation of β-aryl-α-difluoromethyl pyrroles. Furthermore, the influence of the pyrrolic ring substitution pattern upon the reactivity of the α-difluoromethyl functional group is discussed.
Results and discussion
The procedure for the synthesis of pyrrole 5 was repeated according to Scott's method (Scheme 1, middle).24 However, upon N-deprotection of 4 the isolated product corresponded to the N-unprotected aldehyde 8 (75%, Scheme 2), with the two pendant esters of course hydrolysed to the corresponding carboxylic acids. As this result clearly contradicts the literature report,24 the reaction was repeated in deuterated solvents and progress was followed via1H and 19F NMR spectroscopic analysis. The initial N-deprotection of the α-difluoromethyl pyrrole 4 was determined to occur immediately upon addition of 2 M NaOH. The N-deprotected product 5 is unstable under these reaction conditions, forming first the α-dimethylacetal pyrrole 7 which then hydrolyses to the α-formyl pyrrole 8 (Scheme 2 and Fig. 1 in the ESI†). | ||
| Scheme 2 N-Deprotection and hydrolysis of pyrrole bearing a 2-difluoromethyl group. | ||
In order to exclude the possibility that the two alkanoate moieties influence the hydrolytic reactivity of the α-difluoromethyl group, the unsubstituted variant 10 was exposed to the same conditions.26 Xtalfluor-M® was used to fluorinate the 2-formyl pyrrole 9, giving the α-difluoromethyl pyrrole 10 in high yield (90%, Scheme 3). The use of a stabilising N-protecting group proved essential for the fluorination reaction to be successful. To study the N-deprotection, aliquots from the reaction of 10 with 2 M aqueous NaOH in MeOD at 50 °C were analysed using 1H and 19F NMR spectroscopy. As observed previously for 4, the rapid appearance of the N-unprotected material 11 was followed by methoxide displacement of the fluoro substituents, to give the acetal 12, which hydrolysed to the aldehyde 13 (Scheme 3 and Fig. 2 in ESI†).
 | ||
| Scheme 3 Formation and hydrolysis of α-difluoromethyl pyrrole 11. | ||
Although some N-unprotected α-difluoromethyl pyrroles featuring electron-withdrawing substituents on the heterocycle are stable (Scheme 1),19 such systems are clearly different to pyrroles such as 5 and 11, whereupon hydrolysis to result in loss of fluorine is facile. In order to investigate the influence of the electronic properties of substituents about the pyrrolic ring upon the deprotection and hydrolysis reactions, a series of N-(2-nosyl)-β-aryl-substituted pyrroles were synthesised. Moving away from the tosyl and –SO2Ph protecting groups, the 2-nosyl group was selected thus enabling the N-deprotection to be monitored via release of 2-nitrophenyl phenyl sulfide (14, Scheme 4).27,28 Moreover, N-deprotection of the 2-nosyl group would be achieved under anhydrous conditions. However, despite 2,4-dinitrophenol (2,4-DNP) being commonly used as a reporter for measuring reaction rates, we were unable to find reports of monitoring reaction rates through detection of yellow 2-nitrophenyl, 4-nitrophenyl or 2,4-dinitrophenyl phenyl sulfides. Furthermore, although 2-nitrophenyl phenyl sulfide is known,29–31 full characterisation proved elusive in the literature. Compound 14 was, therefore, prepared via deprotection of 15 using PhSH and NEt3. The ejected sulfide 14 was isolated in quantitative yield (Scheme 4), thereby demonstrating that this anhydrous method is viable for the needed N-deprotection of N-(2-nosyl) pyrroles.27,28,32 The absorption maximum of 14 occurs at 368 nm (CH3CN), with an extinction coefficient of 4000 L mol−1 cm−1.
 | ||
| Scheme 4 Synthesis of 2-nitrophenyl phenyl sulfide 14. | ||
α-Difluoromethyl pyrroles 19a–f featuring various aryl groups at the β-position of the ring were synthesised according to Scheme 5. Suzuki coupling between N-(2-nosyl)-3-bromo-2-formyl pyrrole 20 and boronic acids a–f gave the corresponding β-aryl pyrroles 21a–f. Subsequent fluorination yielded the desired α-difluoromethyl pyrroles 19a–f (Scheme 5).
 | ||
| Scheme 5 Synthesis and N-deprotection of 19a–f. | ||
Upon N-deprotection of pyrroles 19a–e with PhSH and NEt3 under anhydrous conditions, the resulting pyrroles (22a–e) proved to be stable providing the material remained under an inert and anhydrous environment (Scheme 5). As such, the deprotection and subsequent hydrolysis reaction could be studied separately. The first step was studied using UV-Vis spectroscopy under pseudo-first order conditions using 400 equivalents of PhSH and 560 equivalents of NEt3 in CH3CN at 0 °C under an inert atmosphere. Despite maintaining a steady flow of nitrogen gas in the sample chamber of the spectrophotometer, the presence of phenyl disulfide (17 in Scheme 4) was noted upon careful inspection of the reaction mixture arising from the N-deprotection of 19a–e. Several pathways to 17 may be at play during the reaction, e.g. thiophenol affords disulfide in the presence of an oxidising agent and a base;33,34 the reaction by-product sulfur dioxide 18 acts as an oxidising agent. As the disulfide 17 also absorbs at 368 nm, the absorbance reading at this35 wavelength is representative of both products. Since the interference from 17 is constant across the sample set, the relative production of 14 (Table 1, in ESI†) represents a qualitative estimation of the N-deprotection rate (Table 1). The starting materials provide no interference at the monitoring wavelength. The production of 14 was monitored via its absorbance at 368 nm (Table 1 in ESI†), and the relative rate constants of deprotection were thus calculated:
| Compound | Substituent (R) | [Starting material] (M) | Rate constantsa (s−1) | Hammett substituent constants (σb) |
|
|---|---|---|---|---|---|
| a Rate constants calculated on two replicates of each substrate using Kaleidagraph v 4.1.3 and error on replicates calculated using XLSTAT v 2015.4.01.21576. b As reported.36 | |||||
| 19a | CF3 | 4.03 × 10−4 | (1.96 ± 0.01) × 10−2 | 0.54 | 0.19 |
| 19b | F | 4.11 × 10−4 | (1.23 ± 0.04) × 10−2 | 0.06 | −0.01 |
| 19c | H | 4.08 × 10−4 | (1.24 ± 0.08) × 10−2 | 0 | 0.000 |
| 19d | CH3 | 4.15 × 10−4 | (1.19 ± 0.03) × 10−2 | −0.17 | −0.02 |
| 19e | OCH3 | 3.57 × 10−4 | (0.81 ± 0.02) × 10−2 | −0.27 | −0.19 |

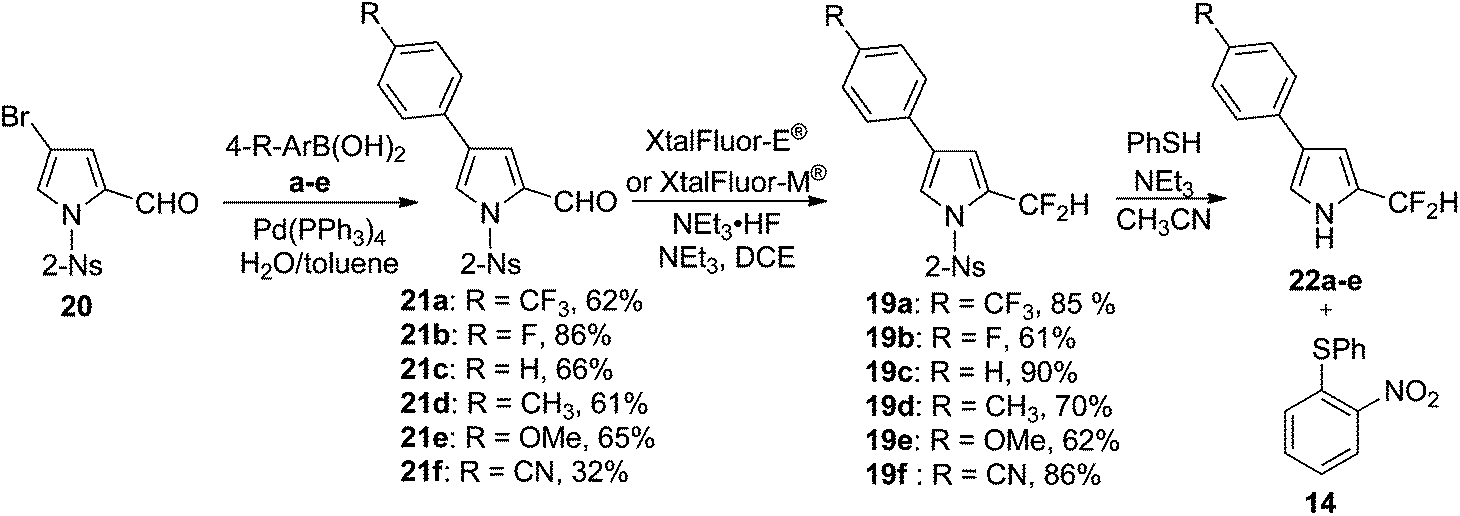
As shown in Table 1, the presence of electron-withdrawing character in the aryl substituent causes an increase in the rate of N-deprotection (19a, R = CF3). In contrast, electron-donating character (19e, R = OCH3) impedes the reaction rate. Fig. 1 shows a Hammett reaction constant (ρ) of 0.4, suggesting an increase of negative charge in the reaction intermediates.
 | ||
| Fig. 1 Hammett plot for N-deprotection of 19a–e. | ||
These features support the reaction mechanism proposed in Scheme 6. After nucleophilic attack by PhSH,37,38 the Meisenheimer-type intermediate 23 presumably collapses to give the reaction intermediate 24 and the development of anionic character in the orthogonal p-orbital of the pyrrolide nitrogen atom.27 The ability of the substituent (R) within the aryl group to stabilise the transition state leading to 24 thus plays an important role in the rate of deprotection. Indeed, electron-withdrawing groups serve to stabilize the forming pyrrolide anion, thereby enhancing the reaction rate, yet electron-withdrawing groups serve to destabilise the transition state en route to 24. As the electron-donating and electron-withdrawing R substituents are somewhat distant from the developing anionic nitrogen atom, their effects in stabilising the transition state are small and thus the ρ value (Fig. 1) is small.
 | ||
| Scheme 6 Proposed mechanism for the N-deprotection of 19. | ||
Having determined successful N-deprotection conditions, attention was turned to the hydrolysis of the α-difluoromethyl group. Hydrolysis was monitored using 19F NMR spectroscopy under pseudo first-order conditions. Pyrroles 19 were first N-deprotected in an NMR tube using 1.8 equivalents of PhSH and 2.5 equivalents of NEt3, dissolved in CD3CN. Subsequently, hydrolysis was initiated through the injection of 1000 equivalents of water at 40 °C. As the hydrolysis reaction progressed, the decreasing intensity of the 19F doublet arising from the –CF2H group, with respect to the inert internal standard (hexafluorobenzene, 0.5 equiv., 0.4 M solution in CD3CN), was measured and the rates of hydrolysis calculated (Table 2). The disappearance of the –CF2H group signal in each case was accompanied by the expected appearance of the HF singlet, which derives from hydrolysis of the –CF2H group (Fig. 3–7 in the ESI†).
| Compound | Substituent (R) | [Starting material] (M) | Rate constanta (s−1) | Hammett substituent constants (σb) |
|
|---|---|---|---|---|---|
| a Rate constants calculated using Kaleidagraph v 4.1.3 and error is calculated using XLSTAT v 2015.4.01.21576. b As reported.36 | |||||
| 22a | CF3 | 1.400 × 10−2 | (1.712 ± 0.092) × 10−3 | 0.54 | 0.27 |
| 22b | F | 1.408 × 10−2 | (0.966 ± 0.108) × 10−3 | 0.06 | 0.02 |
| 22c | H | 1.501 × 10−2 | (0.914 ± 0.094) × 10−3 | 0.00 | 0.00 |
| 22d | CH3 | 1.422 × 10−2 | (1.021 ± 0.013) × 10−3 | −0.17 | 0.05 |
| 22e | OCH3 | 1.450 × 10−2 | (1.195 ± 0.025) × 10−3 | −0.27 | 0.12 |
| 22f | CN | 1.488 × 10−2 | (1.749 ± 0.040) × 10−3 | 0.66 | 0.28 |
Although the variation in the rates of hydrolysis are subtle, the Hammett plot (Fig. 2) resembles a concave up parabola with a minimum at R = H (22c). To confirm the concave up parabola, we synthesized and analysed 22f (R = CN), following the same protocols as previously, and were delighted to observe an excellent fit with the data generated from 22a–22e. The non-linear relationship39,40 revealed on the Hammet plot points to the influence of the electronic features of the substituents on the aryl ring of pyrroles 22. The ρ value for electron-donating groups was found to be −0.4, suggesting a build-up of positive charge (or neutralisation of negative charge, negative slope, Fig. 2). In contrast, the ρ value for electron-withdrawing groups was found to be 0.4, suggesting an increase of the negative charge character in the transition state of the rate-determining step (positive slope, Fig. 2).
 | ||
| Fig. 2 Hammett plot for the hydrolysis of 19a–f following N-deprotection. | ||
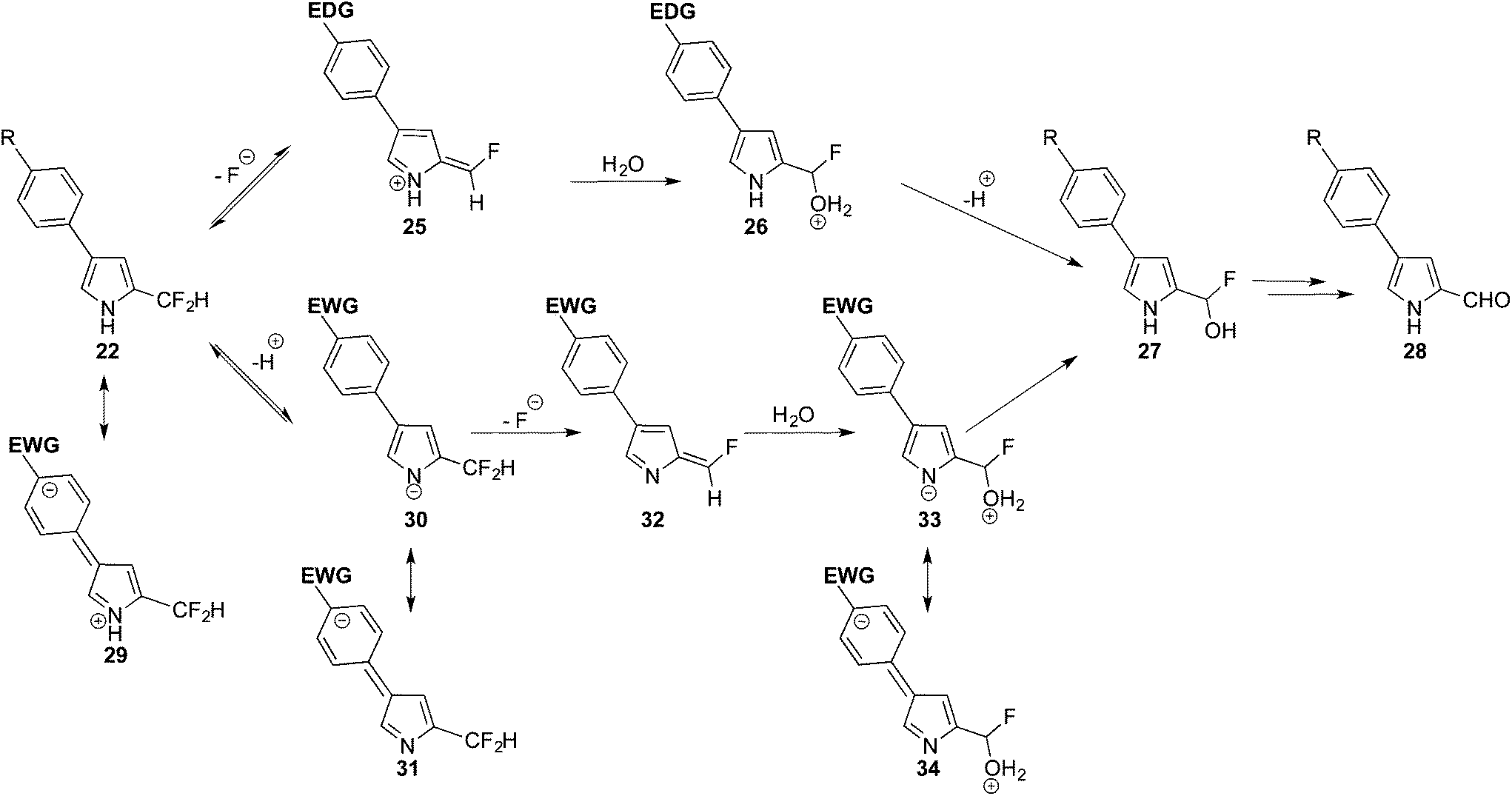
Based on these results, and the concave up parabola of the Hammet plot, two competing reaction pathways are proposed for the hydrolysis of 22 (Scheme 7). In the case of R being an electron-donating group (R = CH3 and OCH3), the rate-determining step is considered to be the loss of fluoride and formation of the positively charged azafulvenium intermediate 25, whose methylene carbon atom is activated towards the fast addition of water and the return of aromaticity to the pyrrole ring in 26.41–44 After proton loss, the intermediate 27 would collapse into the isolated aldehyde 28, again through loss of fluoride via azafulvenium formation. In the case of R being an electron-withdrawing group (R = CF3, F and CN) the generation of 25 would be slow, as appreciated upon consideration of the resonance contributor 29. In this case, loss of a proton, to form pyrrolide 30, is proposed as the rate-determining step. The transition state en route to 30 would be stabilised by electron-withdrawing substituents on the aryl ring. Loss of fluoride would then generate the azaulvene 32, followed by addition of water to give 33. Proton exchange would provide 27, which would collapse into the isolated aldehyde 28, presumably again via initial pyrrolide formation and subsequent generation of the corresponding azafulvene. Akin to the N-deprotection step, as the electron-donating and electron-withdrawing substituents (R) are somewhat distant from the developing azafulvenium and pyrrolide cores, their effects in stabilising the transition states are small and consistent with the small ρ values (Fig. 2) for the two constituent slopes of the concave up parabola.
 | ||
| Scheme 7 Proposed mechanisms for the hydrolysis of 22. | ||
To summarise, the tendency for α-difluoromethyl pyrroles to react with water is significant, particularly in the absence of significant electron-withdrawing N-protection. As such, the electronic nature of substituents about α-difluoromethyl pyrroles influences the reactivity, and hence the stability, of the α-difluoromethyl group. Driven by azafulvenium formation, the loss of fluoride is facile,41 yet a significant electron-donating character on the pyrrolic ring serves to make azafulvenium character (via loss of fluoride) less favourable. In these cases, hydrolysis is proposed to rely upon N-deprotonation, followed by azafulvene formation via loss of fluoride. Therefore, the design of air-stable difluoromethyl pyrroles must consider the incorporation of units with significant pyrrole stabilising ability,42–44 and/or electron-withdrawing capacity via N-protection, in order to avoid hydrolysis.
Experimental
General information
All 1H and 13C NMR spectra were obtained using a 500 MHz NMR spectrometer (operating at 500 MHz and 125 MHz, respectively) and CDCl3 as solvent. Chemical shifts were recorded in parts per million (ppm) with internal reference to CDCl3 (1H NMR at 7.26 ppm, 13C NMR at 77.16 ppm) and to (CD3)2CO (1H NMR at 2.05 ppm, 13C NMR at 29.84 and 206.26 ppm). 19F NMR spectra were obtained using a 500 MHz NMR instrument (operating at 470 MHz) using trifluoroacetic acid (TFA, 19F NMR at −76.55 ppm vrs. CFCl3). Splitting patterns are indicated as follows: a, apparent, br, broad; s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Coupling constants (J) are reported in units of Hertz (Hz). High and low resolution ESI+ mass spectra were recorded using ion trap instruments. Column chromatography was performed using 230–400 mesh ultrapure silica. TLC was performed using silica gel or neutral aluminum oxide plates and were visualised using UV light (254 nm) and/or developed with vanillin stain. UV analysis was carried out with the baseline manually corrected for the solvent used. Moisture-sensitive reactions were performed in flame-dried glassware under a positive pressure of nitrogen. Solutions of air- and moisture-sensitive compounds were introduced via syringe or cannula through a rubber septum. Compounds 4,459,46 and 1547 were prepared according to literature procedures.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O/MeOH (1 mL). The reaction mixture was heated to 50 °C until TLC analysis showed complete consumption of the starting material. The reaction mixture was diluted with H2O (50 mL) and acidified to pH 4 by use of 2 M aq. HCl at 0 °C. The reaction mixture was then extracted with CH2Cl2 (3 × 50 mL), and the combined organic fractions were washed with brine, dried over anhydrous Na2SO4, concentrated in vacuo and purified via column chromatography (50% EtOAc/hexanes). The title compound was isolated as a white solid (0.012 g, 27% yield). 1H (300 MHz, C(O)(CD3)2): δ 10.81 (br s, 1H), 9.70 (s, 1H), 7.07 (as, 1H), 3.85 (s, 2H), 2.78 (t, J = 8.0 Hz, expected 2H but overlaps with water peak), 2.59 (t, J = 8.0 Hz, 2H) ppm; HRMS-ESI (m/z): [M + Na]+ Calcd 248.0529 for C10H11NNaO5; found 248.0528. 13C NMR is not reported because of the instability of 8 in CDCl3, C(O)(CD3)2, and CD3OD for prolonged periods of time and its insolubility in THF-d8 and CD3CN.
:1 H2O/MeOH (1 mL). The reaction mixture was heated to 50 °C until TLC analysis showed complete consumption of the starting material. The reaction mixture was diluted with H2O (50 mL) and acidified to pH 4 by use of 2 M aq. HCl at 0 °C. The reaction mixture was then extracted with CH2Cl2 (3 × 50 mL), and the combined organic fractions were washed with brine, dried over anhydrous Na2SO4, concentrated in vacuo and purified via column chromatography (50% EtOAc/hexanes). The title compound was isolated as a white solid (0.012 g, 27% yield). 1H (300 MHz, C(O)(CD3)2): δ 10.81 (br s, 1H), 9.70 (s, 1H), 7.07 (as, 1H), 3.85 (s, 2H), 2.78 (t, J = 8.0 Hz, expected 2H but overlaps with water peak), 2.59 (t, J = 8.0 Hz, 2H) ppm; HRMS-ESI (m/z): [M + Na]+ Calcd 248.0529 for C10H11NNaO5; found 248.0528. 13C NMR is not reported because of the instability of 8 in CDCl3, C(O)(CD3)2, and CD3OD for prolonged periods of time and its insolubility in THF-d8 and CD3CN.
General procedure (GP1) for fluorination using XtalFluor®
XtalFluor-E® (3 equiv.) or XtalFluor-M® (3 equiv.) was added, as indicated, to a solution of NEt3·3HF (4 equiv.) and NEt3 (2 equiv.) in 1,2-DCE [0.3 M], with stirring at room temperature under N2 atmosphere at 70 °C. The 2-formylpyrrole substrate (1 equiv.) was then added in one portion and the reaction mixture was stirred under N2 at room temperature overnight, before quenching with sat. aqueous NaHCO3 (10 mL) and then stirring for 15 minutes. The reaction mixture was then diluted with CH2Cl2 (30 mL) and washed with H2O (30 mL) and brine (30 mL). The organic phase was dried over anhydrous Na2SO4, concentrated in vacuo and purified via column chromatography.General procedure (GP2) for the Suzuki cross-coupling of 4-bromopyrroles
4-Bromo-1-(2-nitrophenylsulfonyl)-pyrrole-2-carbaldehyde (20) (1 equiv.) was dissolved in toluene [0.1 M], along with a boronic acid a–e (1.2 equiv.) and Pd(PPh3)4 (0.1 equiv.) under N2 atmosphere with stirring. K2CO3 (4 equiv.) was prepared as an aqueous solution [1.1 M], and then purged with N2 for 15 minutes. The K2CO3 aqueous solution was then added to the stirring reaction mixture, which was subsequently heated with stirring at 90 °C overnight. The reaction mixture was then partitioned and the aqueous layer was extracted with CH2Cl2 (equivalent volume to aqueous layer × 3). The combined organic layers were dried over Na2SO4, filtered, concentrated in vacuo and then purified via column chromatography.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Nova Scotia Health Research Foundation (NSHRF). The authors thank Professor Norman P. Schepp (Dalhousie University) for assistance as regards to determining rates of reaction.Notes and references
- T. D. Lash, U. N. Mani, M. A. Drinan, C. Zhen, T. Hall and M. A. Jones, J. Org. Chem., 1999, 64, 464 CrossRef CAS
.
- T. D. Lash, U. N. Mani, E. A. Lyons, P. Thientanavanich and M. A. Jones, J. Org. Chem., 1999, 64, 478 CrossRef CAS
- J. Bachmann and D. G. Nocera, J. Am. Chem. Soc., 2005, 127, 4730 CrossRef CAS PubMed
- C. M. Benton and C. K. Lim, Biomed. Chromatogr., 2012, 26, 1009 CrossRef CAS PubMed
- C. Floriani, Chem. Commun., 1996, 1257 RSC
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1886, 19, 2184 CrossRef
- P. A. Gale, Coord. Chem. Rev., 2001, 213, 79 CrossRef CAS
- P. A. Gale, P. Anzenbacher Jr. and J. L. Sessler, Coord. Chem. Rev., 2001, 222, 57 CrossRef CAS
- P. A. Gale, Acc. Chem. Res., 2011, 44, 216 CrossRef CAS PubMed
- B. Mokhtari and K. Pourabdollah, J. Inclusion Phenom. Macrocyclic Chem., 2013, 77, 23 CrossRef CAS
- S. K. Kim and J. L. Sessler, Acc. Chem. Res., 2014, 47, 2525 CrossRef CAS PubMed
- D. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 532 RSC
- J. Labuta, J. P. Hill, S. Ishihara, L. Hanyková and K. Ariga, Acc. Chem. Res., 2015, 48, 521 CrossRef CAS PubMed
- I. Saha, J. T. Lee and C.-H. Lee, Eur. J. Org. Chem., 2015, 3859 CrossRef CAS
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC
- J. Jaunzems and M. Braun, Org. Process Res. Dev., 2014, 18, 1055 CrossRef CAS
- D. O′Hagan, J. Flourine Chem., 2010, 131, 1071 CrossRef
- M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442 CrossRef CAS PubMed
- Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494 CrossRef CAS PubMed
- X. Lin and Z. Weng, Org. Biomol. Chem., 2015, 13, 3432 CAS
- V. V. Levin, A. L. Trifonov, A. A. Zemtsov, M. I. Struchkova, D. E. Arkhipov and A. D. Dilman, Org. Lett., 2014, 16, 6256 CrossRef CAS PubMed
- N. O. Ilchenko, B. O. A. Tasch and K. J. Szabó, Angew. Chem., Int. Ed., 2014, 53, 12897 CrossRef CAS PubMed
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed
- J. Wang, N. J. Stolowich and A. I. Scott, Tetrahedron Lett., 1995, 36, 2389 CrossRef CAS
- I. S. Kondratov, V. G. Dolovanyuk, N. A. Tolmachova, I. I. Gerus, K. Bergander, R. Frohlich and G. Haufe, Org. Biomol. Chem., 2012, 10, 8778 CAS
- B. A. Merrill and E. LeGoff, J. Org. Chem., 1990, 55, 2904 CrossRef CAS
- T. Fukuyama, C.-K. Jow and M. Cheung, Tetrahedron Lett., 1995, 36, 6373 CrossRef CAS
- A. Thompson, R. J. Butler, M. N. Grundy, A. B. E. Laltoo, K. N. Robertson and T. S. Cameron, J. Org. Chem., 2005, 70, 3753 CrossRef CAS PubMed
- X. Jia, L. Yu, J. Liu, Q. Xu, M. Sickert, L. Chena and M. Lautens, Green Chem., 2014, 16, 3444 RSC
- W. Fu, T. Liu, Z. Fang, Y. Ma, X. Zheng, W. Wang, X. Ni, M. Hu and T. Tang, Chem. Commun., 2015, 51, 5890 RSC
- P. Guan, C. Cao, Y. Liu, Y. Li, P. He, Q. Chen, G. Liu and Y. Shi, Tetrahedron Lett., 2012, 53, 5987 CrossRef CAS
- J. R. Garabatos-Perera, B. H. Rotstein and A. Thompson, J. Org. Chem., 2007, 72, 7382 CrossRef CAS PubMed
- T. Castanheiro, M. Gulea, M. Donnard and J. Suffert, Eur. J. Org. Chem., 2014, 7814 CrossRef CAS
- D. V. Jawale, E. Gravel, V. Geertsen, H. Li, N. Shah, I. N. N. Namboothiri and E. Doris, ChemCatChem, 2014, 6, 719 CrossRef CAS
- R. L. Mauldin III, T. Berndt, M. Sipila, P. Paasonen, T. Petaja, S. Kim, T. Kurten, F. Stratmann, V. M. Kerminen and M. Kulmala, Nature, 2012, 488, 193 CrossRef PubMed
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS
- F. G. Bordwell and D. L. Hughes, J. Org. Chem., 1982, 47, 3224 CrossRef CAS
- I. M. Kolthoff, M. K. J. Chantooni and S. Bhowmik, J. Am. Chem. Soc., 1968, 90, 23 CrossRef CAS
- M. H. Junttila and O. E. O. Hormi, J. Org. Chem., 2007, 72, 2956 CrossRef CAS PubMed
- J. L. Hofstra, B. R. Grassbaugh, Q. M. Tran, N. R. Armada and H. J. P. de Lijser, J. Org. Chem., 2015, 80, 256 CrossRef CAS PubMed
- M. Bergeron, T. Johnson and J. F. Paquin, Angew. Chem., Int. Ed., 2011, 50, 11112 CrossRef CAS PubMed
- A. Thompson, S. Gao, G. Modzelewska, D. S. Hughes, B. Patrick and D. Dolphin, Org. Lett., 2000, 2, 3587 CrossRef CAS PubMed
- J.-R. Schauder, S. Jendrezejewski, L. W. Neidhart, J. G. Hart and R. A. Battersby, J. Chem. Soc., Perkin Trans. 1, 1999, 2691 RSC
- A. D. Abell, B. K. Nabbs and A. R. Battersby, J. Am. Chem Soc., 1998, 120, 1741 CrossRef CAS
- A. R. Battersby, C. J. R. Fookes, M. J. Meegan, E. McDonald and H. K. W. Wurziger, J. Chem. Soc., Perkin Trans. 1, 1981, 2786 RSC
- W. Hinz, R. A. Jones and T. Anderson, Synthesis, 1986, 620 CrossRef CAS
- K. Paulvannan, J. Org. Chem., 2004, 69, 1207 CrossRef CAS PubMed
- Y. Goriya and C. V. Ramana, Tetrahedron, 2010, 66, 7642 CrossRef CAS
- M. Krayer, T. Balasubramanian, C. Ruzié, M. Ptaszek, D. L. Cramer, M. Taniguchi and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2009, 13, 1098 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Data regarding rates of reaction; copies of NMR spectra. See DOI: 10.1039/c6ob01441k |
| This journal is © The Royal Society of Chemistry 2017 |